

Abstract
Background and Purpose
Imatinib mesylate (IM) is a first‐line treatment for chronic myeloid leukaemia (CML) as a specific inhibitor of BCR‐ABL tyrosine kinase. As IM is widely used in CML, in combination with other drugs, the effects of IM on drug‐metabolizing enzymes (DMEs) are crucial to the design of rational drug administration. Carboxylesterases (CESs) are enzymes catalysing the hydrolytic biotransformation of several clinically useful drugs. Although IM is known to inhibit cytochromes P450 (CYPs), its effects on DMEs, and CESs in particular, are still largely undefined.
Experimental Approach
Hepatoma cell lines (HepG2 and Huh7) and primary mouse hepatocytes were used. mRNA and protein expression were evaluated by quantitative RT‐PCR and Western blot analysis. Reporter luciferase activity was determined by transient co‐transfection experiment. Pregnane X receptor (PXR) expression was regulated by overexpression and RNA interference. The activity of CESs was determined by enzymic and toxicological assays. Mice were treated with a range of doses of IM to analyse expression of CESs in mouse liver.
Key Results
The expression and activity of CESs were markedly repressed by IM, along with the down‐regulation of PXR and inhibited expression and activity of CYP3A4 and P‐gp.
Conclusions and Implications
Down‐regulation of PXR mediates IM‐induced suppression of CESs. IM may inhibit expression of other genes targeted by PXR, thus inducing a wide range of potential drug–drug interactions during treatment of CML. The data deserve further elucidation including clinical trials.
Abbreviations
- CES1
human carboxylesterase 1
- CES2
human carboxylesterase 2
- CESs
carboxylesterases
- CML
chronic myeloid leukaemia
- CYP3A4
cytochrome P450 3A4
- DDIs
drug–drug interactions
- DMEs
drug‐metabolizing enzymes
- IM
imatinib mesylate
- P‐gp
P‐glycoprotein
- PXR
pregnane X receptor
Tables of Links
| TARGETS | |
|---|---|
| Nuclear hormone receptors a | Enzymes c |
| Pregnane X receptor | ABL tyrosine kinase |
| Transporters b | CES1, carboxylesterase 1 |
| P‐glycoprotein, MDR1 | CYP3A4 |
| LIGANDS | |
|---|---|
| Clopidogrel | Imatinib |
| Irinotecan | Rhodamine 123 |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a,b,c).
Introduction
Chronic myeloid leukaemia (CML), characterised by the Philadelphia (Ph) chromosome and the Bcr‐Abl fusion gene, is a common haematological malignancy, comprising about 15% of all adult leukemias (Jabbour and Kantarjian, 2014). Imatinib mesylate (IM) is now considered as the first‐line treatment for CML because of its specific inhibition of the BCR‐ABL tyrosine kinase (TKI) (Savage and Antman, 2002; Zhang et al., 2013). IM is also approved for the treatment of gastrointestinal stromal tumours and, in combination with other anti‐cancer drugs, to treat other malignant tumours such as Ph+ acute lymphoblastic leukaemia (Nadal and Olavarria, 2004). It is notable that adverse drug reactions such as thrombocytopenia, nausea and vomiting often occur in CML patients after IM treatment (Kantarjian et al., 2012; Kekale et al., 2015). In addition, a combination of drugs is the most frequent solution to combat drug resistance resulting from IM administration (Cortes et al., 2007; Wang and Li, 2015). Consequently, in clinical practice, IM is used along with other drugs to reduce adverse drug reactions and to overcome resistance. However, the effects of IM on the drug‐metabolizing enzymes (DMEs) and their underlying mechanisms have not been adequately analysed. Such analysis is essential to devise rational drug use, in the context of the clinical use of IM‐based combination therapies.
The DMEs mostly found in the liver play a crucial role in the biotransformation, inactivation and clearance of xenobiotics. The carboxylesterases (CESs, EC.3.1.1.1) are members of the phase I DMEs and hydrolyze substrates containing groups of esters, amides, thioesters and/or carbamates (Zhang et al., 2015a). In humans, there are two well‐characterized CESs. Carboxylesterase 1 (CES1) has a substrate specificity for ester substrates with relatively large acyl moiety and small terminal hydroxyl groups, such as imidapril, cocaine and clopidogrel. Human CES2, in contrast, has a more flexible active site for small acyl groups and large terminal hydroxyl group, as found in irinotecan and heroin (Tsurkan et al., 2013; Zhang et al., 2015a). In mice, Ces1d strongly cross‐reacts with human CES1, while Ces1e resembles human CES2 (Xiao et al., 2012). In the present context, it is highly relevant that about 20% of all therapeutic agents are substrates for the hydrolytic activity of the CESs (Laizure et al., 2013). In addition, the pharmaceutical industry frequently uses ester groups to improve water solubility of clinical leads (Imai et al., 2003). The effectiveness and safety of these agents are also highly likely to be affected by the actions of the CESs.
IM is known to be a potent inhibitor of cytochromeP450 3A4 (CYP3A4), one of the most important DMEs (Filppula et al., 2012). However, the mechanisms underlying this inhibitory action of IM have not been fully elucidated. The most important factor affecting the biotransformation of drugs by the liver is the expression of the DMEs (Parkinson and Ogilvie, 2001; Poso and Honkakoski, 2006). Consequently, the activities of the nuclear receptors, which are known to control transcription of the DMEs, will play essential roles in determining DME activity . The pregnane X receptor (PXR, NR1I2), one of the most important nuclear receptors, is crucial for the biotransformation of many therapeutic agents. When PXR binds to a wide range of structurally different compounds, including drugs (Chen et al., 2012), it translocates to the nucleus and binds to the promoter regions to initiate transcription of its target genes (Buler et al., 2011; Chen et al., 2012). The representative target genes of PXR are the DMEs, such as CESs (Yang and Yan, 2007b) and the CYP3A family in particular, and drug transporters such as P‐glycoprotein (P‐gp), encoded by MDR1 (Chen et al., 2012; Niemira et al., 2013).
On the basis of these results and because of the important contribution of the CESs to the metabolism of clinical drugs, we have focused on the effects of IM on the CESs, aiming to provide data relevant to rational drug use and to the alleviation of adverse drug reactions, in the treatment of CML. Here, we have demonstrated that IM suppressed the expression and the hydrolytic activity of CESs. As PXR is involved in the transcriptional regulation of both CYP3A4 and CESs (Rathod et al., 2014; Yang and Yan, 2007b), we further explored whether PXR mediated IM‐induced suppression of CESs. Our findings contribute to the better understanding of drug–drug interactions (DDIs) in the treatment of CML.
Methods
Animals
All animal care and experimental protocols were approved by the IACUC (Institutional Animal Care and Use Committee) of Nanjing Medical University. Efforts were taken to minimize suffering of the animals. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Male ICR mice (7 weeks old, ~20 g body weight) were obtained from the experimental animal centre of Nanjing (Nanjing, China) and housed in a room under controlled conditions (ambient temperature, 22°C; humidity, 40%) in a 12 h light/dark cycle. After acclimatization for 1 week, the mice were divided into three groups (six mice in each group) at random. Mice were treated with IM (70 or 140 mg·kg−1 body weight) in saline or saline only (control group) injected i.p. every morning for 3 days. Twenty‐four hours after the last injecton, mice were anaesthetized using urethane (2 g·kg−1 body weight). Then, surgery was performed to expose the livers of mice. The livers were perfused with saline through the portal vein and frozen at −80°C for the preparation of S9 fractions.
Group sizes
In order to explore the effects and mechanisms of IM on CESs expression, we performed experiments in vitro and in vivo. In vitro, all experiments were repeated independently at least five times to ensure scientific rigour. For the in vivo study, 18 mice were randomized into three groups: (i) The IM high‐dose group (n = 6) with IM injected i.p. at a dose of 140 mg·kg−1·day−1; (ii) the IM low‐dose group (n = 6) with IM injected i.p.at a dose of 70 mg·kg−1·day−1; and (iii) the control group (n = 6) with the same volume of saline injected. Mice were treated for three successive days.
Validity of animal model
Even though there is divergence in the toxicological responses between humans and mice, mice are still frequently used in investigating altered expression of DMEs and drug transporters (Mao et al., 2011; Xiong et al., 2014a; Zhang et al., 2015a). As it is ethically unacceptable to take liver samples from CML patients treated with IM; we have chosen, in the present study, to use mice to investigate the effect of IM on CESs in the liver.
Randomization
The random number table was used to perform the randomization according to the weights of mice. All mice were weighed and ordered according to their weights from light to heavy over the range of 18–22 g. Then, the lightest mouse was given a number chosen from the random number table. As three groups were needed, we divided the number by 3. If the number could be divided by 3, the mouse was put into the first group, if not, but with remainder 1, the mouse was allocated to the second group. Similarly, remainder 2 meant allocation to the third group. For the second lightest mouse, the number in the random number table on the right of the prior one was given. This method of selection was used until all mice were grouped.
Blinding
Data were collected and analysed by two observers who were not aware of the design and operation of experiments.
Cell culture
The human hepatoma cell lines (HepG2 and Huh7) were purchased from the cell bank of Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences. Cells were cultured in DMEM supplemented with 10% FBS, 100 U·mL−1 penicillin and 100 U·mL−1 streptomycin in a humidified environment with 5% CO2 at 37°C. Mouse hepatocytes were isolated from the livers of male ICR mice using a two‐step perfusion method with some modifications as described previously (Feng et al., 2012).
Cell viability assay
Cell viability was determined by MTT assay. Cells were treated with indicated concentrations of IM for 24 h, then added 20 μL·5 mg·mL−1 MTT, and incubated at 37°C for 4 h. Following, the culture medium was discarded and DMSO was added into each wall to dissolve the formazan crystals. The absorbance was then evaluated at 570 nm.
Preparing the S9 fractions of the mice livers
The frozen livers were homogenized with a Wharton stirrer after being thawed in the homogenization buffer (50 mM Tris–HCl, pH 7.4, 150 mM KCl and 2 mM EDTA). Then, the homogenates were centrifuged at 10000 g for 20 min at 4°C to obtain the S9 fractions, which were assayed for the protein levels of Ces1d and Ces1e.
Quantitative reverse transcription‐polymerase chain reaction
Total RNA was isolated using TRIzol (Invitrogen) and applied to synthesize the first‐strand cDNA at 25°C for 10 min, 42°C for 60 min and 70°C for 15 min with random hexamers and M‐MLV reverse transcriptase (Promega, Madison, WI, USA). The quantitative PCR was performed by the EzOmics SYBR qPCR Kit (Biomics, Jiangsu, China) using a step one real‐time PCR system (Applied Biosystems, Foster City, CA). The Primer sequences were as follows:
GAPDH, Forward 5′‐AAGGTCGGAGTCACCGGATT‐3′, Reverse 5′‐CTGGAAGATGGTGATGGGATT‐3′;
CES1, Forward 5′‐CCAGAGAGAGTCAACCCCTTCT‐3′, Reverse 5′‐TCCTGCTTGTTAATTCCGACC‐3′;
CES2, Forward 5′‐ACCGCAGTGGAGTCAGAGTTTC‐3′, Reverse 5′‐ATGCTGAGGTACAGGCAGTCCT‐3′;
PXR, Forward 5′‐GGCAATCCCAGGTTCTCTTT‐3′, Reverse 5′‐ATGCTTTATGGCAGGTGAGG‐3′;
CYP3A4, Forward 5′‐TTCAGCAAGAAGAACAAGGACAA‐3′, Reverse 5′‐GGTTGAAGAAGTCCTCCTAAGC‐3′;
MDR1, Forward 5′‐GAGGCCAACATACATGCCTTC‐3′, Reverse 5′‐GTCTAACAAGGGCACGAGCTAT‐3′;
Ces1d, Forward 5′‐GAGACCCAAGGCAGTAATAGGA‐3′, Reverse 5′‐GAGTTGAGGCACCAATCTTCA‐3′;
-
Ces1e, Forward 5′‐CCAGTGACAGGGCAAATAGTC‐3′, Reverse 5′‐TCATGCGTAGACAGGACCAGT‐3′.
Gene expression was calculated by 2‐ΔΔCt method, and the values were normalized to GAPDH.
Western blot analysis
The cell lysates (40 μg) or S9 fractions (100 μg) of mouse liver were resolvedon a 8% SDS‐polyacrylamide gel and transferred electrophoretically onto PVDF membranes (Millipore). The membranes were then blocked with 5–10% non‐fat milk for 2 h and incubated with primary antibodies at 4°C overnight against CES1 (1:1000), CES2 (1:1000), CYP3A4 (1:1000), MDR1 (1:3000), PXR (1:3000), Ces1d (1:1000) or Ces1e (1:1000). The immune complexes were then incubated with horseradish peroxidase‐conjugated secondary antibody for 1 h and visualized with an enhanced chemiluminescence detection system.
Transient co‐transfection experiment
The CES2 promoter reporter (−1931/+6) was prepared with the pGL3 basic vector (Wu et al., 2003). The CES1 promoter reporter (−9133/−40) was constructed as previously described (Yang et al., 2007a). Briefly, two fragments (−5155 to −40 and −9224 to −5049) were amplified by PCR. And the sequence (−5155 to −5049) overlapped by these two fragments contained an EcoRV site. Then the whole sequence was cloned into the pGL3 basic vector through MluI and BamHI sites. The promoter sequences of constructs were verified by DNA sequencing. HepG2 cells were plated in 24‐well plates in DMEM with 10% FBS at the density of 7 × 104 cells per well. The transfection mixtures contained 0.64 μg of a reporter plasmid along with 0.16 μg of null‐Renilla reniformis luciferase plasmid. Cells were transfected using lipofectamine 2000 (Invitrogen) and incubated at 37°C for 24 h. The transfected cells were treated with 2 μM IM or the same volume of DMEM (1% FBS) for another 24 h. The luciferase activity was evaluated with Dual‐Luciferase Reporter Assay System, and the firefly luminescence intensity was normalized based on the intensity of Renilla luminescence.
Overall hydrolytic activity assay
The overall hydrolytic activity was determined by using a standard substrate, p‐nitrophenylacetate. HepG2 cells were seeded in 6‐well plates at the density of 3 × 105 cells per well for 24 h and then exposed to IM (0.5, 1 or 2 μM) or same volume of DMEM (1% FBS) for another 24 h. The cells were precipitated and resuspended in 100 μL of 100 mM potassium phosphate buffer (pH 7.4). Then, the cells were sonicated and the cell debris was precipitated by centrifugation at 12 000 g for 15 min at 4°C. Samples of the supernatants were incubated with the substrate and hydrolysis analysed by spectrophotometry, as described previously (Yang and Yan, 2007b).
Cytotoxicity assay
HepG2 cells were plated into 96‐well plates (3000 cells per well) and cultured overnight. Then cells were treated with IM (0 or 2 μM) for another 24 h, washed with PBS once and exposed to clopidogrel (0, 1, 10 or 100 μM) or irinotecan (0, 0.3, 3 or 30 μM). After incubation for 24 h, the medium was replaced with 0.5 mg·mL−1 MTT in fresh DMEM. The OD was determined at 570 nm. Morphological changes were observed by microscopy, before the MTT assay.
Assay of irinotecan and its metabolites
Cells were plated in 12‐well plates at 105 cells per well, incubated for 24 h, then treated with or without IM (2 μM) for another 24 h. After one wash with PBS, the cells were incubated with 30 μM irinotecan for 24 h. The cell pellets were then dissolved in a mixture of methanol‐acetonitrile (1:1 vol:vol; 700 μL), mixed for 30 s by full‐speed vortex and then centrifuged at 12 000 g for 3 min. About 550 μL of the clear supernatant were mixed well with HCl (250 μL; 1 mol·L−1) and used for HPLC assay. Irinotecan and its metabolites SN‐38 were detected and quantified by an HPLC method as previously described (Poujol et al., 2003).
Assay for clopidogrel and its carboxylic acid metabolite (SR26334)
Cells were placed in 12‐well plates at 105 cells per well, incubated for 24 h, then treated with or without IM (2 μM) for another 24 h. After one wash with PBS, the cells were treated with 100 μM clopidogrel for 24 h. Then cell pellets were dissolved and vortex mixed with 1 mL diethyl ether for 3 min and centrifuged at 12000 g for 5 min. The upper organic layer was evaporated to dryness under a stream of nitrogen. The dried residue was dissolved in 300 μL mobile phase, vortex mixed for 1 min and centrifuged at 12000 g for 3 min. Clopidogrel and SR26334 were simultaneously determined using a sensitive LC–MS‐MS method as previously described (Zou et al., 2009).
Assay of oxidative activity of CYP3A4
HepG2 cells were placed into 96‐well plates in DMEM with 10% FBS overnight and exposed to IM (0, 0.5, 1 or 2 μM) for 24 h. Then cells were treated using P450‐Glo™ Luminescent cytochrome P450 3A4 Assay System. Briefly, after cells were washed carefully, a mixture of DMEM and Luciferin‐IPA was added to each well. After incubation for 60 min, 25 μL medium from each well and 25 μL luciferin detection reagent were added to a 96‐well opaque luminometer plate and incubated for 10 min in the dark. The luminescence signal was determined by the spectral scanning multimode reader (Thermo Scientific, Waltham, MA).
Assay for intracellular accumulation of rhodamine 123
The efflux activity of P‐gp was determined by intracellular accumulation of a known P‐gp substrate, rhodamine 123 (Rho123)(Sheu et al., 2014). After the treatment, cells were placed with 5 μg·mL−1 Rho123 in DMEM (10% FBS) to incubate at 37°C, 5% CO2 for 30 min in the dark. Cells were then washed twice with PBS and analysed immediately by flow cytometry (BD FACSCalibur with Cellquest software) or observed under a laser confocal microscope, as described earlier (Xiong et al., 2014a).
Immunofluorescence analysis
To assay the expression of PXR, cells were seeded at 2 × 105 cells per well on glass‐bottom dishes and treated with indicated concentration of IM. At the end of incubation, cells were fixed for 30 min, permeabilized in 0.1% TritonX‐100 for 30 min and blocked with 5% BSA for 1 h. Afterwards, cells were incubated with anti‐PXR primary antibody at 4°C overnight, followed by incubation with FITC‐conjugated secondary antibody (Bioworld, Atlanta, Georgia, USA) in the dark for 1 h. Nuclei were stained with DAPI (Bioworld, St. Louis Park, USA) for 10 min, and cells were examined with laser confocal microscopy.
Regulation of PXR expression by overexpression and RNA interference
The expression vector encoding PXR was constructed by fusing the pFlag‐CMV‐2 vector to the elements of PXR from +1579 to +3997 as described previously (Zhang et al., 1999). The sequences of constructs were verified by DNA sequencing. For the PXR overexpression experiment, HepG2 cells were seeded in 6‐well plates and transfected using X‐tremeGENE 9 DNA transfection reagent (Roche) with 2 μg PXR construct or corresponding vector, which did not encode PXR protein. After 24 h incubation, cells were exposed to 2 μM IM for another 24 h. Cells were then harvested, and the protein was extracted. For the RNA interference experiment, cells were transfected with 50 nM siRNA‐PXR (CACAGAGUUUAUAGUUAAAAA) or control siRNA by using lipofectamine 2000 (Invitrogen). After 24 h incubation, cells were further treated with 2 μM IM for another 24 h. Then the cells were collected for extracting the protein.
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The experimental results were expressed as the mean ± SEM. The following data were normalized: the absorbance in MTT assay and hydrolytic activity assay, quantitative analysis of gene expression by Western blot and qRT‐PCR. For Western blot and qRT‐PCR, all data were adjusted by the values of internal standard (GAPDH or β‐actin). The control mean in the control group was calculated first and then all values were normalized to that mean value of the control group. Appropriate statistical analysis was conducted on these normalized values.
The differences between two groups were analysed using a two‐tailed Student's t‐test. Differences between more than two groups were analysed by one‐way ANOVA, followed by Dunnett's post hoc test. The post hoc tests were run only if an overall statistically significant difference exists in group means, and there was no significant variance in homogeneity. P < 0.05 was considered statistically significant.
Materials
DMEM and FBS were from Gibco (Gaithersburg, MD, USA). IM (purity > 99%) was purchased from Sigma (USA), dissolved in double‐distilled water to 50 mM and stored at −20°C. The concentrations used in the study were 0.5 ~ 8 μM, freshly diluted with DMEM supplemented with 1% FBS (Gibco, Carlsbad, CA) to final concentrations. Irinotecan and p‐nitrophenylacetate were also purchased from Sigma. MTT was from SunShine Biotechnology (Nanjing, China). Dual‐Luciferase Reporter Assay System and P450‐Glo™ Luminescent cytochrome P450 3A4 Assay System were from Promega (Madison, WI, USA). Antibodies used in the present study were as follows: CES1, CES2 and CYP3A4 (Proteintech Technology, Wuhan, China); MDR1 and PXR (Abcam, Cambridge, UK); GAPDH and β‐actin (Bioworld, St. Louis Park, USA); Ces1d (Abcam, Cambridge, UK); and Ces1e (Abgent, San Diego, CA, USA). The goat anti‐rabbit IgG and the goat anti‐mouse IgG conjugated with horseradish peroxidase were from Pierce Chemical (Pierce, Rockford, IL, USA).
Results
IM represses CES1 and CES2 expression in human hepatoma cells
In the beginning, the effects of IM on the expression of CESs were investigated by using human hepatoma HepG2 cells and Huh7 cells. Cells were treated with different concentrations of IM for 24 h. Total RNA and cell lysates were prepared to determine the expression of human CES1 and CES2. At those concentrations that did not affect cell viability (Figure 1A, D), IM significantly reduced the mRNA levels of human CES1 and CES2 in HepG2 and Huh7 cells in a concentration‐dependent manner (Figure 1B, E). Consistent with the decrease in mRNA, the protein level of CESs was also significantly reduced in a concentration‐dependent manner in HepG2 cells and Huh7 cells (Figure 1C, F). As the greatest effect of IM on the expression of CESs was observed at 2 μM, without any obvious cytotoxicity for the HepG2 cells (Figure 1A), we chose this concentration in the following experiments using HepG2 cells.
Figure 1.
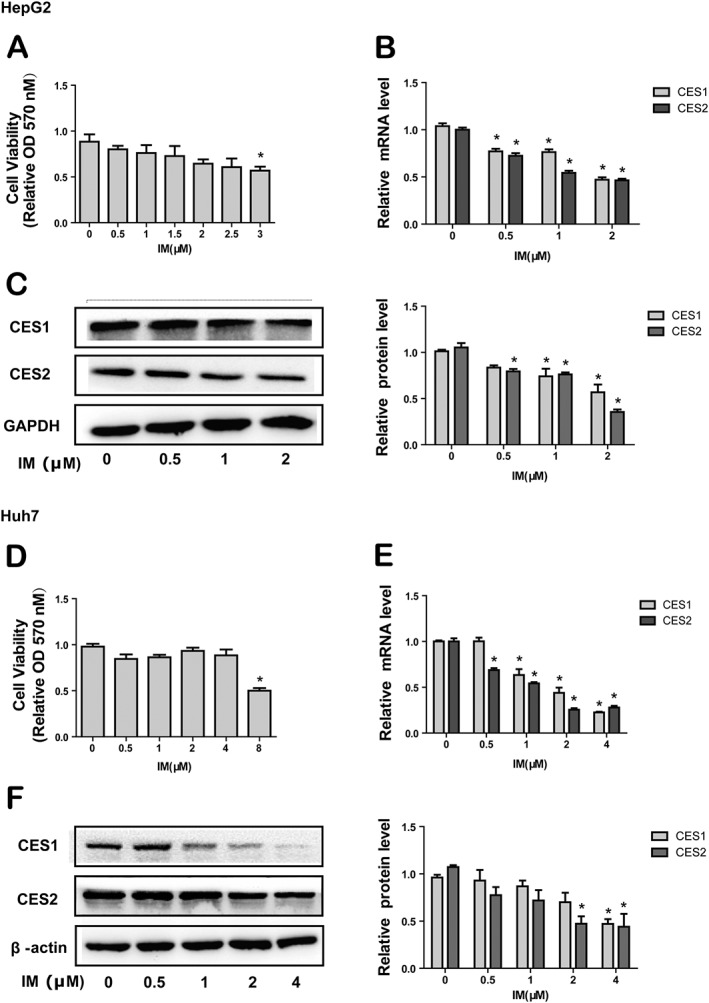
Effect of IM on the expression of CESs in human hepatoma HepG2 cells and Huh7 cells. (1) The cytotoxicity of IM in HepG2 cells (A) and Huh7 cells (D). Hepatocytes were treated with indicated concentrations of IM for 24 h, and MTT assay was used to measure cell viability. (2) The effect of IM on the CES1 and CES2 mRNA expression in HepG2 cells (B) and Huh7 cells (E). Hepatocytes were treated with indicated concentrations of IM for 24 h. Total RNA was isolated and subjected to qRT‐PCR analysis. The signals from each target were normalized to the signal from GAPDH. (3) The effect of IM on the CES1 and CES2 protein expression in HepG2 cells (C) and Huh7 cells (F). Hepatocytes were exposed to indicated concentrations of IM for 24 h, and the expression of CESs was evaluated by Western blot. Data are expressed as mean ± SEM (n = 5). *P < 0.05, significantly different from control.
IM suppresses the hydrolytic activity of CESs in HepG2 cells
We next assessed whether the reduced expression of CES1 and CES2 translated into decreased hydrolytic activity. After treatment of HepG2 cells with a range of concentrations of IM for 24 h, the overall hydrolytic activity in cell lysates was analysed using the standard substrate p‐nitrophenylacetate, which is known to be hydrolyzed by both CES1 and CES2 (Mao et al., 2011). Consistent with the decrease of CES1 and CES2 expression, the hydrolysis of p‐nitrophenylacetate was reduced after treatment with IM, in a concentration‐dependent manner (Figure 2A).
Figure 2.
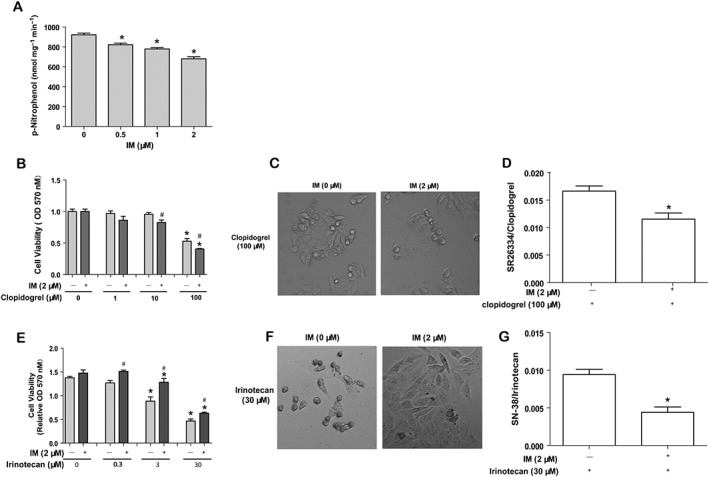
IM suppresses the hydrolytic activity of CESs in HepG2 cells. (1) IM decreases the overall hydrolytic activity of CESs, using the standard substrate p‐nitrophenylacetate (A). HepG2 cells were plated and then exposed to the various concentrations of IM. Cell lysates were prepared and assayed for the overall hydrolytic activity. (2) The effect of IM on the cellular responses to two other ester drugs, clopidogrel (B) and irinotecan (E). HepG2 cells were seeded into 96‐well plates and treated with or without IM (2 μM) for another 24 h, washed with PBS once and treated with clopidogrel (0, 1, 10 or 100 μM) or irinotecan (0, 0.3, 3 or 30 μM) for 24 h. MTT was added to each well for 2 h at 37°C. The OD value was determined at 570 nm. Data are expressed as mean ± SEM (n = 5). *P < 0.05, significantly different from control group (without clopidogrel or irinotecan); #P < 0.05, significantly different from non‐IM‐pretreated group. (3) Morphological changes in HepG2 cells pretreated with IM and exposed to clopidogrel (C) or irinotecan (F). The cells were seeded into 96‐well plates and were treated with or without IM (2 μM) for another 24 h. Then the cells were washed with PBS once and treated with clopidogrel 100 μM or irinotecan 30 μM for another 24 h. The images were taken under bright field (200×) with microscope. (4) The changes in the intracellular ratios of SR26334/clopidogrel or SN‐38/irinotecan after pretreatment of HepG2 cells with IM and exposure to clopidogrel (D) or irinotecan (G). The cells were seeded in 12‐well plates and incubated for 24 h, then treated with or without IM (2 μM) for another 24 h. The cells were washed with PBS once and treated with clopidogrel (100 μM) or irinotecan (30 μM) for 24 h. Cell pellets were collected for analysis. Data are expressed as mean ± SEM (n = 5). *P < 0.05, significantly different from control.
As the hydrolysis of p‐nitrophenylacetate assays the non‐specific, overall hydrolytic activity of all hepatic esterases, including CESs, specific changes in the hydrolytic activity of CES1 and CES2 in cells treated with IM was then evaluated using two carboxylesterase‐specific substrates, clopidogrel and irinotecan. Clopidogrel is specifically hydrolyzed by CES1 to form a less toxic product, whereas irinotecan is hydrolyzed by CES2 to form a more toxic product SN‐38 (Wu et al., 2002; Shi et al., 2006). Thus, we expected that the cytotoxicity of clopidogrel would increase, whereas the cytotoxicity of irinotecan would decrease after pretreatment of the cells with IM. HepG2 cells were first treated with IM (2 μM) for 24 h and then incubated with clopidogrel or irinotecan for 24 h at various concentrations. Treatment with clopidogrel (1 ~ 100 μM) or irinotecan alone (0.3 ~ 30 μM) decreased viability in HepG2 cells (Figure 2B, E). Pretreatment with IM increased the cytotoxicity of clopidogrel, while decreasing the cytotoxicity of irinotecan (Figure 2B, E). These data suggested that IM also suppressed the hydrolytic activity of CES1 and CES2.
Morphological changes were observed before the MTT assay for viability. As shown in Figure 2C, cells pretreated with IM were round and isolated while the cells without IM pretreatment were normal and spread, when exposed to 100 μM clopidogrel. Conversely, when exposed to 30 μM irinotecan, cells without IM pretreatment were isolated and rounded‐up, whereas cells treated with IM were normal and spread (Figure 2F). These morphological change were consistent with the cell viability assay, further confirming that CESs activity was functionally decreased by pretreatment with IM.
We also assayed the intracellular concentrations of clopidogrel and irinotecan, as well as their metabolites SR26334 and SN‐38, in the cells pretreated with IM (2 μM) followed by clopidogrel (100 μM) or irinotecan (30 μM) for 24 h. As expected, the intracellular ratios of SR26334/clopidogrel and SN‐38/irinotecan were decreased when the cells were pretreated with IM (2 μM) (Figure 2D, G). These data also support the inhibiting effects of IM on the hydrolytic activity of CESs.
IM suppresses the expression of CES1 and CES2 at the transcriptional level
HepG2 cells, plated in 6‐well plates, were treated with IM (2 μM) for the times shown and the expression of CES1 and CES2 mRNA and protein was determined by qRT‐PCR and Western blot. Treatment with IM induced a time‐dependent and marked inhibition of the mRNA and protein levels of CES1 and CES2 (Figure 3A, B). Suppression of the mRNA for CES1 and CES2 was significant 6 ~ 12 h after the treatment with IM, whereas expression of CES1 and CES2 protein was inhibited from 12 ~ 24 h of incubation with IM. In order to explore the mechanisms underlying the decrease of CES1 and CES2 mRNA expression by IM, we next investigated whether IM affected the promoters of the expression of CES1 and CES2. Co‐transfection experiments with a CES1 or CES2 promoter luciferase plasmid, showed that treatment with IM (2 μM) resulted in reduced luciferase activity of CES1 and CES2 reporters by nearly 40% (Figure 3C), confirming that IM could suppress the expression of CES1 and CES2 at the transcriptional level.
Figure 3.
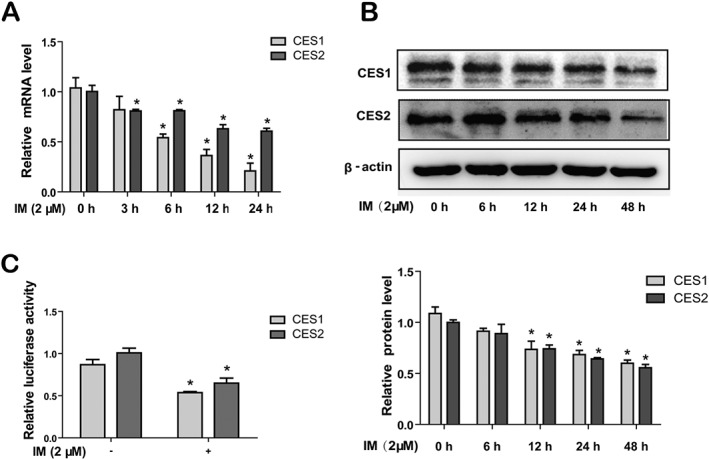
IM suppresses the expression of CES1 and CES2 at transcriptional level. (A) HepG2 cells were treated with 2 μM IM for 0, 3, 6, 12 or 24 h, and the mRNA expression of CES1 and CES2 were evaluated by qRT‐PCR. The signals from each target were normalized based on the signal from GAPDH. (B) After the treatment of 2 μM IM for 0, 6, 12, 24 or 48 h in HepG2 cells, the cells lysates were prepared and then analysed by Western blot. (C) Repression of CES1 and CES2 promoter by IM. HepG2 cells were transfected with CES1 promoter reporter (0.64 μg) or CES2 promoter reporter (0.64 μg) along with 0.16 μg of null‐R. reniformis luciferase plasmid. The transfected cells were treated with 2 μM IM. The luciferase activity was evaluated with Dual‐Luciferase Reporter Assay System. The reporter activity was normalized to that of the null‐R. reniformis luminescence signal. Data are expressed as mean ± SEM (n = 5). *P < 0.05, significantly different from control.
IM suppresses the expression and activity of PXR in HepG2 cells
In order to investigate whether PXR was involved in the suppression of CESs by IM, we determined levels of PXR in HepG2 cells after treatment with IM in different concentrations for 24 h. As shown in Figure 4, the expression of PXR was suppressed by IM concentration‐dependently, in terms of both mRNA (Figure 4A) and protein (Figure 4B). The maximal effect of IM was observed at 2 μM and when the cells were exposed to this concentration for 0, 6, 12, 24 or 48 h, the PXR protein expression was decreased in a time‐dependent manner (Figure 4C). The decrease of PXR protein was first noted at 6 h (Figure 4C), and the suppression of mRNA for CES1 and CES2 was first noted at 12 h after pretreatment with IM (Figure 2A). The data suggested that IM inhibited the expression of CESs by suppressing the expression and activity of PXR. Immunofluorescence analysis was used to verify down‐regulation of PXR by IM. As shown in Figure 4D, PXR was mainly expressed in the nuclei of HepG2 cells, and its fluorescence intensity was reduced after incubation with 2 μM IM for 24 h.
Figure 4.
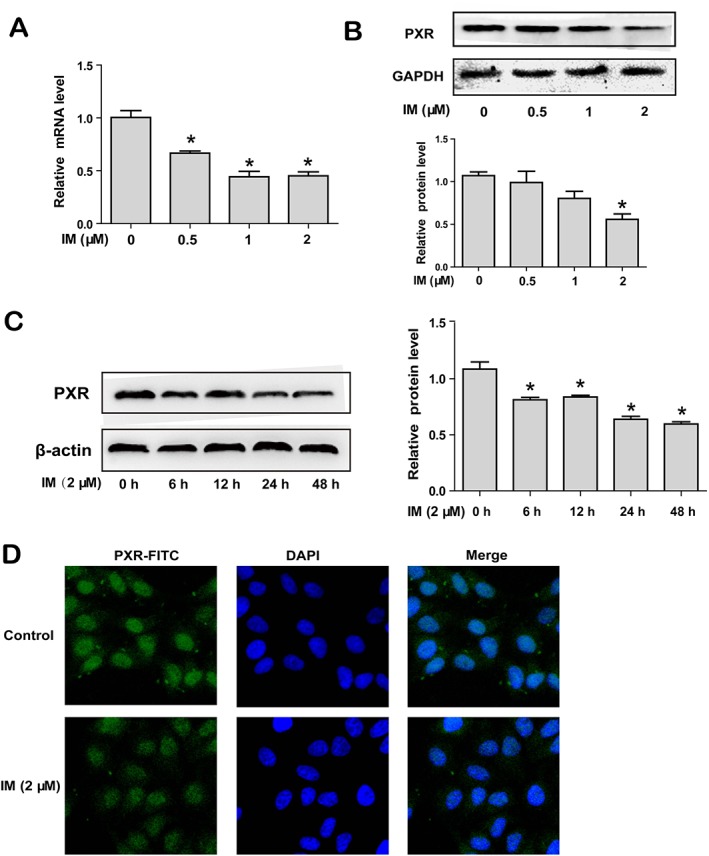
IM down‐regulates the expression of PXR. (A–B) HepG2 cells were treated with IM (0, 0.5, 1 or 2 μM) for 24 h. PXR mRNA levels were investigated by qRT‐PCR (A), and protein expression was evaluated by Western blot (B). (C) HepG2 cells were treated with 2 μM IM for 0, 6, 12, 24 or 48 h, and PXR protein levels were investigated by Western blot. (D) HepG2 cells were treated with or without 2 μM IM for 24 h, and fluorescence intensity of PXR was detected by immunofluorescence analysis (400×). Data are expressed as mean ± SEM (n = 5). *P < 0.05, significantly different from control.
CYP3A4 and P‐gp are classical target genes of PXR (Chen et al., 2012). In order to further explore the mechanism of reduced expression of CESs by IM, we measured expression of CYP3A4 and P‐gp after the HepG2 cells had been exposed to IM. In HepG2 cells treated with IM in different concentrations for 24 h, the expression of CYP3A4 was significantly decreased in a concentration‐ and time‐dependent manner (Figure 5A–C). Measurements of the oxidative activity of CYP3A4 (Figure 5D) also showed a marked decrease in IM‐treated cells, comparable with the decrease of CYP3A4 expression. Effects of IM on P‐gp were similar, with repression of both mRNA level and protein level in the cells treated with IM (Figure 6A–C). Moreover, the efflux activity of P‐gp was analysed by Rho123 intracellular accumulation assay with flow cytometry as well as laser confocal microscopy. In the cells pretreated with IM, intracellular Rho123 accumulation was increased, compared with control cells (Figure 6D, E), suggesting that the efflux activity of P‐gp was significantly reduced. These data suggested that the expression and activity of PXR was repressed by IM.
Figure 5.
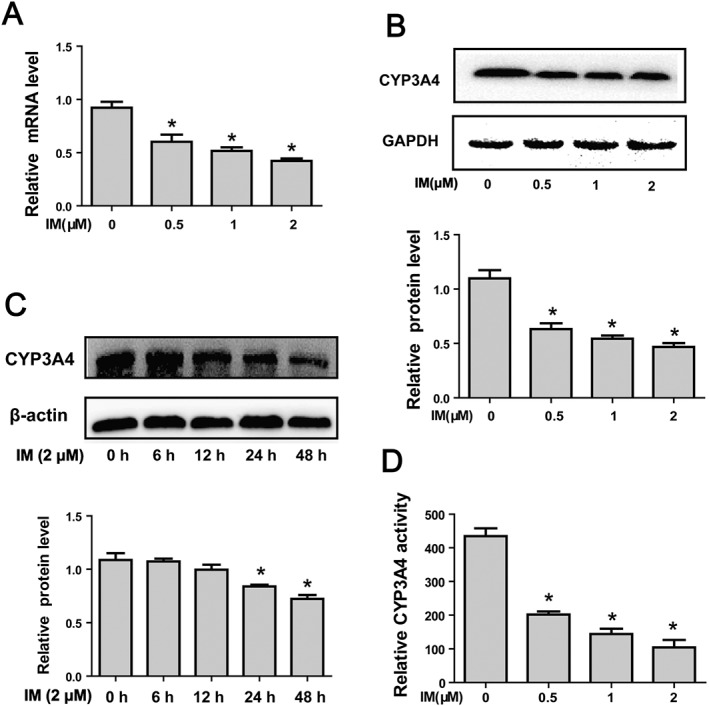
IM represses the expression and oxidative activity of CYP3A4. (A–B) IM represses the expression of CYP3A4 in a concentration‐dependent manner at mRNA levels (A) and at protein levels (B). HepG2 cells were treated with IM (0, 0.5, 1 or 2 μM) for 24 h. CYP3A4 mRNA levels were investigated by qRT‐PCR, and protein levels were determined by Western blot. (C) IM represses the expression of CYP3A4 in a time‐dependent manner on protein level. HepG2 cells were treated with 2 μM IM for 0, 6, 12, 24 or 48 h, and the cells lysates were subjected to Western blot. (D) IM represses the oxidative activity of CYP3A4. HepG2 cells were treated with 0, 0.5, 1 or 2 μM IM for 24 h, and the cells were prepared and assayed for CYP3A4 activity. Data are expressed as mean ± SEM (n = 5). *P < 0.05, significantly different from control.
Figure 6.
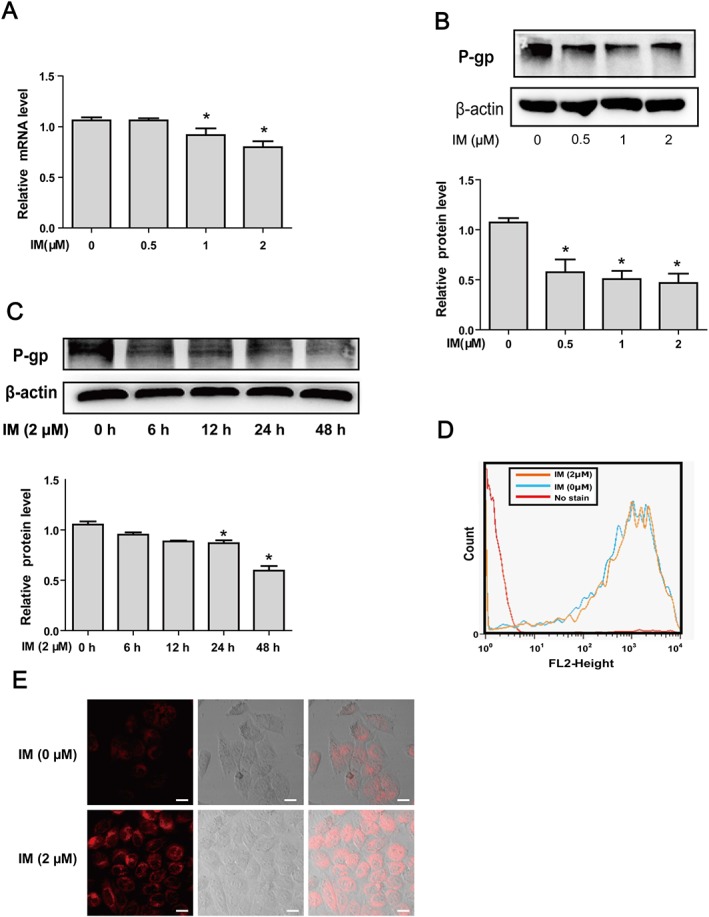
IM suppresses the expression and efflux activity of P‐gp. (A–B) IM down‐regulates the expression of P‐gp in a concentration‐dependent manner at mRNA (A) and protein levels (B). HepG2 cells were treated with IM (0, 0.5, 1 or 2 μM) for 24 h. P‐gp mRNA was analysed by qRT‐PCR, and protein levels were determined by Western blot. (C) IM down‐regulates the expression of P‐gp protein in a time‐dependent manner. HepG2 cells were treated with 2 μM IM for 0, 6, 12, 24 or 48 h, and the cells lysates were analysed by Western blot. (D–E) IM represses the efflux activity of P‐gp. HepG2 cells were treated with or without 2 μM IM for 24 h, and the cells were incubated with DMEM (10% FBS) supplemented with 5 μg·mL−1 Rho123 for 30 min. The efflux activity of P‐gp was analysed immediately on a flow cytometry (D) or observed under a laser confocal microscope (E). Data are expressed as mean ± SEM (n = 5). *P < 0.05, significant effects of IM treatment.
PXR is required for IM‐induced repression of CES1 and CES2
To further confirm the role of PXR in the IM‐induced repression of CES1 and CES2, we performed overexpression and knockdown experiments to selectively modulate the expression of PXR. In the overexpression experiment, HepG2 cells were transfected with the PXR construct or the corresponding vector for 24 h and then treated with 2 μM IM or same volume of DMEM (1% FBS) for another 24 h. The efficiency of transfection was confirmed by analysing the expression of PXR by Western blot (Figure 7A, D). As shown, CES1 and CES2 were decreased by IM in the cells transferred with corresponding vector (Figure 7A, B). Overexpression of PXR increased the basal expression of CES1 and CES2 (Figure 7A, B) and the IM‐induced suppression of CES1 and CES2 was no longer present in the cells transfected with PXR construct. For the knockdown experiment, a siRNA for PXR was used, and the efficiency of gene knockdown was confirmed by analysing expression of PXR by Western blot (Figure 7E, H). The data were consistent with the previous results in that CES1 and CES2 were repressed by IM in the cells transfected with control siRNA (Figure 7E, F). This inhibition of CES1 and CES2 by IM was comparable with the decrease of CESs in the cells transfected with siRNA‐PXR (Figure 7E, F). We also assayed the two other PXR target genes, CYP3A4 and P‐gp, as a positive control (Figure 7A, C, E and G), further confirming the role of PXR in the suppression of CESs by IM. The data confirmed that PXR was essential in the reduced expression of CES1 and CES2 caused by IM.
Figure 7.
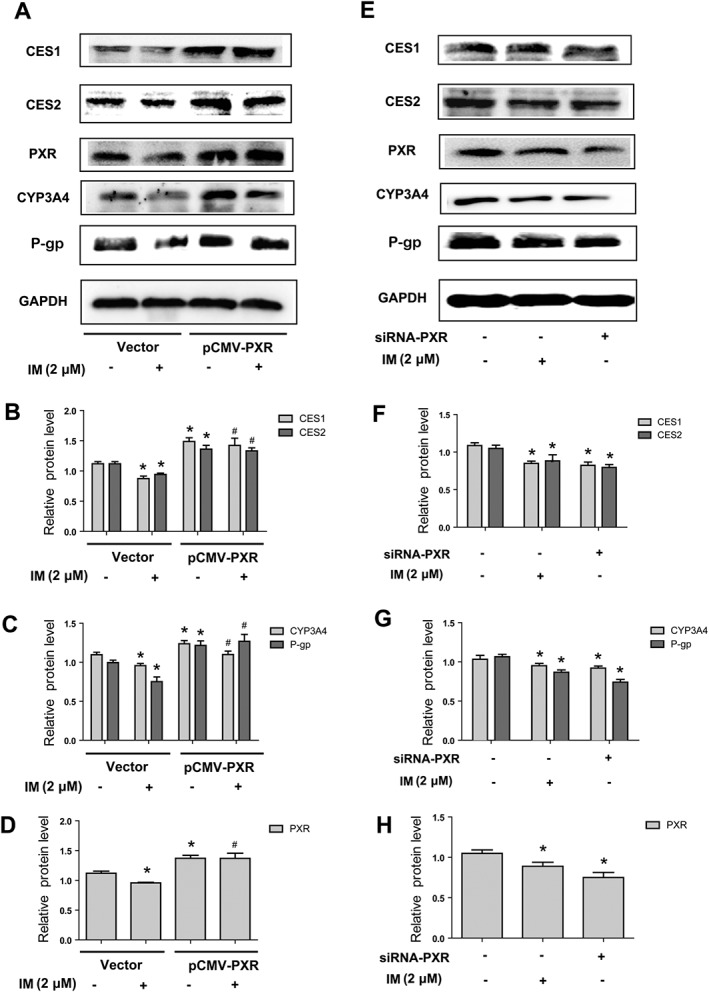
The role of PXR in the IM‐induced suppression of CES1 and CES2 expression. (A–D) The effect of PXR overexpression on the suppression of CES1 and CES2 by IM. HepG2 cells were transfected with PXR construct or the corresponding vector (pFlag‐CMV‐2) for 24 h and then treated with or without 2 μM IM for 24 h. Cell lysates were prepared to analyse the expression of CES1, CES2, PXR, CYP3A4 and P‐gp by Western blot respectively. (E–H) The effect of PXR knockdown on the expression of CES1 and CES2, compared with the effect of IM. HepG2 cells were transfected with siRNA‐PXR or the siRNA‐control for 24 h and then treated with or without 2 μM IM for another 24 h. Cell lysates were prepared and subjected to Western blot to determine the expression of CES1, CES2, PXR, CYP3A4 and P‐gp respectively. Data are expressed as mean ± SEM (n = 5). *P < 0.05, significantly different from control group (vector or siRNA‐control transfected and non‐IM pretreated); #P < 0.05, significantly different from vector‐transfected cells.
IM suppresses the expression of CESs in primary mouse hepatocytes in vitro and mouse liver in vivo
In mouse, Ces1d and Ces1e are close homologues of human CES1 and CES2 respectively (Xiao et al., 2012). We therefore measured the effect of IM on expression of Ces1d and Ces1e in primary mouse hepatocytes cultured and treated with IM in different concentrations. First, the effect of IM on the cell viability of primary mouse hepatocytes was assessed to determine the concentrations that did not affect cell viability (Figure 8A). As shown in Figure 8B, C, the mRNA and protein level of Ces1d and Ces1e were significantly decreased in a concentration‐dependent manner by IM, consistent with the findings in human hepatoma cells.
Figure 8.
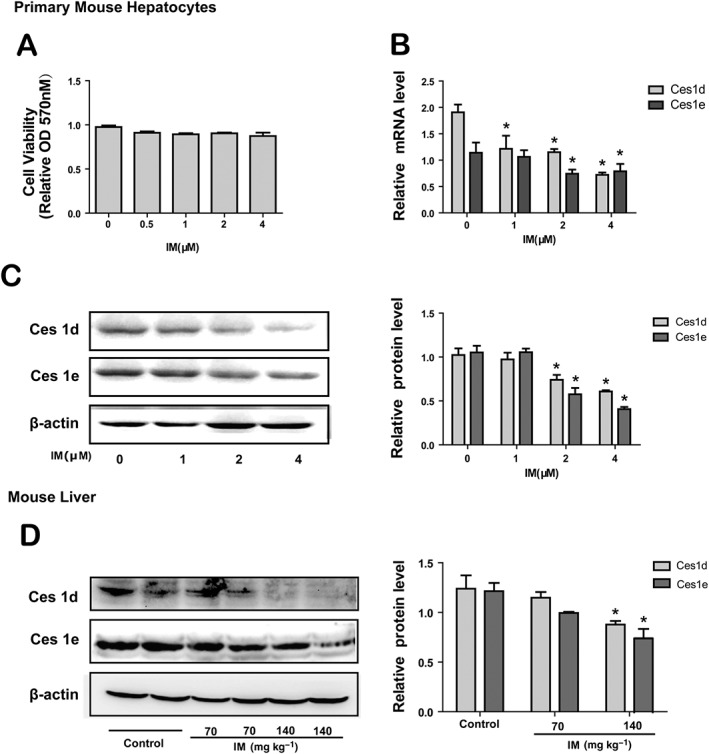
IM down‐regulates the expression of CESs in primary mouse hepatocytes in vitro and mouse liver in vivo. (A) The cytotoxicity of IM in primary mouse hepatocytes. Cells were treated with IM (0, 0.5, 1, 2 or 4 μM) for 24 h, and MTT assay was used to measure cell viability. (B–C) The effect of IM on the Ces1d and Ces1e mRNA expression (B) and protein expression (C) in primary mouse hepatocytes. Cells were treated with indicated concentrations of IM for 24 h. Total RNA was isolated and subjected to qRT‐PCR analysis. The cells lysates were evaluated by Western blot. Data are expressed as mean ± SEM (n = 5). *P < 0.05, significantly different from control group. (D) The effect of IM on the Ces1d and Ces1e protein expression in the mouse liver. Mice in three groups were treated with IM (70 or 140 mg·kg−1) in saline or saline only (control group) intraperitoneally for 3 days. 24 h after last administration, mice were injected with urethane (2 g·kg−1 body weight) for anaesthesia. The liver was perfused with PBS (37°C) through the portal vein to remove blood. The perfused liver was used for preparing S9 fractions for Western analyses (n = 6). The data are expressed as mean ± SEM. *P < 0.05, significantly different from control.
We next studied the regulation of Ces1d and Ces1e expression in mouse liver by IM, in vivo. Male mice were injected i.p. with IM (70 or 140 mg·kg−1) in saline (0.01 mL·g−1 body weight) or the same volume of saline only (control group) daily for 3 days. The doses used in the in vivo experiment were calculated based on the doses for human (7 mg·kg−1·day−1). Mice were killed, and the livers were collected after perfusion. The liver was used for preparing S9 fractions to analyse the expression of Ces1d and Ces1e protein. As shown in Figure 8D, IM decreased the expression of Ces1d and Ces1e, dose‐dependently. IM reduced the Ces1d and Ces1e protein expression by about 40% in high doses (140 mg·kg−1). The results provided evidence that IM suppressed CESs expression in vitro and could do so in vivo.
Discussion
Apart from being the first‐line treatment for CML, IM also has extended clinical applications for the treatment of many other malignant tumours such as gastrointestinal stromal tumours and Ph+ acute lymphoblastic leukaemia (Savage and Antman, 2002). However, the frequent adverse drug reactions and the development of drug resistance require drug combinations with IM as a constant component (Bixby and Talpaz, 2011). Therefore, better understanding of the effects of IM on the expression and activity of DMEs is of considerable clinical importance to IM‐based treatments.
IM is known to increase the concentration of the CYP3A4/5 substrate simvastatin in plasma and is a potent inhibitor of CYP3A4 (Filppula et al., 2012). As simvastatin is also a substrate of CES1 (Laizure et al., 2013), it is possible that inhibition of CESs by IM is another possible cause of the increased plasma concentration of simvastatin. Because the CESs are a major DME , we have focused on these enzymes in the present study of IM‐related DDIs. Human hepatoma HepG2 cells have been used here, as in previous studies of the regulation of DMEs (Wang et al., 2011; Cui et al., 2014; Hu et al., 2014). We found that IM inhibited the expression of human CES1 and CES2 in a concentration‐ and time‐dependent manner (Figures 1A–C 3A, B) in HepG2 cells and also in Huh7 cells (Figure 1D–F). Moreover, IM decreased the expression of mouse Ces1d and Ces1e, which are homologues of human CES1 and CES2, in mouse primary hepatocytes (Figure 8A–C). Our in vivo experiments in mice (Figure 8D) showed that IM also decreased expression of Ces1d and Ces1e in mouse liver. It is one of the major limitations of our present study that human primary hepatocytes, a more relevant in vitro model of human liver biology, were not used. However, since the application of human hepatoma cells is an acceptable alternative to study the regulation of DMEs and the results we obtained using human hepatoma cells are comparable with an earlier study using human liver microsomes (Filppula et al., 2012), our findings are of relevance to the effects of IM‐based drug combinations.
The decreased expression of CESs was translated into decreased hydrolytic activity of CESs, demonstrating the functional outcome of the decreased expression of CESs by IM (Figure 2A–G). Our experiments with two other ester drugs, irinotecan and clopidogrel, showed a reduced cytotoxicity of irinotecan and a increased cytotoxicity of clopidogrel when the cells were pretreated with IM, confirming that CES1 and CES2 enzymic activity was significantly decreased by IM. The data from these drug combination experiments, indicate that DDIs are highly likely when the expression and activity of CESs is decreased by IM. Therefore, DDIs should be considered when IM is administered together with other drugs which are substrates for the CESs.
Treatment with IM decreased the mRNA for CES1 and CES2 (Figure 3A), suggesting two possibilities: (1) IM represses the transcription and/or (2) increases the degradation of mRNA. We used reporter assays to decide between these possibilities and found that the transcriptional activity of CES1 and CES2 promoters was markedly repressed by about 40% by IM (Figure 3C). One of the most important factors affecting CESs gene transcription, is transactivation by nuclear receptors such as the PXR (Rathod et al., 2014). As a crucial transcription factor, PXR controls the regulation of major DMEs, such as CYP2B6, CYP3A4/5 and CYP3A7 (Chen et al., 2012; Rathod et al., 2014). From our experiments, PXR was also involved in the effects of IM on the transcription of CES1 and CES2, as both the mRNA and protein expression of PXR were markedly suppressed by IM in a concentration‐ and time‐dependent manner (Figure 4A–C). This suppression of PXR was also demonstrated by the decreased expression and activity of CYP3A4 (Figure 5A–D) and P‐gp (Figure 6A–E), two well‐established target genes of PXR. It was also highly relevant that the decrease of PXR protein first appeared at 6 h (Figure 4C), and the mRNA suppression of CES1 and CES2 was first apparent at 6 ~ 12 h, after treatment with IM (Figure 3A). These results suggested that IM inhibited the expression of CESs by suppressing the expression and activity of PXR. The crucial role of PXR in the IM‐induced repression of CES1 and CES2 was confirmed by the overexpression and knockdown of PXR. All these data show that PXR is required for IM‐induced repression of CES1 and CES2. Consistent with this proposal, two other compounds, the photochemotherapeutic agent, 8‐methoxypsoralen and the anti‐depressant fluoxetine also modulate the expression of CESs by regulating PXR (Yang and Yan, 2007b; Xiong et al., 2014b; Shang et al., 2016). In primary mouse hepatocytes, the altered expression of CESs is also positively related to PXR as demonstrated before (Xiong et al., 2014a). Taken together, the data show that PXR plays an important role in the activation of CES1 and CES2 gene transcription; although the underlying mechanisms still require further study.
Recently, clinical trials have indicated that 89% of patients exposed to IM show potential DDIs with many drugs, especially paracetamol (77.4% of all DDIs) (Recoche et al., 2016). The combination of paracetamol with IM increased the plasma concentration and toxicity of paracetamol, a substrate of UDP‐glucuronosyltransferase (UGT), which is a well‐known target gene of PXR (Lee et al., 2015). Even though direct inhibition of IM on the activity of UGT has been demonstrated using human liver microsomes and recombinant proteins (Liu et al., 2011; Zhang et al., 2015b), the increase of paracetamol levels and toxicity suggest both inhibited activity and suppressed expression of UGT by IM. However, none of the earlier studies were designed to investigate the mechanisms of this DDI. Our present findings could provide a novel mechanistic explanation of IM‐induced DDIs by demonstrating PXR‐mediated suppression of DMEs and drug transporters by IM (Figures 4, 5, 6). The risk of the DDIs caused by drug combination with IM may change the curative effect and induce adverse reactions and therefore deserve serious attention. Prescribers should be aware of possible DDIs in patients treated with IM and dose adjustment has to be considered when using IM together with other drugs, especially those which are substrates for CESs.
According to clinical data, IM is well‐tolerated and can be given orally with bioavailability of 98%, in the treatment of CML (Martins et al., 2011). This treatment with IM achieves a complete haematological remission rate of above 95% and a major cytogenetic response rate above 80% (Singh et al., 2009). The standard dose of IM is 400 mg·day−1 for patients with chronic‐phase CML and 600 mg·day−1 for accelerated phase or blast crisis of CML (Martins et al., 2011). The efficacy of the threshold plasma trough concentration (C0) of IM must be set above 1000 ng·mL−1 (1.7 μM) to achieve optimized activity (Martins et al., 2011). Plasma levels of IM ranged from 181 to 2947 ng·mL−1 (0.3 ~ 5 μM), with a mean of 1058 ± 557 ng·mL−1 (about 1.8 μM) for 400 mg·day−1 and 1447 ± 710 ng·mL−1 (about 2.5 μM) for 600 mg·day−1 from a French study (Picard et al., 2007). Besides, the mean steady‐state IM C0 is reached 24 h after taking a 400 mg daily dose, ranged from 900 to 1400 ng·mL−1 (1.5 ~ 2.4 μM) (Takahashi and Miura, 2011). Currently, the total plasma concentration of IM is monitored becuase of its large inter‐individual pharmacokinetic variability with consistent concentration–efficacy and concentration–toxicity relationships (Picard et al., 2007). At relevant clinical concentrations, about 95% of IM binds to α1 acid glycoprotein (Widmer et al., 2008). However, higher intracellular accumulation of IM is observed in patients, as intracellular concentrations of IM are close to total plasma concentrations (Widmer et al., 2006). In our present study, another limitation is the absence of data from clinical studies undertaken in CML patients in vivo. But our results show that 2 μM IM (comparable with the plasma concentrations of IM in CML patients) significantly suppressed the expression and activity of CES1 and CES2 in vitro (Figures 1A–F and 2A–G). The in vivo experiments further confirmed the suppression of CESs by IM in mouse liver (Figure 8D). Therefore, our findings suggest the possible suppression of CESs by IM in the livers of CML patients. The present study will help to design an effective and safe treatment regimen in CML by indicating the possible IM‐related DDIs.
As a well‐known target gene of PXR, the expression and oxidative activity of CYP3A4 were also decreased by IM (Figure 5A–D), consistent with previous studies (Filppula et al., 2012). CYP3A4 is one of the most important human CYPs in the liver, responsible for the oxidative metabolism of more than 60% of all therapeutic drugs (Chen et al., 2012). The data of the present study strongly suggest that the efficacy and toxicity of drugs metabolized by CYP3A4 would be altered significantly when they are co‐administered with IM. This suggestion is supported by the report that IM increased the Cmax and AUC of simvastatin, a known CYP3A4 substrate, by 2‐fold and 3.5‐fold respectively (O'Brien et al., 2003). We also demonstrated prolonged and potent suppression of P‐gp expression and efflux activity by IM (Figure 6A–E). IM is also known to directly inhibit the efflux activity of P‐gp in a dose‐dependent fashion after a short incubation (30 min ~ 1 h) (Hamada et al., 2003). P‐gp plays a role in drug transportation in many tissues such as liver, kidney and intestines (Demeule et al., 2002) and IM also promotes the delivery and increases tumour concentrations of the P‐gp substrate, doxorubicin (Vlahovic et al., 2007). Therefore, the inhibition by IM of the expression and activity of P‐gp could be of significance in reversing multi‐drug resistance (Dharmapuri et al., 2015). P‐gp is also involved in the transport of substances out of the CNS, through the blood–brain barrier (Demeule et al., 2002; Ebinger and Uhr, 2006; Ueno et al., 2010), so IM could possibly increase drug concentrations in the CNS by preventing efflux.
As the most prominent example of selective inhibitors of TKIs, IM has led to the development of novel TKIs in the treatment of a wide range of haematological malignancies and solid tumours, to overcome drug resistance and achieve better therapeutic effects (Haouala et al., 2009). Novel IM analogues are synthesized by constructing a number of non‐aromatic structural motifs in place of the original phenyl moiety (Nicolaou et al., 2016). In this study, we demonstrate that the PXR‐targets, DMEs and P‐gp, are suppressed by IM (Figures 1A–F, 5A–D, 6A–E). It is therefore necessary to evaluate the influence of IM on the metabolism of other drugs to avoid DDI‐induced adverse reactions, in the development of new IM analogues.
In summary, our work points to three important conclusions. First, IM induced marked decreases of CES1 and CES2 expression in vitro and in vivo, as well as suppression of hydrolytic activity. Second, suppressed PXR expression is crucially involved in the decrease of CES1 and CES2 induced by IM. Third, IM also suppresses the expression and activity of CYP3A4 and P‐gp, which are also targets of PXR (see diagram of the present study in Figure 9). The findings suggest that IM could exert a widespread suppression of the expression of many DMEs by regulating PXR and such widespread effects are of importance in predicting potential DDIs and avoiding adverse drug reactions in the clinical treatment of CML. Further evaluation, especially in clinical trials, is needed to explore the influence of IM on the pharmacokinetic and pharmacodynamic properties of drugs used in CML patients.
Figure 9.
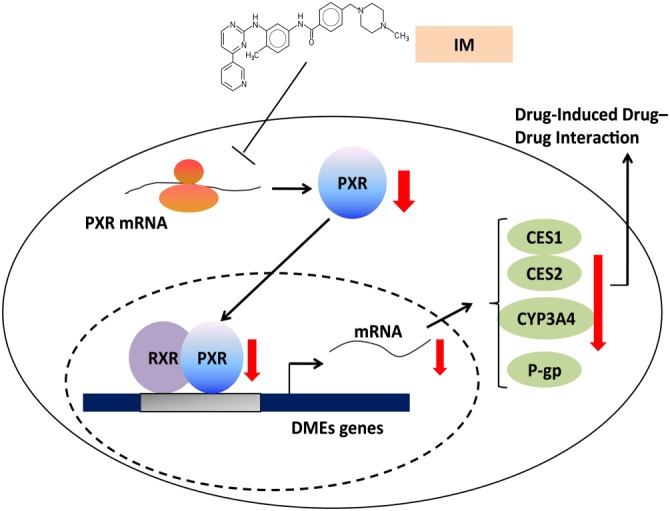
Diagram of the actions and mechanisms of IM in the present study. Imatinib inhibits the expression of PXR, leading to the suppressed expression of CESs, as well as other PXR target genes, CYP3A4 and P‐gp. The findings of the present study are highly relevant to the assessment of potential drug–drug interactions (DDI) in the clinical therapy of CML.
Author contributions
L.W., X.J., X.Y., Z.X., Z.F., L.C. and F.H. performed the research; X.J. and X.T. designed the research study; X.J. and X.T. contributed essential reagents or tools; L.W. and X.J. analysed the data; and X.J. and L.W. wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
The study was supported by the Natural Science Foundation of China (no. 81302855), the Natural Science Foundation of Jiangsu Province of China (no. BK2012446) and the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions.
Luo, W. , Xin, Y. , Zhao, X. , Zhang, F. , Liu, C. , Fan, H. , Xi, T. , and Xiong, J. (2017) Suppression of carboxylesterases by imatinib mediated by the down‐regulation of pregnane X receptor. British Journal of Pharmacology, 174: 700–717. doi: 10.1111/bph.13731.
[Correction note: The layout of the Primer sequences on page 3 and the clarity of the y‐axis of Figure 3B were corrected during issue compilation, after first publication online Early View.]
Contributor Information
Tao Xi, Email: xitao18@hotmail.com.
Jing Xiong, Email: xiong.jing@njmu.edu.cn.
References
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bixby D, Talpaz M (2011). Seeking the causes and solutions to imatinib‐resistance in chronic myeloid leukemia. Leukemia 25: 7–22. [DOI] [PubMed] [Google Scholar]
- Buler M, Aatsinki SM, Skoumal R, Hakkola J (2011). Energy sensing factors PGC‐1alpha and SIRT1 modulate PXR expression and function. Biochem Pharmacol 82: 2008–2015. [DOI] [PubMed] [Google Scholar]
- Chen Y, Tang Y, Guo C, Wang J, Boral D, Nie D (2012). Nuclear receptors in the multidrug resistance through the regulation of drug‐metabolizing enzymes and drug transporters. Biochem Pharmacol 83: 1112–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes J, Quintas‐Cardama A, Garcia‐Manero G, O'Brien S, Jones D, Faderl S et al. (2007). Phase 1 study of tipifarnib in combination with imatinib for patients with chronic myelogenous leukemia in chronic phase after imatinib failure. Cancer 110: 2000–2006. [DOI] [PubMed] [Google Scholar]
- Cui HM, Zhang QY, Wang JL, Chen JL, Zhang YL, Tong XL (2014). In vitro studies of berberine metabolism and its effect of enzyme induction on HepG2 cells. J Ethnopharmacol 158 (Pt A): 388–396. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeule M, Regina A, Jodoin J, Laplante A, Dagenais C, Berthelet F et al. (2002). Drug transport to the brain: key roles for the efflux pump P‐glycoprotein in the blood–brain barrier. Vascul Pharmacol 38: 339–348. [DOI] [PubMed] [Google Scholar]
- Dharmapuri G, Doneti R, Philip GH, Kalle AM (2015). Celecoxib sensitizes imatinib‐resistant K562 cells to imatinib by inhibiting MRP1‐5, ABCA2 and ABCG2 transporters via Wnt and Ras signaling pathways. Leuk Res 39: 696–701. [DOI] [PubMed] [Google Scholar]
- Ebinger M, Uhr M (2006). ABC drug transporter at the blood–brain barrier: effects on drug metabolism and drug response. Eur Arch Psychiatry Clin Neurosci 256: 294–298. [DOI] [PubMed] [Google Scholar]
- Feng XM, Xiong J, Qin H, Liu W, Chen RN, Shang W et al. (2012). Fluoxetine induces hepatic lipid accumulation via both promotion of the SREBP1c‐related lipogenesis and reduction of lipolysis in primary mouse hepatocytes. CNS Neurosci Ther 18: 974–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filppula AM, Laitila J, Neuvonen PJ, Backman JT (2012). Potent mechanism‐based inhibition of CYP3A4 by imatinib explains its liability to interact with CYP3A4 substrates. Br J Pharmacol 165: 2787–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada A, Miyano H, Watanabe H, Saito H (2003). Interaction of imatinib mesilate with human P‐glycoprotein. J Pharmacol Exp Ther 307: 824–828. [DOI] [PubMed] [Google Scholar]
- Haouala A, Zanolari B, Rochat B, Montemurro M, Zaman K, Duchosal MA et al. (2009). Therapeutic drug monitoring of the new targeted anticancer agents imatinib, nilotinib, dasatinib, sunitinib, sorafenib and lapatinib by LC tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 877: 1982–1996. [DOI] [PubMed] [Google Scholar]
- Hu X, Zhang J, Jiang Y, Lei Y, Lu L, Zhou J et al. (2014). Effect on metabolic enzymes and thyroid receptors induced by BDE‐47 by activation the pregnane X receptor in HepG2, a human hepatoma cell line. Toxicol In Vitro 28: 1377–1385. [DOI] [PubMed] [Google Scholar]
- Imai T, Yoshigae Y, Hosokawa M, Chiba K, Otagiri M (2003). Evidence for the involvement of a pulmonary first‐pass effect via carboxylesterase in the disposition of a propranolol ester derivative after intravenous administration. J Pharmacol Exp Ther 307: 1234–1242. [DOI] [PubMed] [Google Scholar]
- Jabbour E, Kantarjian H (2014). Chronic myeloid leukemia: 2014 update on diagnosis, monitoring, and management. Am J Hematol 89: 547–556. [DOI] [PubMed] [Google Scholar]
- Kantarjian HM, Shah NP, Cortes JE, Baccarani M, Agarwal MB, Undurraga MS et al. (2012). Dasatinib or imatinib in newly diagnosed chronic‐phase chronic myeloid leukemia: 2‐year follow‐up from a randomized phase 3 trial (DASISION). Blood 119: 1123–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kekale M, Peltoniemi M, Airaksinen M (2015). Patient‐reported adverse drug reactions and their influence on adherence and quality of life of chronic myeloid leukemia patients on per oral tyrosine kinase inhibitor treatment. Patient Prefer Adherence 9: 1733–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laizure SC, Herring V, Hu Z, Witbrodt K, Parker RB (2013). The role of human carboxylesterases in drug metabolism: have we overlooked their importance? Pharmacotherapy 33: 210–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Lee JY, Kim YM, Kim SK, Oh SJ (2015). Expression of hepatic cytochrome P450s and UDP‐glucuronosyltransferases in PXR and CAR double humanized mice treated with rifampicin. Toxicol Lett 235: 107–115. [DOI] [PubMed] [Google Scholar]
- Liu Y, Ramirez J, Ratain MJ (2011). Inhibition of paracetamol glucuronidation by tyrosine kinase inhibitors. Br J Clin Pharmacol 71: 917–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Li Y, Peng Y, Luan X, Gui H, Feng X et al. (2011). Lipopolysaccharide down‐regulates carbolesterases 1 and 2 and reduces hydrolysis activity in vitro and in vivo via p38MAPK‐NF‐kappaB pathway. Toxicol Lett 201: 213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins DH, Wagner SC, Dos Santos TV, Lizot Lde L, Antunes MV, Capra M et al. (2011). Monitoring imatinib plasma concentrations in chronic myeloid leukemia. Rev Bras Hematol Hemoter 33: 302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadal E, Olavarria E (2004). Imatinib mesylate (Gleevec/Glivec) a molecular‐targeted therapy for chronic myeloid leukaemia and other malignancies. Int J Clin Pract 58: 511–516. [DOI] [PubMed] [Google Scholar]
- Nicolaou KC, Vourloumis D, Totokotsopoulos S, Papakyriakou A, Karsunky H, Fernando H et al. (2016). Synthesis and biopharmaceutical evaluation of imatinib analogues featuring unusual structural motifs. ChemMedChem 11: 31–37. [DOI] [PubMed] [Google Scholar]
- Niemira M, Dastych J, Mazerska Z (2013). Pregnane X receptor dependent up‐regulation of CYP2C9 and CYP3A4 in tumor cells by antitumor acridine agents, C‐1748 and C‐1305, selectively diminished under hypoxia. Biochem Pharmacol 86: 231–241. [DOI] [PubMed] [Google Scholar]
- O'Brien SG, Meinhardt P, Bond E, Beck J, Peng B, Dutreix C et al. (2003). Effects of imatinib mesylate (STI571, Glivec) on the pharmacokinetics of simvastatin, a cytochrome p450 3A4 substrate, in patients with chronic myeloid leukaemia. Br J Cancer 89: 1855–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson A, Ogilvie BW (2001). Biotransformation of Xenobiotics. McGraw‐Hill Companies: New York. [Google Scholar]
- Picard S, Titier K, Etienne G, Teilhet E, Ducint D, Bernard MA et al. (2007). Trough imatinib plasma levels are associated with both cytogenetic and molecular responses to standard‐dose imatinib in chronic myeloid leukemia. Blood 109: 3496–3499. [DOI] [PubMed] [Google Scholar]
- Poso A, Honkakoski P (2006). Ligand recognition by drug‐activated nuclear receptors PXR and CAR: structural, site‐directed mutagenesis and molecular modeling studies. Mini Rev Med Chem 6: 937–947. [DOI] [PubMed] [Google Scholar]
- Poujol S, Pinguet F, Malosse F, Astre C, Ychou M, Culine S et al. (2003). Sensitive HPLC‐fluorescence method for irinotecan and four major metabolites in human plasma and saliva: application to pharmacokinetic studies. Clin Chem 49: 1900–1908. [DOI] [PubMed] [Google Scholar]
- Rathod V, Jain S, Nandekar P, Sangamwar AT (2014). Human pregnane X receptor: a novel target for anticancer drug development. Drug Discov Today 19: 63–70. [DOI] [PubMed] [Google Scholar]
- Recoche I, Rousseau V, Bourrel R, Lapeyre‐Mestre M, Chebane L, Despas F et al. (2016). Drug–drug interactions with imatinib: an observational study. Medicine 95: e5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage DG, Antman KH (2002). Imatinib mesylate – a new oral targeted therapy. N Engl J Med 346: 683–693. [DOI] [PubMed] [Google Scholar]
- Shang W, Liu J, Chen R, Ning R, Xiong J, Liu W et al. (2016). Fluoxetine reduces CES1, CES2, and CYP3A4 expression through decreasing PXR and increasing DEC1 in HepG2 cells. Xenobiotica 46: 393–405. [DOI] [PubMed] [Google Scholar]
- Sheu MJ, Teng YN, Chen YY, Hung CC (2014). The functional influences of common ABCB1 genetic variants on the inhibition of P‐glycoprotein by Antrodia cinnamomea extracts. PLoS One 9: e89622. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Shi D, Yang J, Yang D, LeCluyse EL, Black C, You L et al. (2006). Anti‐influenza prodrug oseltamivir is activated by carboxylesterase human carboxylesterase 1, and the activation is inhibited by antiplatelet agent clopidogrel. J Pharmacol Exp Ther 319: 1477–1484. [DOI] [PubMed] [Google Scholar]
- Singh N, Kumar L, Meena R, Velpandian T (2009). Drug monitoring of imatinib levels in patients undergoing therapy for chronic myeloid leukaemia: comparing plasma levels of responders and non‐responders. Eur J Clin Pharmacol 65: 545–549. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Miura M (2011). Therapeutic drug monitoring of imatinib for chronic myeloid leukemia patients in the chronic phase. Pharmacology 87: 241–248. [DOI] [PubMed] [Google Scholar]
- Tsurkan LG, Hatfield MJ, Edwards CC, Hyatt JL, Potter PM (2013). Inhibition of human carboxylesterases hCE1 and hiCE by cholinesterase inhibitors. Chem Biol Interact 203: 226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M, Nakagawa T, Wu B, Onodera M, Huang CL, Kusaka T et al. (2010). Transporters in the brain endothelial barrier. Curr Med Chem 17: 1125–1138. [DOI] [PubMed] [Google Scholar]
- Vlahovic G, Ponce AM, Rabbani Z, Salahuddin FK, Zgonjanin L, Spasojevic I et al. (2007). Treatment with imatinib improves drug delivery and efficacy in NSCLC xenografts. Br J Cancer 97: 735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ, Li YH (2015). Inhibition of human chronic myelogenous leukemia K562 cell growth following combination treatment with resveratrol and imatinib mesylate. Genet Mol Res 14: 6413–6418. [DOI] [PubMed] [Google Scholar]
- Wang Q‐L, Wu Q, Tao Y‐Y, Liu C‐H, El‐Nezami H (2011). Salvianolic acid B modulates the expression of drug‐metabolizing enzymes in HepG2 cells. Hepatobiliary Pancreat Dis Int 10: 502–508. [DOI] [PubMed] [Google Scholar]
- Widmer N, Decosterd LA, Csajka C, Leyvraz S, Duchosal MA, Rosselet A et al. (2006). Population pharmacokinetics of imatinib and the role of alpha‐acid glycoprotein. Br J Clin Pharmacol 62: 97–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmer N, Decosterd LA, Leyvraz S, Duchosal MA, Rosselet A, Debiec‐Rychter M et al. (2008). Relationship of imatinib‐free plasma levels and target genotype with efficacy and tolerability. Br J Cancer 98: 1633–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MH, Yan B, Humerickhouse R, Dolan ME (2002). Irinotecan activation by human carboxylesterases in colorectal adenocarcinoma cells. Clin Cancer Res 8: 2696–2700. [PubMed] [Google Scholar]
- Wu MH, Chen P, Remo BF, Cook EH Jr, Das S, Dolan ME (2003). Characterization of multiple promoters in the human carboxylesterase 2 gene. Pharmacogenetics 13: 425–435. [DOI] [PubMed] [Google Scholar]
- Xiao D, Chen YT, Yang D, Yan B (2012). Age‐related inducibility of carboxylesterases by the antiepileptic agent phenobarbital and implications in drug metabolism and lipid accumulation. Biochem Pharmacol 84: 232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J, Shang W, Wu L, Chen R, Liu W, Ning R et al. (2014a). Glucose dominates the regulation of carboxylesterases induced by lipopolysaccharide or interleukin‐6 in primary mouse hepatocytes. Life Sci 112: 41–48. [DOI] [PubMed] [Google Scholar]
- Xiong J, Yang H, Wu L, Shang W, Shan E, Liu W et al. (2014b). Fluoxetine suppresses AMP‐activated protein kinase signaling pathway to promote hepatic lipid accumulation in primary mouse hepatocytes. Int J Biochem Cell Biol 54: 236–244. [DOI] [PubMed] [Google Scholar]
- Yang J, Yan B (2007b). Photochemotherapeutic agent 8‐methoxypsoralen induces cytochrome P450 3A4 and carboxylesterase HCE2: evidence on an involvement of the pregnane X receptor. Toxicol Sci 95: 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Shi D, Yang D, Song X, Yan B (2007a). Interleukin‐6 alters the cellular responsiveness to clopidogrel, irinotecan, and oseltamivir by suppressing the expression of carboxylesterases HCE1 and HCE2. Mol Pharmacol 72: 686–694. [DOI] [PubMed] [Google Scholar]
- Zhang H, LeCulyse E, Liu L, Hu M, Matoney L, Zhu W et al. (1999). Rat pregnane X receptor: molecular cloning, tissue distribution, and xenobiotic regulation. Arch Biochem Biophys 368: 14–22. [DOI] [PubMed] [Google Scholar]
- Zhang H, Chang G, Wang J, Lin Y, Ma L, Pang T (2013). CUEDC2 sensitizes chronic myeloid leukemic cells to imatinib treatment. Leuk Res 37: 1583–1591. [DOI] [PubMed] [Google Scholar]
- Zhang C, Xu Y, Gao P, Lu J, Li X, Liu D (2015a). Down‐regulation of carboxylesterases 1 and 2 plays an important role in prodrug metabolism in immunological liver injury rats. Int Immunopharmacol 24: 153–158. [DOI] [PubMed] [Google Scholar]
- Zhang N, Liu Y, Jeong H (2015b). Drug–drug interaction potentials of tyrosine kinase inhibitors via inhibition of UDP‐glucuronosyltransferases. Sci Rep 5: 17778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou JJ, Fan HW, Guo DQ, Li YB, Lin S, Zhu YB et al. (2009). Simultaneous determination of clopidogrel and its carboxylic acid metabolite (SR26334) in human plasma by LC‐ESI‐MS‐MS: application to the therapeutic drug monitoring of clopidogrel. Chromatographia 70: 1581–1586. [Google Scholar]