

Abstract
Background and Purpose
We and others have shown that inhibiting phosphatase and tensin homolog deleted on chromosome 10 (PTEN) or activating ERK1/2 confer neuroprotection. As bisperoxovanadium compounds are well‐established inhibitors of PTEN, we designed bisperoxovandium (pyridin‐2‐squaramide) [bpV(pis)] and determined whether and how bpV(pis) exerts a neuroprotective effect in cerebral ischaemia–reperfusion injury.
Experimental Approach
Malachite green‐based phosphatase assay was used to measure PTEN activity. A western blot assay was used to measure the phosphorylation level of Akt and ERK1/2 (p‐Akt and p‐ERK1/2). Oxygen–glucose deprivation (OGD) was used to injure cultured cortical neurons. Cell death and viability were assessed by LDH and MTT assays. To verify the effects of bpV(pis) in vivo, Sprague–Dawley rats were subjected to middle cerebral artery occlusion, and brain infarct volume was measured and neurological function tests performed.
Key Results
bpV(pis) inhibited PTEN activity and increased p‐Akt in SH‐SY5Y cells but not in PTEN‐deleted U251 cells. bpV(pis) also elevated p‐ERK1/2 in both SH‐SY5Y and U251 cells. These data indicate that bpV(pis) enhances Akt activation through PTEN inhibition but increases ERK1/2 activation independently of PTEN signalling. bpV(pis) prevented OGD‐induced neuronal death in vitro and reduced brain infarct volume and promoted functional recovery in stroke animals. This neuroprotective effect of bpV(pis) was blocked by inhibiting Akt and/or ERK1/2.
Conclusions and Implications
bpV(pis) confers neuroprotection in OGD‐induced injury in vitro and in cerebral ischaemia in vivo by suppressing PTEN and activating ERK1/2. Thus, bpV(pis) is a bi‐target neuroprotectant that may be developed as a drug candidate for stroke treatment.
Abbreviations
- bpV(pis)
bisperoxovandium (pyridin‐2‐squaramide)
- MCAO
middle cerebral artery occlusion
- OGD
oxygen–glucose deprivation
- PTEN
phosphatase and tensin homolog deleted on chromosome 10
Tables of Links
| LIGANDS |
|---|
| Akt Inhibitor IV |
| LY294002 |
| U0126 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is a dual phosphatase that dephosphorylates both protein and lipid substrates (Schmid et al., 2004). PTEN displays its lipid phosphatase activity by antagonizing Akt‐dependent cell survival‐promoting signalling via dephosphorylation of PIP3 (phosphatidylinositol (3,4,5)‐trisphosphate) (Maehama and Dixon, 1998; Yamada and Araki, 2001; Milella et al., 2015). Akt is a multi‐isoform serine/threonine kinase and a direct downstream target of PI3K (Schmid et al., 2004; Chang et al., 2007), and plays a pivotal role in promoting cell survival and growth (Chang et al., 2007). There is substantial evidence indicating that inhibiting PTEN confers neuroprotection in ischaemia–reperfusion injury (Ning et al., 2004; Chang et al., 2007; Zhang et al., 2007; Liu et al., 2010a; Shi et al., 2011; Zheng et al., 2012). PTEN deletion or down‐regulation has also been found to enhance axon regeneration after spinal cord injury (Liu et al., 2010b; Zukor et al., 2013; Lewandowski and Steward, 2014; Ohtake et al., 2014; Singh et al., 2014; Geoffroy et al., 2015).
ERK1/2 is believed to mediate cell survival, neuronal plasticity and migration (Roux and Blenis, 2004; Kim et al., 2005; Segarra et al., 2006; Drosten et al., 2010; Hao and Rockwell, 2013). In both in vitro and in vivo ischaemia stroke models, ERK1/2 signalling exerts neuroprotective effects in cerebral ischaemia–reperfusion injury (Chang et al., 2004; Kilic et al., 2005; Meller et al., 2005; Zhang and Chen, 2008; Atif et al., 2013; Nakano et al., 2014; Buendia et al., 2015; Koh, 2015).
In this study, we synthesized a new compound, bisperoxovandium (pyridin‐2‐squaramide) [bpV(pis)] and showed that it confers neuroprotection in ischaemia–reperfusion injury through inhibiting PTEN and activating ERK1/2.
Methods
The synthesis of bpV(pis)
V2O5 (1.43 g, 7.84 mmol) was dissolved in a solution of KOH (1.04 g, 18.53 mmol) in water (15 mL) and stirred for 5 min. Then 2 mL of a 30% (w.v‐1) aqueous solution of H2O2 was added to the resulting green solution, which resulted in effervescence. The mixture was next stirred for 25 min, which produced a progressive change in colour to orange and the complete dissolution of all solids. A further 12 mL of a 30% (w.v‐1) aqueous solution of H2O2 was added, and the reaction mixture was stirred for 15 min. At this point, a solution of pyridine‐2‐squaramide (2.98 g, 15.68 mmol) in a mixture of water (20 mL) and ethanol (5 mL) was added to the reaction mixture, which was stirred at room temperture (RT) for 30 min, after which consistent cloudiness was noted. The resulting pale yellow precipitate was filtered through a glass microfibre filter paper, washed with cold water and diethyl ether and dried overnight under reduced pressure (Rosivatz et al., 2006).
PTEN activity assay
The ability of bpV(pis) to inhibit PTEN activity was assessed using a Malachite green‐based phosphatase assay kit (Echelon Biosciences, Salt Lake City, UT, USA) following the manufacturer's instruction. PTEN enzyme and PIP3 substrate were purchased from Echelon Biosciences (Salt Lake City, UT). This assay is based on the quantification of the phosphate liberated from PI(3,4,5)P3, which forms a coloured complex with molybdate/malachite green after 20 min at room temperature; this is quantified by reading the absorbance at 620 nm. The % conversion of PIP3 was determined by using the formula according to the manufacturer's instruction. Control wells containing PIP3 only were set at 0% PTEN activity, and wells containing PTEN + PIP3 were set at 100% PTEN activity. The IC50 value was calculated from the concentration–response curves generated using GraphPad Prism 5 software.
ERK1/2 activity assay
Recombinant ERK1/2 protein was purchased from Active Motif (Carlsbad, CA, USA). The ERK1/2 activity was tested using an ERK1/2 Assay Kit (Merck Millipore, Billerica, MA, USA) following the manufacturer's instructions. The activity of recombinant ERK1/2 was measured in assay dilution buffer I, pH 7.2, containing 75 mM magnesium chloride, 500 μM ATP, 2 mg·mL−1 dephosphorylated myelin basic protein (MBP) and bpV(pis) (0–100 μM). After a 20–30 min incubation at 30°C, 2.5 μL of the reaction mixture was removed and placed in a centrifuge tube. TBS and laemmli sample buffer were added. The sample was loaded for SDS‐PAGE and immunoblot analysis, and probed with a monoclonal phospho‐specific MBP antibody. The ERK activity is based on the phosphorylation of MBP. The control group, which did not contain bpV(pis) was set at 100% of ERK1/2 activity. All experiments were repeated in triplicate. The quantification of immunoblot data was performed using ImageJ software.
Detection of bpV(pis) in SH‐SY5Y cells
The SH‐SY5Y cells were maintained at 8 × 105 cells per dish in 60 mm dishes. When the confluence of the SH‐SY5Y cells reached 80–90%, the cells were washed three times in extracellular solution (ECS) (137 mM NaCl, 2.0 mM Ca2+, 5.4 mM KCl, 1.0 mM MgCl2, 25 mM HEPES and 33 mM glucose, titrated to pH 7.4 with an osmolarity of 300–320 mOsm) and then treated with bpV(pis) for 30 min. Excess bpV(pis) was removed by washing for five times with ECS, and then cells were collected for MS analysis.
The cells were incubated for 1 min with 0.25% trypsin, resuspended in 2 mL of DMEM and centrifuged at 1000 x g for 3 or 5 min. The precipitate was resuspended in 200 μL of chilled saline solution (0.9%) and ultrasonicated. The mixture was then centrifuged at 14000× g for 20 min at 4°C. The supernatant was mixed with 2 × volume of methanol and centrifuged to remove protein. [13C]‐bpV(pis) (purchased from Beita Pharmatech co. , LTD, Jiangyin, China) was dissolved in a mixture of acetonitrile and water (1:1, v:v) to a concentration of 5 μM. The supernatant (20 μL) was mixed with 5 μM [13C]‐bpV(pis) solution (20 μL) in a 500 μL centrifuge tube. The mixture (5 μL) was injected into the MS system. The signal intensity of [13C]‐bpV(pis) and bpV(pis) in the mixture was measured, and the intensity ratios of the signals produced by bpV(pis) relative to that of [13C]‐bpV(pis) (internal standard) were calculated. These ratios were assumed to reflect the relative proportions of the various compounds contained in the mixture. The relative intensity of [13C]‐bpV(pis) was 100%. The higher the ratio, the more bpV(pis) had crossed through the cell membrane.
Cell culture, transfection and treatment
For cell culture, SH‐SY5Y and U251 cells were seeded in a 6‐well plate (8 × 105 cells per well) in DMEM supplemented with 10% heat‐inactivated FBS, penicillin G (100 U·mL−1), streptomycin (100 mg·mL−1) and L‐glutamine (2.0 mM) and incubated at 37°C in a humidified atmosphere containing 5% CO2 and 95% air (Wang et al., 2015). When the confluence of cells reached 80–90% on the day of treatment, cells were washed with standard ECS for 60 min and then treated with bpV(pis) for 30 min. The cells were then collected for western blot analysis.
When the confluence of SH‐SY5Y cells reached 60–70% on the treatment day, cells were transfected with human PTEN siRNA (siRNApten) and non‐targeting control siRNA (NsiRNA) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 8 h. The medium was then replaced with normal growth medium for 24 h. On the following day, the cells were treated with standard ECS for 60 min and then treated with bpV(pis) for 30 min. The cells were then collected for western blot analysis.
Cortical neuron culture and OGD insult
The cortical neuronal cultures were prepared from Sprague–Dawley (SD) rats at gestation day 17 as described previously (Brewer et al., 1993; Shan et al., 2009). The pregnant rats were anaesthetized with 4% isoflurane in 70% N2O and 30% O2 and killed by cervical dislocation. The rats were sprayed with 70% ethanol, and the embryos were removed. The embryos were quickly decapitated, and the cortices were placed in ice‐cold plating medium (Neurobasal medium, 2% B‐27 supplement, 0.5% FBS, 0.5 mM L‐glutamax and 25 mM glutamic acid) following the removal of the meninges. The cortical neurons were suspended in plating medium and plated on Petri dishes coated with poly‐D‐lysine. Half of the plating medium was removed and replaced with maintenance medium (Neurobasal medium, 2% B‐27 supplement and 0.5 mM L‐glutamine) in the same manner every 3 days. After 12 days, the cultured neurons were used for the experiments (Brewer et al., 1993; Shan et al., 2009).
For the oxygen–glucose deprivation (OGD) challenge, cells were transferred to deoxygenated, glucose‐free, extracellular solution (in mM: 116 NaCl, 5.4 KCl, 0.8 MgSO4, 1.0 NaH2PO4, 1.8 CaCl2 and 26 NaHCO3), placed in a specialized, humidified chamber (Plas‐Labs, Lansing, USA), and maintained at 37°C in 85% N2/10% H2/5% CO2 for 60 min (Liu et al., 2006; 2010a). Then the medium was replaced with fresh maintenance medium containing an appropriate concentration of reagents for 24 h during the recovery period in a 95% O2/5% CO2 incubator. The control cultures were firstly transferred to another extracellular solution (in mM: 116 NaCl, 5.4 KCl, 0.8 MgSO4, 1.0 NaH2PO4, 1.8 CaCl2, 26 NaHCO3 and 33 glucose), and then placed in the humidified chamber, which was maintained at 37°C in 95% O2/5% CO2 for 60 min (Liu et al., 2006; 2010a). Then the medium was replaced with fresh maintenance medium for the whole period at 37°C in a 95% O2/5% CO2 incubator.
Western blot
Western blotting was performed as described previously (Ning et al., 2004). Briefly, the PVDF membrane (Millipore, USA) was incubated with a primary antibody against phospho‐ERK1/2 (Thr202/Tyr204), ERK1/2 from Cell Signaling Technology (Boston, MA) and an antibody against PTEN, phospho‐Akt (Ser 473), Akt, GAPDH from Santa Cruz Biotechnology (Santa Cruz, CA). Primary antibodies were labelled with horseradish peroxidase‐conjugated secondary antibody, and protein bands were imaged using SuperSignal West Femto Maximum Sensitivity Substrate (Pierce, USA). The EC3 Imaging System (UVP, USA) was used to obtained blot images directly from the PVDF membrane. The western blot data were quantified using ImageJ software.
Neuronal viability assays
Lactate dehydrogenase (LDH) is a cytoplasmic enzyme retained by viable cells with intact plasma membranes and released from cells with damaged membranes. LDH release was measured using a CytoTox 96 Cytotoxicity kit by following the manufacturer's instructions (Promega, USA). The levels of maximal LDH release were measured by treating the cultures with 10 × lysis solution (provided by the manufacturer) to yield complete lysis of the cells. Absorbance data were obtained using a 96‐well plate reader (Molecular Devices, USA) at 490 nm. According to the manufacturer's instructions, the LDH release (%) was measured by calculating the ratio of experimental LDH release to maximal LDH release (Shan et al., 2009).
The viability of the cells in the neuronal cultures was assessed by their ability to take up thiazolyl blue tetrazolium bromide (MTT). The cells were incubated with MTT for 1 h, then lysed with DMSO and left at room temperature in the dark overnight. The lysates were then read on a plate reader (PowerWave X, Bio‐Tek, Winooski, State, USA) at an absorbance wavelength of 540 nm (Qu et al., 2009; Chien et al., 2015).
Animals
Adult male SD rats, weighing 250 to 300 g, were group‐housed with 2–3 rats per cage on a 12 h light/dark cycle in a temperature‐controlled room (23–25°C) with free access to food and water. Animals were allowed at least 3 days to acclimatize before experimentation. The total number of male rats used in our in vivo experiments was 224. Adult pregnant female rats (n = 10) and 36 embryos were used for cortical neuronal cultures. All animal use and experimental protocols were approved and carried out in compliance with the IACUC guidelines and the Animal Care and Ethics Committee of Wuhan University School of Medicine. Randomization was used to assign samples to the experimental groups and to collect and process data. The experiments were performed by investigators blinded to the groups for which each animal was assigned. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Focal cerebral ischaemia and infarct measurement
Transient focal cerebral ischaemia was induced using the suture occlusion technique (Longa et al., 1989). Male SD rats weighing 250–300 g were anaesthetized with 4% isoflurane in 70% N2O and 30% O2 by using a mask. A midline incision was made in the neck, the right external carotid artery (ECA) was carefully exposed and dissected and a 3–0 monofilament nylon suture was inserted from the ECA into the right internal carotid artery to occlude the origin of the right middle cerebral artery (approximately 22 mm). After 90 min of occlusion, the suture was removed to allow reperfusion, the ECA was ligated and the wound was closed. Sham‐operated rats underwent identical surgery and/or i.c.v. injections except that the suture was inserted and withdrawn immediately. Rectal temperature was maintained at 37.0 ± 0.5°C using a heating pad and heating lamp. At 24 h after middle cerebral artery occlusion (MCAO), rats (n=24) were decapitated after being reperfused with ice‐cold 0.9% saline after anaesthetized with 4% isoflurane in 70% N2O and 30% O2, and the brains were rapidly removed for western blot analysis and 2,3,5‐triphenyltetrazolium chloride (TTC) staining.
The peri‐infarct tissues of ipsilateral hemispheres were homogenized in RIPA buffer using a tissue grinder on ice for 30 min. Then, the tissue lysates were centrifuged at 12000× g for 15 min at 4 C, and the total protein concentrations were assessed with a bicinchoninic acid protein assay kit.
Other groups of rats (n = 136) were killed, and the brains were removed for TTC staining to evaluate cerebral infarct volumes (Wexler et al., 2002). The brain was placed in a cooled matrix, and 2 mm coronal sections were cut. Individual sections were placed in 10 cm petri dishes and incubated for 30 min in a solution of 2% TTC in PBS at 37°C. The slices were fixed in 4% paraformaldehyde at 4°C. All image collection, processing and analysis were performed in a blind manner and under controlled environmental lighting. The scanned images were analysed using ImageJ software, and the infarct data for all groups were expressed as the ratios of the infarcted areas to the total brain section areas (Wexler et al., 2002).
Technique for i.c.v. administration
First the rat was anaesthetized with a mixture of 4% isoflurane in 70% N2O and 30% O2 in a sealed perspective plastic box. Once the rat was deeply anaesthetized, its head was shaved and secured in position in a stereotaxic frame using the ear bars and upper incisor bar; the rat was continuously under anaesthesia with 4% isoflurane using a mask. Then, a small mid‐sagittal incision was made, and bregma was located as the anatomical reference point. Drug infusion, at a rate of 1.0 μL·min−1, to the cerebral ventricle (from the bregma: anteroposterior, −0.8 mm; lateral, 1.5 mm; depth, 3.5 mm) was performed using a 23 gauge needle attached via polyethylene tubing to a Hamilton microsyringe. Proper needle placement was verified via withdrawing a few microlitres of clear cerebrospinal fluid into the Hamilton microsyringe.
Administration of bpV(pis) i.p.
bpV(pis) (20 mM) was dissolved in sterile saline containing DMSO and i.p. administered to rats at 1.0 h after ischaemia–reperfusion at concentrations of 20, 200 and 2000 μg·kg−1. Rats in the vehicle group received i.p. injections of the same volume of sterile saline containing a corresponding proportion of DMSO solution without bpV(pis).
Neurobehavioral tests
A total of 56 rats were used for neurobehavioral tests.
Neurological severity scores: The rats were subjected to a modified neurological severity score test as reported previously (Chen et al., 2001). These tests are a battery of motor, sensory, reflex and balance tests, which are similar to the contralateral neglect tests in humans. Neurological function was graded on a scale of 0 to 18 (normal score, 0; maximal deficit score, 18).
Beam walk test: The beam walk test measures the animals' complex neuromotor function (Aronowski et al., 1996; Petullo et al., 1999). The animal was timed as it walked a (100 × 2 cm) beam. A box for the animal to feel safe was placed at one end of the beam. A loud noise was created to stimulate the animal to walk towards and into the box (Aronowski et al., 1996; Petullo et al., 1999). Scoring was based upon the time it took the rat to go into the box (Petullo et al., 1999). The higher the score, the more severe the neurological deficit.
Adhesive‐removal test: A modified sticky‐tape test was performed to evaluate forelimb function (Sughrue et al., 2006). A sleeve was created using a 3.0 × 1.0 cm piece of yellow paper tape and was subsequently wrapped around the forepaw so that the tape attached to itself and allowed the digits to protrude slightly from the sleeve. The typical response is for the rat to vigorously attempt to remove the sleeve by either pulling at the tape with its mouth or brushing the tape with its contralateral paw. The rat was placed in its cage and observed for 30 s. Two timers were started: the first ran without interruption and the second was turned on only while the animal attempted to remove the tape sleeve. The ratio of the left (affected)/right (unaffected) forelimb performance was recorded. The contralateral and ipsilateral limbs were tested separately. The test was repeated three times per test day, and the best two scores of the day were averaged. The lower the ratio, the more severe the neurological deficit.
Statistics
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis (Curtis et al., 2015). The group size per experiment was based on a power analysis (Charan and Kantharia, 2013). The criterion for significance (α) was set at 0.05. The power of 80% was considered to yield a statistically significant result. Student's t‐test or ANOVA was used where appropriate to examine the statistical significance of the differences between groups of data. Bonferroni tests were used for post hoc comparisons when appropriate. All results are presented as mean ± SEM. Significance was placed at P < 0.05.
Materials
bpV(pis) was synthesized in the Faculty of Pharmacy, Wuhan University School of Medicine. The PI3K inhibitor LY294002 was purchased from Cell Signaling Technology (Boston, MA). Akt Inhibitor IV was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The ERK1/2 inhibitor U0126 was purchased from Selleckchem (Houston, TX).
Results
bpV(pis) enhances Akt activation through PTEN inhibition
To find a potential neuroprotectant that exerts its effect through PTEN inhibition, we designed and synthesized bpV(pis). The chemical equation and structure of bpV(pis) are shown in Figure 1A. Compound characterization of bpV(pis) was tested by infrared spectra analysis. The result obtained was shown as IR v (KBr) in cm−1: v (NH2) 3194,v (CN) 1617,v (VO) 960,v (OO) 816 cm−1 (Figure 1B). The molecular mass of bpV(pis) was tested by MS analysis. The result obtained was: MS (ESI) calcd for C9H5KN2O8V [M + H]+ 359.2, found 359.2; yield 76%.
Figure 1.
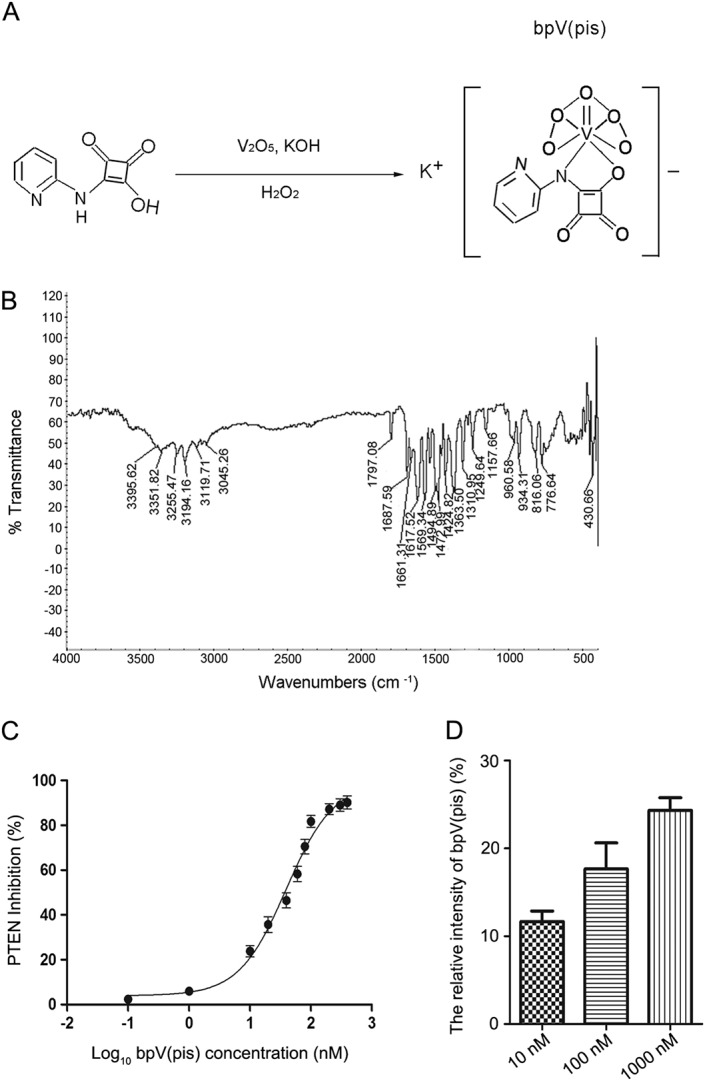
Characterization of bisperoxovandium (pyridin‐2‐squaramide) [bpV(pis)]. (A) The chemical equation and structure of bpV(pis). (B) Compound characterization of bpV(pis) was tested by infrared spectra analysis. The result was IR v (KBr) in cm−1: v (NH2) 3194,v (CN) 1617,v (VO) 960,v (OO) 816 cm−1. (C) Malachite green‐based phosphatase assay showing the percentage of inhibition of PTEN activity by bpV(pis). The IC50 value of bpV(pis) for PTEN was 39.44 ± 5.92 nM. Three independent tests were performed. (D) bpV(pis) crosses through the cell membrane of SH‐SY5Y cells. We used a MS system to detect the signal intensity of [13C]‐bpV(pis) and bpV(pis) from the cell extracts of SH‐SY5Y cells treated with bpV(pis) (10, 100 and 1000 nM). The intensity ratios of these signals relative to that of [13C]‐bpV(pis) (internal standard) are shown; n = 6 independent cultures.
We then measured the percentage inhibition of PTEN activity by using the Malachite green‐based phosphatase assay. The IC50 value of bpV(pis) for PTEN activity was 39.44 ± 5.92 nM, indicating that bpV(pis) is markedly potent as inhibiting PTEN (Figure 1C).
Before testing the effects of bpV(pis) in cell and animal experiments, we examined whether bpV(pis) is able to traverse the cell membrane. As described in the Methods section, we injected [13C]‐bpV(pis) and the SH‐SY5Y cell extracts treated with bpV(pis) into a MS system. The signal intensity of [13C]‐bpV(pis) and bpV(pis) in the cell extracts was measured, and the intensity ratios of these signals relative to that of [13C]‐bpV(pis) (internal standard) were calculated. The higher the ratio, the more bpV(pis) passed through the cell membrane. The results showed that the signal intensity was detected in the cells treated with 10, 100 and 1000 nM bpV(pis), indicating that bpV(pis) can traverse the cell membrane (Figure 1D).
As PTEN negatively regulates Akt activation (Maehama and Dixon, 1998), we also tested the effect of bpV(pis) on PTEN inhibition by measuring Akt activation. Akt activation was quantified by measuring Akt phosphorylation (p‐Akt) on Ser473 in a western blot assay (Shan et al., 2009). The human neuroblastoma SH‐SY5Y cells were washed with standard ECS for 60 min and then treated with bpV(pis) for 30 min. We showed that bpV(pis) markedly increased the level of p‐Akt in SH‐SY5Y cells (Figure 2A). Using a PTEN knockdown approach (Figure 2B), we showed that PTEN suppression by siRNApten increased the level of p‐Akt (Figure 2C). However, bpV(pis) did not cause a further increase in p‐Akt in SH‐SY5Y cells transfected with siRNApten [siRNApten + bpV(pis) group in Figure 2C] compared with bpV(pis) treated SH‐SY5Y cells transfected with the NsiRNA [NsiRNApten + bpV(pis) group in Figure 2C]. These results suggest that the bpV(pis)‐induced increase in Akt activation in SH‐SY5Y cells is mediated by the inhibition of PTEN.
Figure 2.
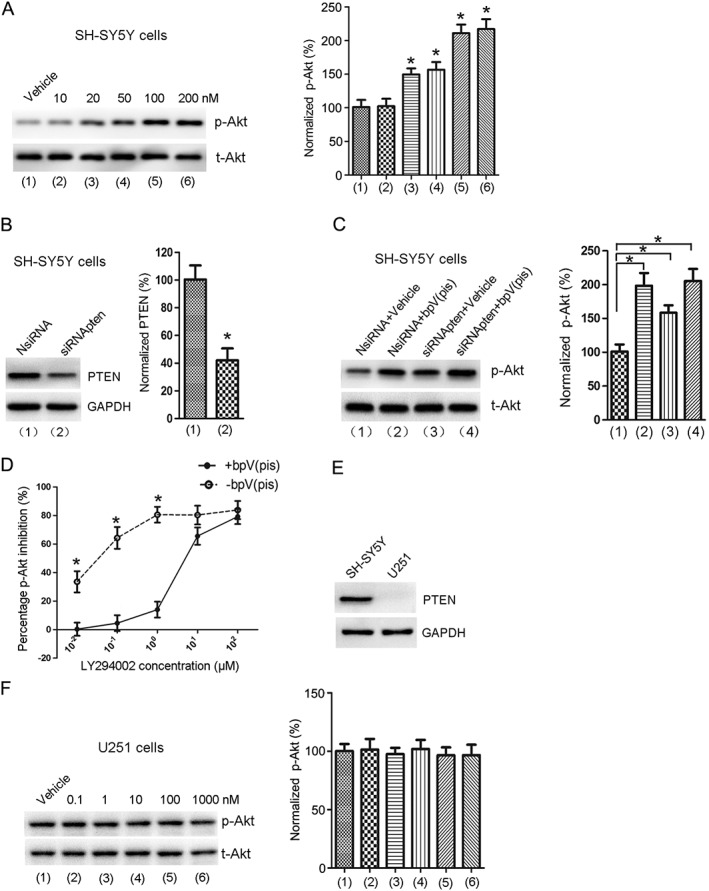
bpV(pis) enhances Akt phosphorylation through PTEN inhibition. (A) The level of p‐Akt was increased in SH‐SY5Y cells treated with bpV(pis) (20–200 nM). Data from each group are normalized to that of Vehicle group; n = 7 independent cultures, *P < 0.05 versus Vehicle, one‐way ANOVA test, followed by the Bonferroni post hoc test. (B) Treatment with siRNApten suppresses PTEN expression in SH‐SY5Y cells. n = 6 independent cultures, *P < 0.05 versus NsiRNA, Student's t‐test. (C) The effect of bpV(pis) (200 nM) on p‐Akt in SH‐SY5Y cells transfected with siRNApten or NsiRNA; n = 6 independent cultures, *P < 0.05 versus NsiRNA + Vehicle; one‐way ANOVA test, followed by Bonferroni post hoc test. (D) bpV(pis) (200 nM) counteracts LY294002‐induced inhibition of p‐Akt in SH‐SY5Y cells; n = 7 independent cultures, *P < 0.05 versus bpV(pis), two‐way ANOVAs, followed by Bonferroni post hoc test. (E) PTEN protein expression in SH‐SY5Y and U251 cells. (F) p‐Akt is not changed in U251 cells treated with bpV(pis) (0.1–1000 nM); n = 6 independent cultures.
To provide further evidence for the inhibition of PTEN by bpV(pis), SH‐SY5Y cells were treated with LY294002 (10−2–102 μM) and incubated with or without 200 nM bpV(pis) for 30 min, followed by washing with standard ECS for 60 min. Western blotting assay was used to measure the percentage of p‐Akt inhibition. As expected, bpV(pis) attenuated LY294002 (<10 μM)‐induced Akt inhibition in SH‐SY5Y cells (Figure 2D).
We also tested the effect of bpV(pis) on Akt phosphorylation in PTEN‐deficient human glioblastoma U251 cells. We first confirmed that there was no PTEN expression in U251 cells (Figure 2E). We then showed that bpV(pis) (0.1, 1, 10, 100 and 1000 nM) did not affect Akt phosphorylation in U251 cells (Figure 2F). Together, these results indicate that bpV(pis) inhibits PTEN to enhance Akt activation.
bpV(pis) increases ERK1/2 activation independently of PTEN inhibition
bpV(pis) induced a marked elevation of ERK1/2 activation in SH‐SY5Y cells (Figure 3A). The activation of ERK1/2 was measured by quantifying ERK1/2 phosphorylation (p‐ERK1/2) on Thr202/Tyr204 in a western blot assay (Figure 3A). We also measured the effect of PTEN knockdown on ERK1/2 phosphorylation in SH‐SY5Y cells. PTEN suppression by siRNApten had no effect on p‐ERK1/2 (Figure 3B). The bpV(pis)‐induced increase in ERK1/2 phosphorylation was not affected by siRNApten transfection (Figure 3B). These data suggest that the elevation in ERK1/2 phosphorylation induced by bpV(pis) is independent of PTEN inhibition.
Figure 3.
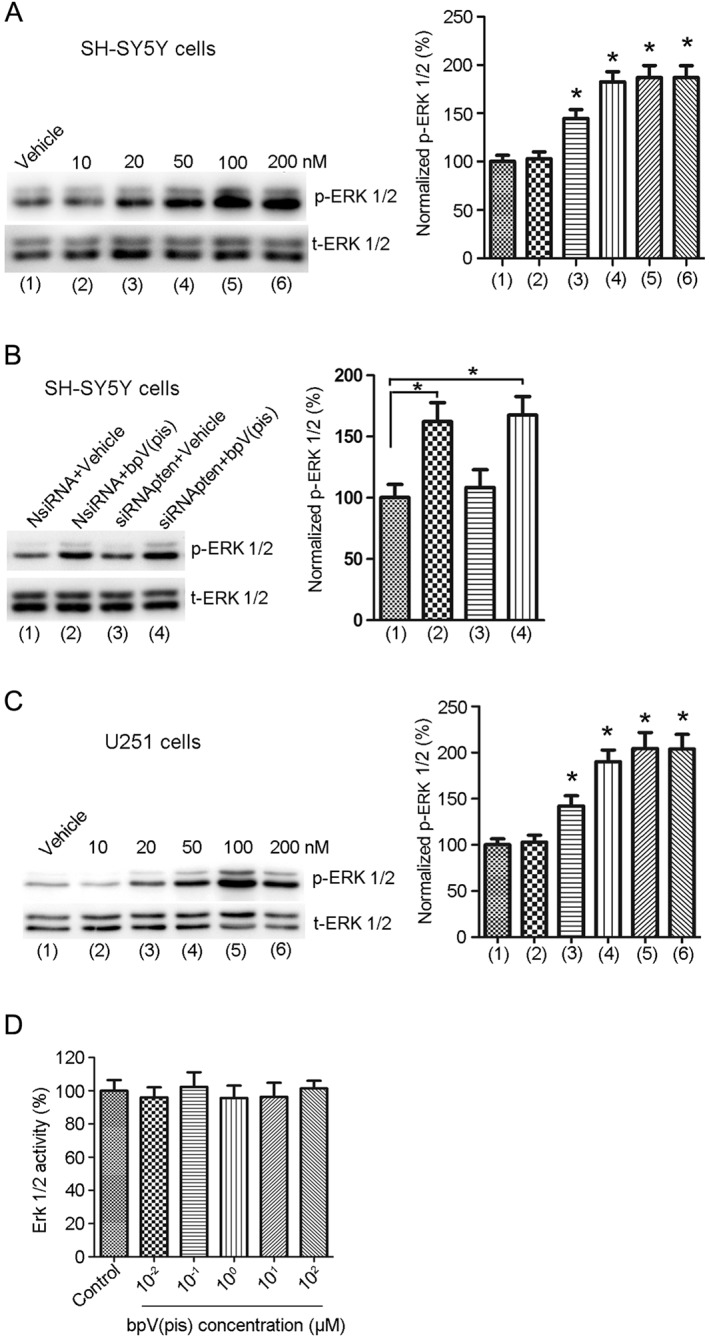
bpV(pis) increases p‐ERK1/2 independently of PTEN inhibition. (A) The p‐ERK1/2 is increased in SH‐SY5Y cells treated with bpV(pis) (20–200 nM); n = 7 independent cultures, *P < 0.05 versus Vehicle, one‐way ANOVA, followed by the Bonferroni post hoc test. (B) The effect of bpV(pis) (200 nM) on p‐ERK1/2 in SH‐SY5Y cells transfected with siRNApten; n = 6 independent cultures, *P < 0.05 versus NsiRNA + Vehicle; one‐way ANOVA, followed by the Bonferroni post hoc test. (C) p‐ERK1/2 is increased in U251 cells treated with bpV(pis) (20–200 nM); n = 6 independent cultures, *P < 0.05 versus Vehicle, one‐way ANOVA, followed by the Bonferroni post hoc test. (D) bpV(pis) does not activate ERK1/2 in vitro; n = 6 independent cultures.
We further tested the effect of bpV(pis) (10, 20, 50, 100 and 200 nM) on ERK1/2 phosphorylation in PTEN‐deficient U251 cells; bpV(pis) (20, 50, 100 and 200 nM) increased ERK1/2 phosphorylation in these cells (Figure 3C), supporting the notion that bpV(pis) activates ERK1/2 independently of PTEN inhibition.
To verify whether bpV(pis) directly activates ERK1/2, we tested the effect of bpV(pis) on the activity of recombinant ERK1/2 in vitro. bpV(pis) did not alter the ERK1/2 activity (Figure 3D), indicating that another signalling pathway different from PTEN mediates the enhancement of ERK1/2 activation by bpV(pis) in SH‐SY5Y and U251 cells.
bpV(pis) prevents the reduction in Akt and ERK1/2 phosphorylation in injured cortical neurons
The observed increase in p‐Akt and p‐ERK1/2 induced by bpV(pis) in normal conditions led us to determine whether bpV(pis) modulates Akt and ERK1/2 phosphorylation in injury conditions. To address this, we first tested the effect of bpV(pis) on p‐Akt and p‐ERK1/2 in OGD‐induced neuronal injury, an in vitro model that mimics ischaemic neuronal injury. Both p‐Akt and p‐ERK1/2 were suppressed at 24 h after OGD injury in cortical neurons (Figure 4A, B). However, treatment with bpV(pis) at 30 min after re‐oxygenation attenuated the reduction in p‐Akt and p‐ERK1/2 in OGD‐treated neurons (Figure 4A, B).
Figure 4.
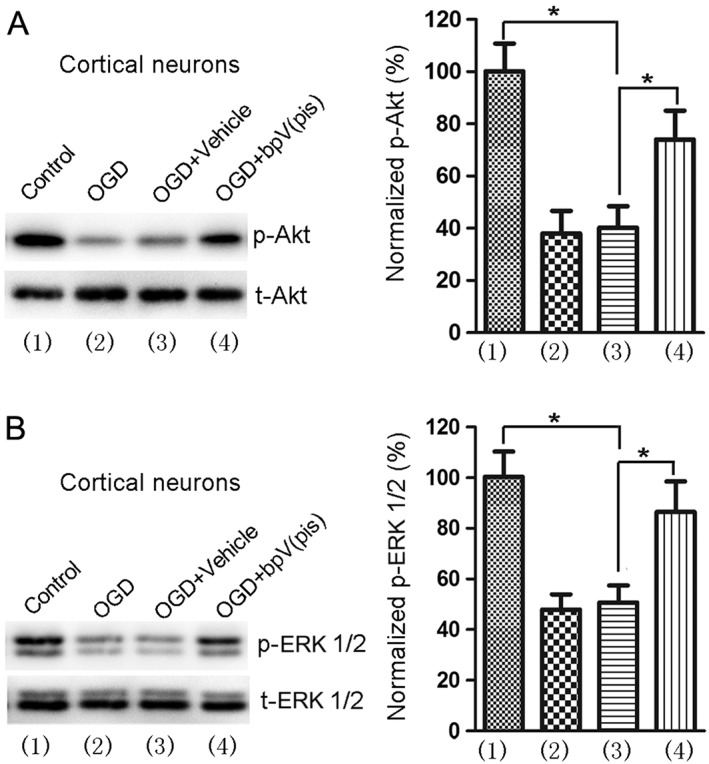
OGD‐induced decrease in p‐Akt and p‐ERK1/2 is reduced by bpV(pis). (A), (B) bpV(pis) (200 nM) attenuates OGD‐induced reduction of p‐Akt and p‐ERK1/2 in cortical neurons; n = 6 independent cultures, *P < 0.05, one‐way ANOVA, followed by the Bonferroni post hoc test.
Both PTEN/Akt and ERK1/2 signalling mediate bpV(pis)‐induced neuroprotection in OGD insult
According the above findings (Figure 4A, B), we hypothesized that bpV(pis) might exert a neuroprotective effect in OGD‐induced neuronal injury. We measured neuronal death/viability by LDH and MTT assay 24 h after OGD injury. Treatment with bpV(pis) (20, 50, 100 and 200 nM) 30 min after re‐oxygenation decreased LDH release and increased the viability of the cells (Figure 5A, B). The beneficial effects of bpV(pis) (200 nM) were impaired after pretreatment with an Akt, Inhibitor IV (1.0 μM), and/or ERK1/2 inhibitor, U0126 (10 μM) (Figure 5C, D). Thus, both PTEN inhibition‐mediated Akt activation and ERK1/2 activation contribute to bpV(pis)‐induced neuroprotection.
Figure 5.
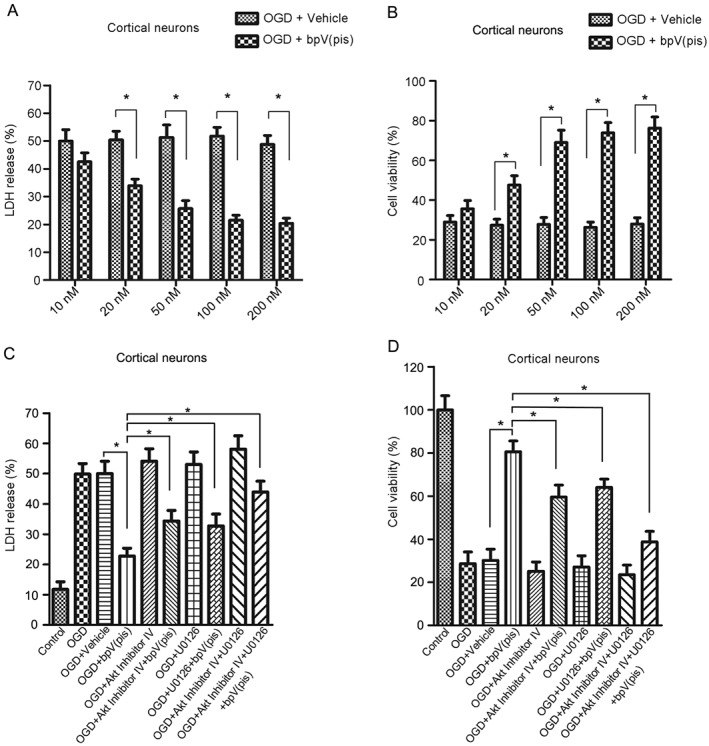
bpV(pis) protects against OGD‐induced neuronal death through PTEN inhibition and ERK1/2 activation. (A, B) bpV(pis) (20–200 nM) decreased LDH release (A) and increased cell viability (B) during OGD‐induced neuronal death; n = 7 independent cultures. The tests were performed in quintuplicate. *P < 0.05 versus OGD + Vehicle, two‐way ANOVA, followed by the Bonferroni post hoc test. (C, D) Treatment with bpV(pis) (200 nM) at 30 min after re‐oxygenation in cultured cortical neurons decreased LDH release (C) and increased cell viability (D), and the neuroprotective effect of bpV(pis) was blocked by Akt Inhibitor IV (1.0 μM) and ERK1/2 inhibitor U0126 (10 μM); n = 7 independent cultures. The tests were performed in quintuplicate. *P < 0.05, one‐way ANOVA, followed by the Bonferroni post hoc test. The cortical neurons were treated with Akt Inhibitor IV and U0126 at 20 min before bpV(pis) treatment.
bpV(pis) prevents the reduction in phosphorylation of Akt and ERK1/2 in rat cerebral ischaemia–reperfusion injury
To provide in vivo evidence for the role of bpV(pis) in neuroprotection, we tested the effect of bpV(pis) on p‐Akt and p‐ERK1/2 in the peri‐infarct tissues of ischaemic brain in a rat MCAO model (Figure 6A) (Ashwal et al., 1998; Sun et al., 2003). At 1.0 h following ischaemia–reperfusion, bpV(pis) (100 μM, 5 μL) was administered into rat lateral ventricles. The phosphorylation of Akt and ERK1/2 was suppressed at 24 h after ischaemia onset (Figure 6B, C), whereas bpV(pis) treatment reduced the decrease in p‐Akt and p‐ERK1/2 in the ischaemic brain (Figure 6B, C).
Figure 6.
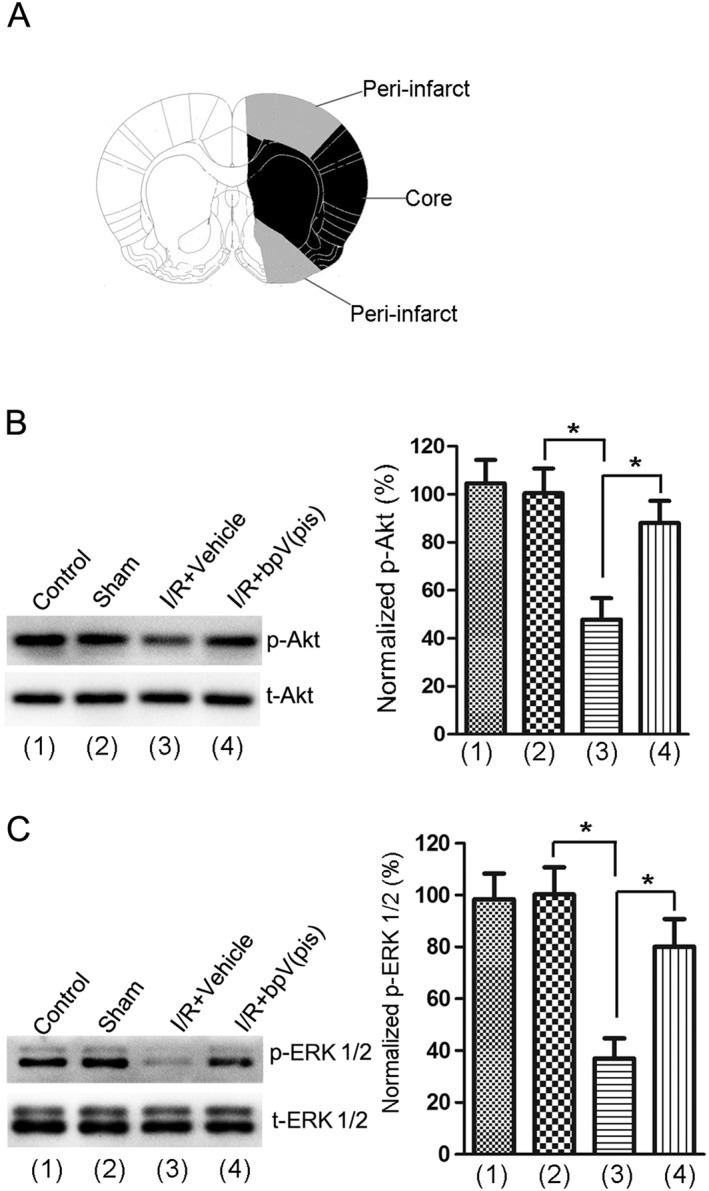
bpV(pis) blocked the reduction in p‐Akt and p‐ERK1/2 in rat cerebral ischaemia–reperfusion injury. (A) Diagram shows the dissected ischaemic peri‐infarct. (B, C) At 1.0 h following ischaemia–reperfusion, treatment with bpV(pis) (100 μM, 5 μL) prevents the decrease in p‐Akt (B) and p‐ERK1/2 (C) at 24 h after ischaemia onset in the ischaemic peri‐infarct (n = 8 for each group; *P < 0.05, one‐way ANOVA, followed by the Bonferroni post hoc test). I/R, ischaemia–reperfusion.
Both PTEN/Akt and ERK1/2 signalling mediate bpV(pis)‐induced neuroprotection in cerebral ischaemia–reperfusion injury
To test whether bpV(pis) conferred neuroprotection in ischaemic stroke. We measured the effect of bpV(pis) on ischaemic neuronal death in rats subjected to MCAO. The experimental procedures are shown in Figure 7A. bpV(pis), (100 μM, 5 μL, i.c.v.) injected 1.0 h after ischaemia–reperfusion, decreased the infarct volume at 24 h after ischaemia onset (Figure 7B). Animals treated with bpV(pis) (20, 200 and 2000 μg·kg−1) by i.p. injections 1.0 h after ischaemia–reperfusion also had a significantly reduced infarct volume (Figure S1).
Figure 7.
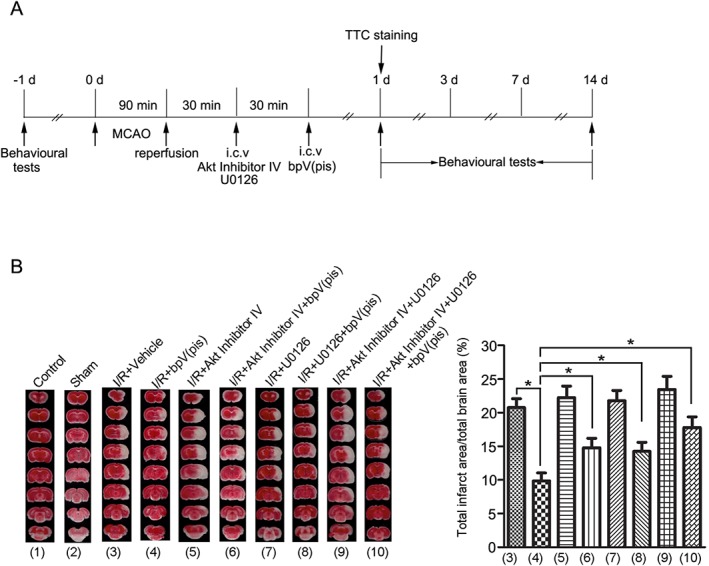
bpV(pis) reduces the infarct volume in MCAO rats through PTEN inhibition and ERK1/2 activation. (A) Diagram shows the experimental procedures in MCAO rats. All groups of animals were subjected to two i.c.v. injections. Akt Inhibitor IV (100 μM, 2 μL) or U0126 (500 μM, 2 μL)] was given at 30 min after reperfusion as the first injection, and bpV(pis) (100 μM, 5 μL) was given at 60 min after reperfusion as the second injection.(B) Sample images and the summarized data of TTC staining indicate that bpV(pis) reduces the infarct volume in the rat brain 24 h after ischaemia onset and that pretreatment with Akt Inhibitor IV and/or U0126 block the bpV(pis)‐induced reduction in infarct volume (n = 10 for each group; *P < 0.05, one‐way ANOVA, followed by the Bonferroni post hoc test). I/R, ischaemia–reperfusion.
To investigate whether the decrease in infarct volume induced by bpV(pis) was mediated through both the PTEN/Akt and/or ERK1/2 signalling pathway, we suppressed the activity of Akt and ERK1/2 by injecting Akt Inhibitor IV (100 μM, 2 μL) and/or U0126 (500 μM, 2 μL) into the lateral ventricles 30 min before the bpV(pis) injection (Pignataro et al., 2008; Liu et al., 2013). This pre‐injection of Akt Inhibitor IV and/or U0126 prevented the bpV(pis)‐induced decrease of infarct volume (Figure 7B).
We also performed a battery of neurobehavioral tests to evaluate the functional effect of bpV(pis). As illustrated in Figure 8A–C, bpV(pis) improved the functional outcome of stroke animals by effects mediated through both the PTEN/Akt and ERK1/2 signalling pathways.
Figure 8.
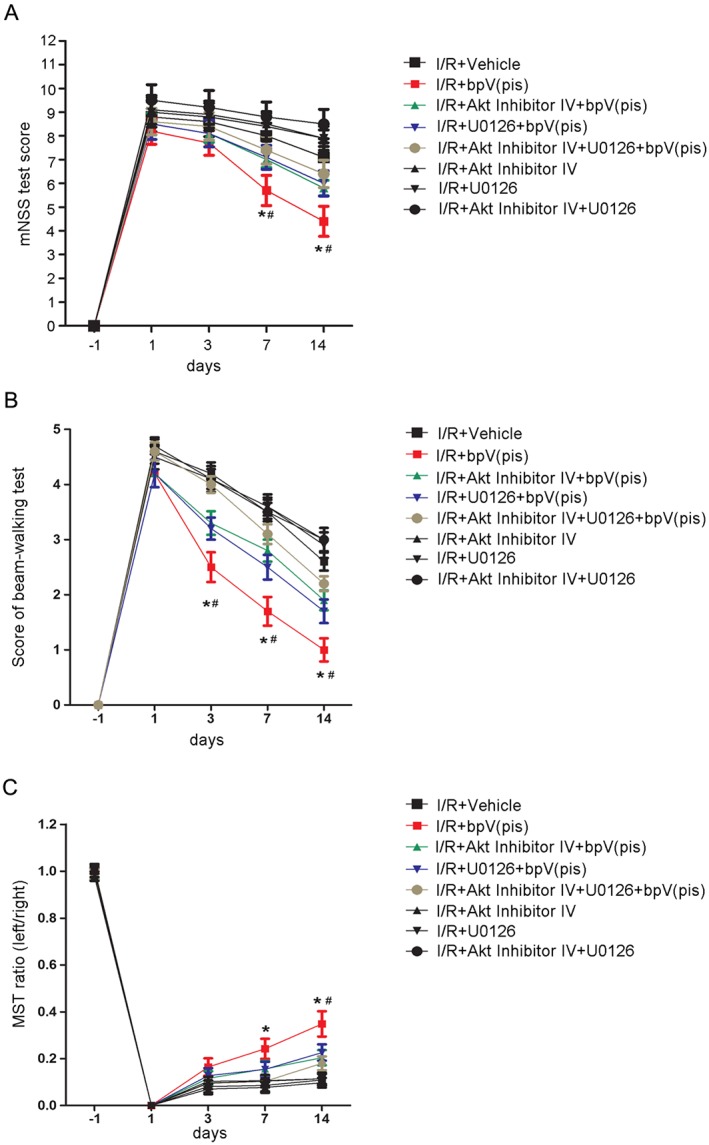
bpV(pis) improves neurological function in ischaemia–reperfusion injury by inhibiting PTEN and activating ERK1/2. (A) Animals treated with bpV(pis) have lower scores of modified neurological severity score (mNSS) test at days 7 and 14 after MCAO. Animals pre‐injected with Akt inhibitor IV and/or U0126 have higher scores of mNSS test at days 7 and 14 after MCAO [n = 7 for each group; *P < 0.05, vs. I/R + Vehicle, # P < 0.05, vs. I/R + bpV(pis), two‐way ANOVA, followed by the Bonferroni post hoc test]. (B) Animals treated with bpV(pis) have lower scores in the beam‐walking test at days 3, 7 and 14 after MCAO. Animals pre‐injected with Akt inhibitor IV and/or U0126 have higher scores in the beam‐walking test at days 3, 7 and 14 after MCAO [n = 7 for each group; *P < 0.05, vs. I/R + Vehicle, # P < 0.05, vs. I/R + bpV(pis), two‐way ANOVA, followed by the Bonferroni post hoc test]. (C) Animals treated with bpV(pis) have a higher ratio in the modified sticky‐tape (MST) test at days 7 and 14 after MCAO. Animals pre‐injected with Akt inhibitor IV and/or U0126 have lower ratio in the MST test at day 14 after MCAO [n = 7 for each group; *P < 0.05, vs. I/R + Vehicle, # P < 0.05, vs. I/R + bpV(pis), two‐way ANOVA, followed by the Bonferroni post hoc test]. I/R, ischaemia–reperfusion.
Discussion
PTEN is a tumour suppressor but plays an important role in neurological diseases (Endersby and Baker, 2008; Kreis et al., 2014; Weston et al., 2014; Stavarache et al., 2015). In our previous study, we demonstrated that suppressing the lipid phosphatase activity of PTEN activates Akt and suppressing the protein phosphatase activity of PTEN inhibits extrasynaptic GluN2B‐containing NMDA receptors, which lead to the protection against ischaemic neuronal death (Ning et al., 2004). We further showed that activation of GluN2A‐containing NMDA receptors exerts neuroprotective effects through suppression of PTEN and subsequent increase in nuclear TDP‐43 (TAR DNA‐binding protein‐43) (Zheng et al., 2012). We also showed that inhibiting PTEN, by enhancing the expression and function of the GABAA receptor, protects against neuronal death in the in vitro OGD and in vivo ischaemic stroke models (Liu et al., 2010a). Other studies have provided substantial evidence to support the notion that inhibiting PTEN confers neuroprotection in ischaemia–reperfusion injury (Howitt et al., 2012; Zhang et al., 2013). The results of the present study suggest that bpV(pis) inhibits PTEN to confer neuroprotection.
ERK1/2 activation has been reported to have a neuroprotective effect. A number of growth factors are shown to reduce ischaemic damage by up‐regulating ERK1/2 activity (Zhu et al., 2002; Buisson et al., 2003; Sansone et al., 2013). Oestrogen, osteopontin, leptin, melatonin derivative Neu‐P11 and apelin‐13 are also known to confer protection through ERK1/2 activation (Guerra et al., 2004; Meller et al., 2005; Zhang and Chen, 2008; Yang et al., 2014; Buendia et al., 2015). Our findings indicate that bpV(pis) protects against ischaemic neuronal death by activating ERK1/2, providing evidence to support the neuroprotective role of ERK1/2 in ischaemic stroke.
However, the activation of ERK1/2 by bpV(pis) is neither through PTEN inhibition nor via direct activation. MAP kinase is a family of serine/threonine protein kinases. The Raf protein, an up‐stream activator of MAP kinase, activates MEK1/2 through phosphorylation and then phosphorylated MEK1/2 phosphorylates and activates ERK1/2 (Lake et al., 2016). It is possible that the activation of ERK1/2 by bpV(pis) might depend upon the Raf/MEK/ERK1/2 and other signalling pathways. The detailed mechanisms remain to be further explored.
Bisperoxovanadium compounds belong to the class of vanadate derivatives that are well‐established inhibitors of protein tyrosine phosphatases and PTEN (Schmid et al., 2004). However, the mechanisms underlying the effect of bisperoxovanadium compounds have not been clarified. Squaramide is a functional group existing in many bioactive compounds. Some squaramide‐based compounds have anti‐leishmanial activity (Marin et al., 2016). Both in vitro and in vivo studies have confirmed that N,N′‐squaramides are effective against Chagas disease (Olmo et al., 2014). The squaric acid‐containing glutamate analogue exhibits a high affinity for NMDA receptors (Chan et al., 1995) and squaric acid dibutylester is a topical sensitizing agent utilized for the treatment of alopecia areata (Hill et al., 2015). Hence, because of its functional activity, stability, low cost of starting materials and straightforward synthesis, squaramide is an appropriate molecule for the development of affordable agents. We therefore designed and synthesized the compound, bpV(pis), in the presence of pyridine‐2‐squaramide and V2O5.
In conclusion, the results of the present study demonstrate for the first time that a new compound bpV(pis) confers neuroprotection both through suppressing PTEN and increasing the activation of ERK1/2 in cerebral ischaemia–reperfusion injury. These findings provide experimental evidence to support the development of bpV(pis) as a potential drug candidate for ischaemic stroke.
Author contributions
Z.F.Z., J.C., X.H., Y.Z., H.B.L., R.X.L. and Z.F.W. conducted the experiments, acquired and analysed the data. Z.Q.L., W.J.L., F.L., H.B.Z. and Q.W. conceived the project and designed the experiments. Z.F.Z., J.C. and Q.W. wrote the manuscript. Z.Q.L., W.J.L., F.L. and H.B.Z. reviewed and edited the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Sample images (above) and the summarized data (below) of TTC staining show that intraperitoneal injections of bpV(pis) (20–2000 μg/kg) at 1.0 h after I/R reduce the infarct volume at 24 h after ischaemia onset (n = 8 for each group; *P < 0.05 vs. I/R + Vehicle, two‐way ANOVA test, followed by Bonferroni post hoc test). Vehicle rats received intraperitoneal injections of corresponding proportion of DMSO solution dissolved in 0.9% saline. I/R: ischaemia–reperfusion.
{kind=link}
Acknowledgements
This work was supported by China Key Project of Basic Research (‘973’ Project; 2014CB541606), Natural Science Foundation of China (NSFC; 81470599) and Fund of Collaborative Innovation Center for Brain Science to Q.W.
Zhang, Z.‐F. , Chen, J. , Han, X. , Zhang, Y. , Liao, H.‐B. , Lei, R.‐X. , Zhuang, Y. , Wang, Z.‐F. , Li, Z. , Chen, J.‐C. , Liao, W.‐J. , Zhou, H.‐B. , Liu, F. , and Wan, Q. (2017) Bisperoxovandium (pyridin‐2‐squaramide) targets both PTEN and ERK1/2 to confer neuroprotection. British Journal of Pharmacology, 174: 641–656. doi: 10.1111/bph.13727.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronowski J, Samways E, Strong R, Rhoades HM, Grotta JC (1996). An alternative method for the quantitation of neuronal damage after experimental middle cerebral artery occlusion in rats: analysis of behavioral deficit. J Cereb Blood Flow Metab 16: 705–713. [DOI] [PubMed] [Google Scholar]
- Ashwal S, Tone B, Tian HR, Cole DJ, Pearce WJ (1998). Core and penumbral nitric oxide synthase activity during cerebral ischemia and reperfusion. Stroke 29: 1037–1046 .discussion 1047 [PubMed] [Google Scholar]
- Atif F, Yousuf S, Sayeed I, Ishrat T, Hua F, Stein DG (2013). Combination treatment with progesterone and vitamin D hormone is more effective than monotherapy in ischemic stroke: the role of BDNF/TrkB/ERK1/2 signaling in neuroprotection. Neuropharmacology 67: 78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ (1993). Optimized survival of hippocampal neurons in B27‐supplemented Neurobasal, a new serum‐free medium combination. J Neurosci Res 35: 567–576. [DOI] [PubMed] [Google Scholar]
- Buendia I, Gomez‐Rangel V, Gonzalez‐Lafuente L, Parada E, Leon R, Gameiro I et al. (2015). Neuroprotective mechanism of the novel melatonin derivative Neu‐P11 in brain ischemia related models. Neuropharmacology 99: 187–195. [DOI] [PubMed] [Google Scholar]
- Buisson A, Lesne S, Docagne F, Ali C, Nicole O, MacKenzie ET et al. (2003). Transforming growth factor‐beta and ischemic brain injury. Cell Mol Neurobiol 23: 539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PC, Roon RJ, Koerner JF, Taylor NJ, Honek JF (1995). A 3‐amino‐4‐hydroxy‐3‐cyclobutene‐1,2‐dione‐containing glutamate analogue exhibiting high affinity to excitatory amino acid receptors. J Med Chem 38: 4433–4438. [DOI] [PubMed] [Google Scholar]
- Chang SH, Poser S, Xia Z (2004). A novel role for serum response factor in neuronal survival. J Neurosci 24: 2277–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang N, El‐Hayek YH, Gomez E, Wan Q (2007). Phosphatase PTEN in neuronal injury and brain disorders. Trends Neurosci 30: 581–586. [DOI] [PubMed] [Google Scholar]
- Charan J, Kantharia ND (2013). How to calculate sample size in animal studies? J Pharmacol Pharmacother 4: 303–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li Y, Wang L, Zhang Z, Lu D, Lu M et al. (2001). Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke 32: 1005–1011. [DOI] [PubMed] [Google Scholar]
- Chien L, Chen WK, Liu ST, Chang CR, Kao MC, Chen KW et al. (2015). Low‐dose ionizing radiation induces mitochondrial fusion and increases expression of mitochondrial complexes I and III in hippocampal neurons. Oncotarget 6: 30628–30639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten M, Dhawahir A, Sum EY, Urosevic J, Lechuga CG, Esteban LM et al. (2010). Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J 29: 1091–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endersby R, Baker SJ (2008). PTEN signaling in brain: neuropathology and tumorigenesis. Oncogene 27: 5416–5430. [DOI] [PubMed] [Google Scholar]
- Geoffroy CG, Lorenzana AO, Kwan JP, Lin K, Ghassemi O, Ma A et al. (2015). Effects of PTEN and Nogo codeletion on corticospinal axon sprouting and regeneration in mice. J Neurosci 35: 6413–6428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra B, Diaz M, Alonso R, Marin R (2004). Plasma membrane oestrogen receptor mediates neuroprotection against beta‐amyloid toxicity through activation of Raf‐1/MEK/ERK cascade in septal‐derived cholinergic SN56 cells. J Neurochem 91: 99–109. [DOI] [PubMed] [Google Scholar]
- Hao T, Rockwell P (2013). Signaling through the vascular endothelial growth factor receptor VEGFR‐2 protects hippocampal neurons from mitochondrial dysfunction and oxidative stress. Free Radic Biol Med 63: 421–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill ND, Bunata K, Hebert AA (2015). Treatment of alopecia areata with squaric acid dibutylester. Clin Dermatol 33: 300–304. [DOI] [PubMed] [Google Scholar]
- Howitt J, Lackovic J, Low LH, Naguib A, Macintyre A, Goh CP et al. (2012). Ndfip1 regulates nuclear PTEN import in vivo to promote neuronal survival following cerebral ischemia. J Cell Biol 196: 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilic E, Kilic U, Soliz J, Bassetti CL, Gassmann M, Hermann DM (2005). Brain‐derived erythropoietin protects from focal cerebral ischemia by dual activation of ERK‐1/‐2 and Akt pathways. FASEB J 19: 2026–2028. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M (2005). Differential roles of NR2A‐ and NR2B‐containing NMDA receptors in Ras–ERK signaling and AMPA receptor trafficking. Neuron 46: 745–760. [DOI] [PubMed] [Google Scholar]
- Koh PO (2015). Ferulic acid attenuates the down‐regulation of MEK/ERK/p90RSK signaling pathway in focal cerebral ischemic injury. Neurosci Lett 588: 18–23. [DOI] [PubMed] [Google Scholar]
- Kreis P, Leondaritis G, Lieberam I, Eickholt BJ (2014). Subcellular targeting and dynamic regulation of PTEN: implications for neuronal cells and neurological disorders. Front Mol Neurosci 7: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake D, Correa SA, Muller J (2016). Negative feedback regulation of the ERK1/2 MAPK pathway. Cell Mol Life Sci 73: 4397–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowski G, Steward O (2014). AAVshRNA‐mediated suppression of PTEN in adult rats in combination with salmon fibrin administration enables regenerative growth of corticospinal axons and enhances recovery of voluntary motor function after cervical spinal cord injury. J Neurosci 34: 9951–9962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Liao M, Mielke JG, Ning K, Chen Y, Li L et al. (2006). Ischemic insults direct glutamate receptor subunit 2‐lacking AMPA receptors to synaptic sites. J Neurosci 26: 5309–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Li L, Zhang Q, Chang N, Wang D, Shan Y et al. (2010a). Preservation of GABAA receptor function by PTEN inhibition protects against neuronal death in ischemic stroke. Stroke 41: 1018–1026. [DOI] [PubMed] [Google Scholar]
- Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears‐Kraxberger I et al. (2010b). PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci 13: 1075–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Xu Q, Wang H, Wang R, Hou XY (2013). Neuroprotection of ischemic postconditioning by downregulating the postsynaptic signaling mediated by kainate receptors. Stroke 44: 2031–2035. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R (1989). Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20: 84–91. [DOI] [PubMed] [Google Scholar]
- Maehama T, Dixon JE (1998). The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5‐trisphosphate. J Biol Chem 273: 13375–13378. [DOI] [PubMed] [Google Scholar]
- Marin C, Ximenis M, Ramirez‐Macias I, Rotger C, Urbanova K, Olmo F et al. (2016). Effective anti‐leishmanial activity of minimalist squaramide‐based compounds. Exp Parasitol 170: 36–49. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller R, Stevens SL, Minami M, Cameron JA, King S, Rosenzweig H et al. (2005). Neuroprotection by osteopontin in stroke. J Cereb Blood Flow Metab 25: 217–225. [DOI] [PubMed] [Google Scholar]
- Milella M, Falcone I, Conciatori F, Cesta Incani U, Del Curatolo A, Inzerilli N et al. (2015). PTEN: multiple functions in human malignant tumors. Front Oncol 5: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano Y, Suzuki Y, Takagi T, Kitashoji A, Ono Y, Tsuruma K et al. (2014). Glycoprotein nonmetastatic melanoma protein B (GPNMB) as a novel neuroprotective factor in cerebral ischemia–reperfusion injury. Neuroscience 277: 123–131. [DOI] [PubMed] [Google Scholar]
- Ning K, Pei L, Liao M, Liu B, Zhang Y, Jiang W et al. (2004). Dual neuroprotective signaling mediated by downregulating two distinct phosphatase activities of PTEN. J Neurosci 24: 4052–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtake Y, Park D, Abdul‐Muneer PM, Li H, Xu B, Sharma K et al. (2014). The effect of systemic PTEN antagonist peptides on axon growth and functional recovery after spinal cord injury. Biomaterials 35: 4610–4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmo F, Rotger C, Ramirez‐Macias I, Martinez L, Marin C, Carreras L et al. (2014). Synthesis and biological evaluation of N,N′‐squaramides with high in vivo efficacy and low toxicity: toward a low‐cost drug against Chagas disease. J Med Chem 57: 987–999. [DOI] [PubMed] [Google Scholar]
- Petullo D, Masonic K, Lincoln C, Wibberley L, Teliska M, Yao DL (1999). Model development and behavioral assessment of focal cerebral ischemia in rats. Life Sci 64: 1099–1108. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Meller R, Inoue K, Ordonez AN, Ashley MD, Xiong Z et al. (2008). In vivo and in vitro characterization of a novel neuroprotective strategy for stroke: ischemic postconditioning. J Cereb Blood Flow Metab 28: 232–241. [DOI] [PubMed] [Google Scholar]
- Qu Y, Mao M, Zhao F, Zhang L, Mu D (2009). Proapoptotic role of human growth and transformation‐dependent protein in the developing rat brain after hypoxia‐ischemia. Stroke 40: 2843–2848. [DOI] [PubMed] [Google Scholar]
- Rosivatz E, Matthews JG, McDonald NQ, Mulet X, Ho KK, Lossi N et al. (2006). A small molecule inhibitor for phosphatase and tensin homologue deleted on chromosome 10 (PTEN). ACS Chem Biol 1: 780–790. [DOI] [PubMed] [Google Scholar]
- Roux PP, Blenis J (2004). ERK and p38 MAPK‐activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 68: 320–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansone L, Reali V, Pellegrini L, Villanova L, Aventaggiato M, Marfe G et al. (2013). SIRT1 silencing confers neuroprotection through IGF‐1 pathway activation. J Cell Physiol 228: 1754–1761. [DOI] [PubMed] [Google Scholar]
- Schmid AC, Byrne RD, Vilar R, Woscholski R (2004). Bisperoxovanadium compounds are potent PTEN inhibitors. FEBS Lett 566: 35–38. [DOI] [PubMed] [Google Scholar]
- Segarra J, Balenci L, Drenth T, Maina F, Lamballe F (2006). Combined signaling through ERK, PI3K/AKT, and RAC1/p38 is required for met‐triggered cortical neuron migration. J Biol Chem 281: 4771–4778. [DOI] [PubMed] [Google Scholar]
- Shan Y, Liu B, Li L, Chang N, Li L, Wang H et al. (2009). Regulation of PINK1 by NR2B‐containing NMDA receptors in ischemic neuronal injury. J Neurochem 111: 1149–1160. [DOI] [PubMed] [Google Scholar]
- Shi GD, OuYang YP, Shi JG, Liu Y, Yuan W, Jia LS (2011). PTEN deletion prevents ischemic brain injury by activating the mTOR signaling pathway. Biochem Biophys Res Commun 404: 941–945. [DOI] [PubMed] [Google Scholar]
- Singh B, Singh V, Krishnan A, Koshy K, Martinez JA, Cheng C et al. (2014). Regeneration of diabetic axons is enhanced by selective knockdown of the PTEN gene. Brain 137 (Pt 4): 1051–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavarache MA, Musatov S, McGill M, Vernov M, Kaplitt MG (2015). The tumor suppressor PTEN regulates motor responses to striatal dopamine in normal and Parkinsonian animals. Neurobiol Dis 82: 487–494. [DOI] [PubMed] [Google Scholar]
- Sughrue ME, Mocco J, Komotar RJ, Mehra A, D'Ambrosio AL, Grobelny BT et al. (2006). An improved test of neurological dysfunction following transient focal cerebral ischemia in rats. J Neurosci Methods 151: 83–89. [DOI] [PubMed] [Google Scholar]
- Sun Y, Jin K, Xie L, Childs J, Mao XO, Logvinova A et al. (2003). VEGF‐induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Invest 111: 1843–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Ma M, Teng J, Zhang J, Zhou S, Zhang Y et al. (2015). Chronic exposure to cerebrospinal fluid of multiple system atrophy in neuroblastoma and glioblastoma cells induces cytotoxicity via ER stress and autophagy activation. Oncotarget 6: 13278–13294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston MC, Chen H, Swann JW (2014). Loss of mTOR repressors Tsc1 or Pten has divergent effects on excitatory and inhibitory synaptic transmission in single hippocampal neuron cultures. Front Mol Neurosci 7: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler EJ, Peters EE, Gonzales A, Gonzales ML, Slee AM, Kerr JS (2002). An objective procedure for ischemic area evaluation of the stroke intraluminal thread model in the mouse and rat. J Neurosci Methods 113: 51–58. [DOI] [PubMed] [Google Scholar]
- Yamada KM, Araki M (2001). Tumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosis. J Cell Sci 114 (Pt 13): 2375–2382. [DOI] [PubMed] [Google Scholar]
- Yang Y, Zhang X, Cui H, Zhang C, Zhu C, Li L (2014). Apelin‐13 protects the brain against ischemia/reperfusion injury through activating PI3K/Akt and ERK1/2 signaling pathways. Neurosci Lett 568: 44–49. [DOI] [PubMed] [Google Scholar]
- Zhang F, Chen J (2008). Leptin protects hippocampal CA1 neurons against ischemic injury. J Neurochem 107: 578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QG, Wu DN, Han D, Zhang GY (2007). Critical role of PTEN in the coupling between PI3K/Akt and JNK1/2 signaling in ischemic brain injury. FEBS Lett 581: 495–505. [DOI] [PubMed] [Google Scholar]
- Zhang S, Taghibiglou C, Girling K, Dong Z, Lin SZ, Lee W et al. (2013). Critical role of increased PTEN nuclear translocation in excitotoxic and ischemic neuronal injuries. J Neurosci 33: 7997–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Liao M, Cui T, Tian H, Fan DS, Wan Q (2012). Regulation of nuclear TDP‐43 by NR2A‐containing NMDA receptors and PTEN. J Cell Sci 125 (Pt 6): 1556–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Yang GY, Ahlemeyer B, Pang L, Che XM, Culmsee C et al. (2002). Transforming growth factor‐beta 1 increases bad phosphorylation and protects neurons against damage. J Neurosci 22: 3898–3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zukor K, Belin S, Wang C, Keelan N, Wang X, He Z (2013). Short hairpin RNA against PTEN enhances regenerative growth of corticospinal tract axons after spinal cord injury. J Neurosci 33: 15350–15361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Sample images (above) and the summarized data (below) of TTC staining show that intraperitoneal injections of bpV(pis) (20–2000 μg/kg) at 1.0 h after I/R reduce the infarct volume at 24 h after ischaemia onset (n = 8 for each group; *P < 0.05 vs. I/R + Vehicle, two‐way ANOVA test, followed by Bonferroni post hoc test). Vehicle rats received intraperitoneal injections of corresponding proportion of DMSO solution dissolved in 0.9% saline. I/R: ischaemia–reperfusion.