

Abstract
Inhibition of cyclin dependent kinases (CDKs) 4 and 6 prevent cells from entering the synthesis phase of the cell cycle. CDK4 and 6 are therefore important drug targets in various cancers. The selective CDK4/6 inhibitor palbociclib is approved for the treatment of breast cancer and has shown activity in a cellular model of mixed lineage leukaemia (MLL)‐rearranged acute myeloid leukaemia (AML). We studied the interactions of palbociclib and CDK6 using molecular dynamics simulations. Analysis of the simulations suggested several interactions that stabilized the drug in its binding site and that were not observed in the crystal structure of the protein‐drug complex. These included a hydrogen bond to His 100 that was hitherto not reported and several hydrophobic contacts. Evolutionary‐based bioinformatic analysis was used to suggest two mutants, D163G and H100L that would potentially yield drug resistance, as they lead to loss of important protein–drug interactions without hindering the viability of the protein. One of the mutants involved a change in the glycine of the well‐conserved DFG motif of the kinase. Interestingly, CDK6‐dependent human AML cells stably expressing either mutant retained sensitivity to palbociclib, indicating that the protein‐drug interactions are not affected by these. Furthermore, the cells were proliferative in the absence of palbociclib, indicating that the Asp to Gly mutation in the DFG motif did not interfere with the catalytic activity of the protein.
Keywords: resistance mutations, molecular dynamics, protein–drug interactions, DFG motif
Short abstract
PDB Code(s): 2EUF
Abbreviations
- AML
acute myeloid leukaemia
- CDK
cyclin dependent kinase
- ER
oestrogen receptor
- KI
kinase inhibitor
- MD
molecular dynamics
- MLL
mixed lineage leukaemia
- MSA
multiple sequence alignment
- SNV
single nucleotide variance
Introduction
Targeted cancer therapies are drugs that affect specific molecules in the cancer cell (molecular targets), and thereby limit the progression of the tumour, or even lead to a diminishment of the tumour's size. These drugs are more specific than chemotherapy, which kills dividing cells and is often associated with severe side effects. Many targeted therapies are kinase inhibitors (KIs); they inhibit certain kinases, which are necessary for the signalling network of the tumour. The interest in kinases started with the development of imatinib mesylate (Glivec®) as a treatment for chronic myeloid leukaemia. Imatinib mesylate was approved by the U S Food and Drug Administration (FDA) in 2001. Since then, about 30 KIs have been approved, mostly as drugs against cancer. Unfortunately, many of these drugs become ineffective after months or even years of use due to the development of resistance within the tumour cells.1 Resistance often develops by somatic mutations where a single nucleotide variance (SNV) between the original and mutated drug target leads to a missense mutation that reduces the binding affinity for the protein–drug complex.
CDKs are serine/threonine protein kinases which are activated by the binding of regulatory cyclins, and in some cases phosphorylation by various protein kinases. CDK1, CDK2, CDK4, and CDK6 are involved in cell‐cycle control and have therefore been considered as targets for cancer therapy. The highly homologous proteins CDK4 and CDK6 regulate entry into the cell cycle. Palbociclib (PD0332991, now marketed as Ibrance®), an inhibitor of CDK4 and CDK6 has been approved by the FDA on February 2015 for use in combination with letrozole (an aromatase inhibitor) for the treatment of postmenopausal women with oestrogen receptor (ER)‐positive, human epidermal growth factor receptor 2 (HER2)‐negative advanced (metastatic) breast cancer. The drug has recently been approved in Europe as well, and is subject to clinical trials in patients suffering from other cancers.
The structure of the CDK6‐palbociclib complex was solved by X‐ray crystallography (PDB code 2EUF 2). Other complexes of CDK6 that were solved by X‐ray crystallography include CDK6 with endogenous inhibitors,3, 4 with a flavonol inhibitor,5 with pyrazol‐pyramidine inhibitors,6 with azabenzimidazoles7 and with FLX925 (also known as AMG925), which is a dual CDK4/6 and FLT3 inhibitor.8 The structure of CDK4 is yet to be solved but is presumably very similar since the proteins' sequences are 64.1% identical and 73.9% similar. The proteins are comprised almost exclusively of a catalytic core. The core is built of two lobes with the ATP and substrate (or inhibitor) binding between them. The structural features of CDK6 (Fig. 1) include the two lobes and a short hinge region between them. The specificity of palbociclib is due to interactions with the hinge region.2 A viral cyclin interacts with CDK6 via multiple contacts to the N and C lobes, including contacts to the catalytically important activation loop.
Figure 1.
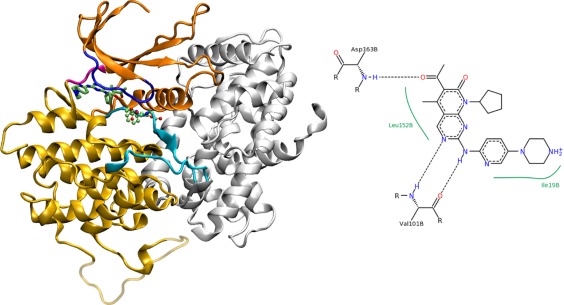
The structure of palbociclib‐bound CDK6 (PDB code 2EUF). The kinase (color) is bound to the activator V‐cyclin (white). The DFG loop adopts an active (DFG‐in) conformation. The interface region includes parts of the kinase activation loop (cyan), but not the DFG motif (displayed in a ball‐and‐stick representation). The drug palbociclib (stick model) is bound between the C‐domain (light orange), the glycine‐rich loop (blue) and the hinge region (magenta). The N‐lobe is colored dark orange. Residues of the C‐lobe that were missing in the X‐ray structure are transparent. A 2D representation of protein‐drug interactions (adapted from the PDB) is also shown.
Palbocilib is a pyridopyrimidine derivative. When bound to CDK6, its piperazine moiety is water‐accessible. According to the reported crystal structure, it binds to CDK6 through three hydrogen bonds (two to the backbone of Val 101 and one to the backbone of Asp 163, see Fig. 1) and several hydrophobic contacts, altogether leading to very tight binding (IC50 10 nM for CDK4 and CDK6 in complex with cyclin D9). Ribociclib (LEE011) and Abemaciclib (LY2835219) are CDK4/CDK6 dual inhibitors that are similar to palbocilib in structure and presumably bind the target in almost the same way.
Whereas the crystal structure of palbociclib‐bound CDK6 provides valuable information, there is a need for a better understanding of the protein–drug interactions. The resolution of the structure (3 Å) may be insufficient to account for all possible interactions. It should also be noted that it is not clear to what extent the complex presented in the X‐ray structures (CDK6 with a viral cyclin) is similar to complexes with human cyclin D proteins. Moreover, flexibility of the binding site and the drug can play a role in binding but such effects are often not captured within crystal structures. In the absence of solution structures, computer simulations and modeling offer an alternative to study protein–drug interactions. In particular, atomistic molecular dynamics (MD) simulations have proven very useful for drug discovery10 and cancer research.11 Such simulations can provide data on alternative binding sites, conformations, or contacts that are not observed in the crystal. Accordingly, MD simulations were used to study CDKs. Bártová et al. carried out MD simulations of CDK2 in various states to compare between active, partially active and inactive conformations.12 The same group later studied the mechanism of inhibition of CDK2 by phosphorylation,13, 14 compared the dynamics of CDK2 and CDK5,15 and identified flexible and rigid regions in the protein by simulations.16 In another interesting application of MD to the study of CDKs, Andrews et al. used simulations in the process of inhibitor design.17 Several groups used simulations to rationalize the activity and/or specificity of CDK inhibitors.18, 19, 20 These studies exemplify the utility of MD simulations to understand the dynamics of kinases, and CDKs in particular.
We used MD simulations of the CDK6‐palbociclib complex in an effort to model how the protein and drug interact in vitro. We further employed evolutionary‐based bioinformatic analysis to suggest potential mutations that could lead to palbociclib resistance. One of these mutants was studied by MD simulations as well, and the results were compared to those obtained with the wild‐type protein. Finally, two mutants were tested for their ability to hinder the effects of palbociclib in AML cells.
Results
Dynamics of palbociclib‐bound CDK6 in the absence of cyclin
The reported crystal structure of CDK6 was solved in its cyclin‐bound form. Because the cyclin activator does not interfere with the binding of the drug, we simulated the protein in the absence of cyclin and followed closely on its overall dynamics, to make sure that the sampling is correct. Indeed, the protein maintained its overall shape and secondary structure during the simulations (Fig. 2). Clustering analysis revealed some modifications in the activation and glycine‐rich loops (Fig. 3, see also the molecular movie in the Supporting Information). The unstructured region of the activation loop which is located at the cyclin interface is very mobile, whereas the DFG (Asp‐Phe‐Gly) motif and nearby C helix maintain their structure and location.
Figure 2.

Overall structural features calculated during four individual simulations of CDK6 inhibited by palbociclib. The solvent accessible surface area, radius of gyration and its principle components (PC1, PC2, and PC3), number of residues with a regular secondary structure and Cα root mean square deviation with respect to the crystal structure are shown. The colors represent individual simulations.
Figure 3.

Clustering analysis of the activation and glycine rich loops. All clusters of the simulations were superimposed on the structure of the protein, revealing the dynamics of the loops. The color code is the same as in Figure 1. Clustering analysis identified 2246 configurations in total (from 40000 frames) of which 186 contained 50 snapshot structures or more.
One of the interesting features of the structure is the hydrogen bond between Asp 163 of the DFG motif and Ala 23 of the glycine‐rich loop. This hydrogen bond was present in 88% of the simulation time over all simulations. A closer look at individual simulations revealed that hydrogen bonds between Asp 163 and Ala 23 were present throughout all of the simulation time in two simulations. In one simulation these bonds broke down after >40 ns, and in the fourth simulation they broke down but established again (Supporting Information, Fig. S1). Structurally, a hydrogen bond between Asp 163 and Ala 23 keeps the activation and glycine‐rich loops close to each other. This, in turn, makes binding of the drug so tight.2 A similar hydrogen bond is present in structures of other CDKs. The existence and persistence of this bond appears to depend on the presence and identity of an inhibitor. No such bond was evident in reference simulations of the protein without an inhibitor.
Drug‐receptor interactions between palbociclib and CDK6
The number of contacts between the protein and the drug is reported in Supporting Information Table SI. There were more contacts between the drug and the protein in the crystal structure than in the simulation. This might be expected, as palbociclib is partially solvent‐exposed. It is quite common that, when solubilized in water at room temperature, some of the protein‐drug contacts observed in the crystal structures are not constantly maintained and others may form. Here, the distal amino nitrogen (N4) of the piperazine ring is fully exposed to the solvent, whereas the proximal one (N1) is not involved in any hydrogen bonds with the protein. Moreover, the crystal structure of the complex was solved to a resolution of 3.0 Å, which makes the analysis of contacts somewhat inaccurate, as noted by the authors who published the structure.2
Whereas the number of contacts between the protein and the drug was larger in the X‐ray than in the simulations, the number of residues that were in contact with the drug was larger in the simulations, though not all of them maintained contact with the drug at any specific time. There were 17 residues that were involved in protein–drug contacts in the crystal structure and 24 that formed protein–drug contacts in all four simulations. These include the original 17 residues, namely Ile 19, Gly 20, Val 27, Lys 43, Val 77, Phe 98, Glu 99, Val 101, Asp 102, Gln 103, Asp 104, Thr 107, Gln 149, Asn 150, Leu 152, Ala 162, and Asp 163. The residues that were identified in the MD simulations as interacting (Fig. 4) are located at the glycine‐rich loop (Glu 18, Glu 21, Gly 22), the N‐terminal lobe (Ala 41), the N‐terminal helix (Glu 61), hinge region (His 100), and activation loop (Phe 164). Analysis of the interaction energies between these residues and the drug (Supporting Information Table SII) revealed that the residues that contribute mostly to the binding energy are Phe 164 (−24.4 kcal mol−1), Leu 152 (−18.5 kcal mol−1), Ile 19 (−8.5 kcal mol−1), Val 101 (−7.2 kcal mol−1), His 100 (−6.4 kcal mol−1), and Asp 163 (−5.8 kcal mol−1).
Figure 4.

Residues that make contact with the drug during the simulations but not in the X‐ray structure are displayed on the crystal structure in ball‐and stick representation and colored according to residue type (white—hydrophobic, green—polar, blue—positive or His, and red—negative). The color code for the protein structure is the same as in Figure 1.
Palbociclib was involved in three hydrogen‐bonds with the protein, based on donor–acceptor distances as observed in the crystal structure. Two were with the carbonyl oxygen and amino nitrogen of the backbone of Val 101 and one with the amino nitrogen of the backbone of Asp 163 of the DFG motif. There were also almost three hydrogen bonds on average during the simulations (Supporting Information Table SI). The two hydrogen bonds with Val 101 and the hydrogen bond with Asp 163 were present in most of the simulation time. Additionally, a hydrogen bond between His 100 and the pyridine ring nitrogen was present in 53% of the simulation time (average over all simulations).
Can resistance mutations lead to palbociclib‐resistant CDK6?
Resistance mutations are SNVs that lead to reduced affinity between the drug target and the drug. Whereas resistance mutations were observed for different kinases and inhibitors, none has hitherto been reported for palbociclib. Because the drug is fairly new in the market, it is too early to predict if such mutations emerge. The binding mode of palbociclib is rather robust in this respect, as its piperazine moiety is solvent‐exposed and it is hydrogen bonded only to backbone atoms, so that mutations of the hydrogen‐bonding residues are not expected to lead to the breaking of any of the hydrogen bonds.
Examination of three other kinases has shown that resistance mutations are seldom novel with respect to the evolutionary development of kinases.21 In other words, when a resistance mutation occurs, a residue is modified to another residue that can be observed at the same position in a homologous or orthologous protein. Palbociclib interacted with the side‐chain of His 100, to which it was hydrogen bonded throughout most of the simulation time. We carried out multiple sequence alignment (MSA) of proteins that show sequence homology to CDK6. Interestingly, analysis of the MSA revealed that variations in the position of His 100 are common. His was present in only 9% of the aligned sequences. Tyr (44%) and Phe (36%) were more common whereas Leu (8%) and Asn (1%) were also found. Examination of the DNA codon revealed that SNVs can modify His 100 to Tyr, Asn or Leu. The side chains of Tyr and Asn can be hydrogen‐bonded to the drug, but that of Leu cannot. Thus, an H100L mutation could potentially perturb the binding of palbociclib. However, since the hydrogen‐bond between His 100 and the drug is not observed in the crystal structure, it is likely that the drug can bind the protein irrespective of the presence of this hydrogen bond.
Another factor that can affect the binding of the drug is the interaction between the glycine‐rich and activation loops (Fig. 1). The glycine‐rich loop sits on top of the drug and aids in its fitting to its binding place. Its location is dictated by a key hydrogen bond between Asp 163 and the backbone of Ala 23. Ala 23 is not conserved and since it is its backbone nitrogen that interacts with Asp 163 a mutation of the residue does not necessarily lead to weakening of the hydrogen bond. Asp 163 is highly conserved, as part of the DFG motif that is a hallmark of many kinases. Interestingly, Gly substitutes Asp in the sequence of mouse peripheral plasma membrane protein CASK that has been identified in our search for homologues. Needleman–Wunsch alignment confirms the variation and the overall homology between the two proteins (28.5% indentity, 47.5% similarity, for the alignment see Fig. 5). Examination of CASK proteins from other organisms (including human) revealed that their kinase domain indeed includes a GFG motif rather than the more usual DFG, i.e., this is not a sequencing error or an error in the sequence database. Structural and kinetic studies of CASK confirmed that it is an atypical but catalytically active kinase.22 This suggests that kinases can be active even in the absence of the conserved DFG motif. Interestingly, some CDKs also carry a modified activation loop motif (DMG in human CDK8 and CDK19).
Figure 5.

Needelman–Wunsch alignment between human CDK6 (CDK6_HUMAN, uniprot code Q00534) and mouse CASK (CSKP_MOUSE, uniprot code O70589). Note that the CASK carries a GFG sequence instead of the more common DFG motif (red box).
The CDK6 mutants H100L and D163G retain sensitivity to 100 nM palbociclib in THP‐1 cells
Sequence analysis indicated that the H100L and D163G mutants are viable in spite of the high conservation of the DFG motif. To examine this, THP‐1 cells, which were established from a patient with MLL‐rearranged AML, were transduced with lentiviral vectors encoding either wildtype CDK6 or one of the two mutants. Western blot analysis demonstrated expression of either mutant, albeit at slightly lower levels than endogenous CDK6 [Fig. 6(A)]. Palbociclib was recently shown to induce expression of the myeloid differentiation marker CD11b on the surface of MLL‐rearranged AML cells.23 Flow cytometric analysis demonstrated that stable expression of CDK6 H100L or CDK6 D163G did not impair the capacity of 100 nM palbociclib to induce CD11b expression in THP‐1 cells [Fig. 6(B)]. Similarly, neither of the two mutants mitigated the anti‐proliferative effect of palbociclib treatment in THP‐1 cells [Fig. 6(C)]. Of note, cells containing the CDK6 mutants maintained proliferation in the absence of the drug [Fig. 6(C)], which indicates that the CDK6 protein was active (due to difficulty in expressing the protein a kinetic analysis of the enzyme was not performed and the catalytic activity was deduced from cell viability).
Figure 6.

Evaluation of the viability of CDK6 mutants in THP‐1 cells in the presence and absence of palbociclib. Expression of HA‐tagged CDK6 wildtype and mutant constructs was verified by western blot analysis 11 days after transduction (A). The CDK6 blot shows both the endogenous (lower band) and the exogenous HA‐tagged protein (upper band). Neither phosphorylation nor expression levels of RB1 changed as a result of CDK6 (wildtype or mutant) overexpression. Band intensities were quantified using ImageJ software (version 1.46r; right panel). Flow cytometric analysis of the myeloid differentiation marker CD11b after 7 days of treatment (B) and assessment of proliferation by cell counting (C) revealed no differences between the constructs upon addition of palbociclib. Data of four independent experiments are shown.
Discussion
Simulations of cyclin‐free palbociclib‐bound CDK6 were carried out, and indicated that the protein maintained its structure during multiple 100 ns long simulations in the absence of cyclin. The drug was bound through a combination of hydrogen‐bonds and hydrophobic interactions, whereas its charged moiety was stabilized through interactions with the solvent. This enabled some conformational freedom at the piperizine ring even when the drug was bound. Some interactions between the drug and the protein were observed in the simulations but not in the crystal structure. This may be ascribed to the low resolution of the latter. Of these, hydrogen bonding to atom of the His 100 seemed to be the most important.
Bioinformatic analysis indicated two potential SNV mutations that would be viable and could potentially lead to drug resistance, namely H100L and D163G. Asp 163 is highly conserved, as part of the DFG‐motif. However, the presence of a GFG motif in peripheral plasma membrane protein CASK suggested that the mutant would potentially be active. We note for example the G2032R mutation of the kinase ROS1 which mutates a very conserved glycine residue and leads to resistance against crizotinib.24
Though the simulations suggested that protein–drug hydrogen bonds would be lost in the H100L and D163G mutants, we did not observe unbinding of the drug in the simulations. Experiments with a cellular model of MLL‐rearranged AML confirmed that the proteins were active, whereas transduced cells remained sensitive to the drug. The binding mode of palbociclib thus appears to be quite robust. Although the mutations led to fewer hydrogen bonds and contacts between the drug and the protein, palbociclib retained the ability to inhibit CDK6 at a concetration of 100 nM, which is within the range of plasma concentrations observed in patients.25
In our opinion, the sensitivity of the H100L and D163G mutant proteins to palbociclib does not contradict the observations of the MD simulations. Rather, it shows that a small reduction in the number of protein–drug contacts and the lack of a single hydrogen‐bond (on average) were not enough to make the protein resistance to binding of the drug. Additional studies of resistant mutants will be necessary to enable better predictions of resistance based on MD simulations.
Materials and Methods
Preparation of the structure for simulations
The structure of the complex between CDK6, palbociclib and a viral cyclin was downloaded from the protein data bank (code 2EUF). Missing residues were modelled by use of Swissmodel.26 Force‐field parameters for the drug were generated by the SwissParam server.27 Palbociclib's piperazin moiety was protonated at both nitrogen atoms. Protein residues were modelled in their standard state, that is, Glu and Asp were nonprotonated, Arg and Lys fully protonated, and His mono‐protonated. In particular, residue His100 was protonated at of the imidazole side chain. Reference simulations of the noninhibited (apo) protein were performed starting from the same structure after removing the drug molecule.
Molecular dynamics simulations
All simulations and preparations were performed with the Gromacs simulation package,28, 29 version 4.6.7. Energies and forces were calculated by use of the CHARMM22 force field,30 as implemented in Gromacs.31 The protein was solvated in a cubic box so that the minimum distance between any protein atom and the edge of the box was 1.2 nm. The water molecules were modelled with the TIP3P model.32 K+ and Cl‐ ions were added in order to neutralize the charge and mimic an ionic strength of 0.1 M. Before each MD simulation, internal constraints were relaxed by energy minimization, until the maximal force on individual atoms was smaller than 100 kJ mol−1 nm−1. After the minimization, restrained MD runs were performed for 20 ps. During the restrained simulations, protein heavy atoms were fixed to their initial positions with a force constant of 1000 kJ mol−1 nm−2. The restraints were released, and the system was equilibrated for 5 ns before data collection for analysis. During the MD runs, the LINCS algorithm33 was used to constrain the lengths of bonds, while water molecules were kept rigid by use of the SETTLE algorithm.34
The temperature was kept constant (T = 300 K) by use of the velocity‐rescaling algorithm35 (τ T = 0.1 ps). The pressure was coupled to an external bath with Berendsen's coupling algorithm36 (P ref = 1 bar, τ P = 1 ps) during the equilibration and by the Parrinello–Rahman algorithm37 afterwards. Van der Waals forces were truncated at 1.0 nm with a plain cutoff. Long‐range electrostatic forces were treated using the particle mesh Ewald method.38 Dispersion correction was applied for the energy and pressure. Four independent 100 ns simulations were run for each system.
Analysis of the simulations
Analysis was performed in Gromacs. Secondary structure analysis was performed with the DSSP algorithm.39 Hydrogen bonds were calculated along the simulation trajectories using the Gromacs program g_hbond with default parameters. A drug‐protein contact is defined whenever a pair of heavy (non‐hydrogen) atoms of the two molecules is located within 0.4 nm of each other. Clustering analysis employed the algorithm developed by Daura et al.,40 with a cutoff of 0.1 nm. Analysis of the interaction energy between palbociclib and the residues that form contacts with it was performed by summing up the short‐range Coulomb and Lennard–Jones interactions from a simulation of the complex.
Graphical presentations
Protein figures were generated by use of the VMD program.41
Analysis of sequence variations in homologues of CDK6
Analysis of the sequence variations in homologues and orthologues of CDK6 was performed as before.21 Briefly, the protein sequence was downloaded, and 2000 sequences that are most similar to it were identified, downloaded and screened for uniqueness. The remaining 1319 sequences were aligned together by use of the FFT‐NS‐2 method within the MAFFT program.42
Cell culture
AML THP‐1 cells were maintained under standard conditions. Cell line identity and purity were verified using the multiplex cell authentication and contamination tests (Multiplexion). Palbociclib was obtained from Selleck.
Plasmids and viral transduction
The CDK6 complementary DNA (cDNA) was obtained from Open Biosystems and introduced into the lentiviral vector pLenti6.2/V5‐DEST using Gateway technology43 for stable expression in human cells. The CDK6 mutants were generated using the QuikChange XL Site‐Directed vector Mutagenesis Kit (Stratagene). pLenti6.2/V5 DEST 1x FLAG was used as empty vector control. Generation of viral supernatants and viral transduction were performed as described previously.44
Immunoblotting
Immunoblotting was performed using standard procedures with the following antibodies: mouse anti‐CDK6 (DCS83, Cell Signaling), rabbit anti‐HA‐Tag (C29F4, Cell Signaling) and rabbit anti‐HSP90 (H‐114, Santa Cruz Biotechnology). Whole‐cell protein extracts were prepared using lysis buffer containing 20 mM Tris, 1% Triton X‐100, 150 mM NaCl, 1 mM EDTA, and 10% glycerol supplemented with Halt Protease and Phosphatase Inhibitor Cocktail (Pierce), and 50 of protein were subjected to SDS‐PAGE and immunoblotting.
Determination of viable cell numbers
Viable cell numbers in suspension culture were determined by trypan‐blue staining followed by cell counting using a Neubauer chamber.
Flow cytometry analysis
To determine the expression of surface antigens associated with myelomonocytic differentiation, cells were stained with PE‐conjugated anti‐CD11b or IgG1κ isotype control (BD Biosciences). Samples were analyzed on a BD LSRII flow cytometer after 7 days.
Supporting information
Supporting Information 1
Supporting Information 2
Acknowledgments
The authors thank Stefanie Reinhart for excellent technical assistance.
References
- 1. Friedman R (2016) Drug resistance in cancer: molecular evolution and compensatory proliferation. Oncotarget 7:11746–11755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lu H, Schulze‐Gahmen U (2006) Toward understanding the structural basis of cyclin‐dependent kinase 6 specific inhibition. J Med Chem 49:3826–3831. [DOI] [PubMed] [Google Scholar]
- 3. Brotherton DH, Dhanaraj V, Wick S, Brizuela L, Domaille PJ, Volyanik E, Xu X, Parisini E, Smith BO, Archer SJ, Serrano M, Brenner SL, Blundell TL, Laue ED (1998) Crystal structure of the complex of the cyclin D‐dependent kinase Cdk6 bound to the cell‐cycle inhibitor p19INK4d. Nature 395:244–250. [DOI] [PubMed] [Google Scholar]
- 4. Russo AA, Tong L, Lee JO, Jeffrey PD, Pavletich NP (1998) Structural basis for inhibition of the cyclin‐dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature 395:237–243. [DOI] [PubMed] [Google Scholar]
- 5. Lu H, Chang DJ, Baratte B, Meijer L, Schulze‐Gahmen U (2005) Crystal structure of a human cyclin‐dependent kinase 6 complex with a flavonol inhibitor, fisetin. J Med Chem 48:737–743. [DOI] [PubMed] [Google Scholar]
- 6. Cho YS, Borland M, Brain C, Chen CH, Cheng H, Chopra R, Chung K, Groarke J, He G, Hou Y, Kim S, Kovats S, Lu Y, O'Reilly M, Shen J, Smith T, Trakshel G, Vogtle M, Xu M, Xu M, Sung MJ (2010) 4‐(Pyrazol‐4‐yl)‐pyrimidines as selective inhibitors of cyclin‐dependent kinase 4/6. J Med Chem 53:7938–7957. [DOI] [PubMed] [Google Scholar]
- 7. Cho YS, Angove H, Brain C, Chen CH, Cheng H, Cheng R, Chopra R, Chung K, Congreve M, Dagostin C, Davis DJ, Feltell R, Giraldes J, Hiscock SD, Kim S, Kovats S, Lagu B, Lewry K, Loo A, Lu Y, Luzzio M, Maniara W, McMenamin R, Mortenson PN, Benning R, O'Reilly M, Rees DC, Shen J, Smith T, Wang Y, Williams G, Woolford AJ, Wrona W, Xu M, Yang F, Howard S (2012) Fragment‐based discovery of 7‐azabenzimidazoles as potent, highly selective, and orally active CDK4/6 inhibitors. ACS Med Chem Lett 3:445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li Z, Wang X, Eksterowicz J, Gribble MW, Alba GQ, Ayres M, Carlson TJ, Chen A, Chen X, Cho R, Connors RV, DeGraffenreid M, Deignan JT, Duquette J, Fan P, Fisher B, Fu J, Huard JN, Kaizerman J, Keegan KS, Li C, Li K, Li Y, Liang L, Liu W, Lively SE, Lo MC, Ma J, McMinn DL, Mihalic JT, Modi K, Ngo R, Pattabiraman K, Piper DE, Queva C, Ragains ML, Suchomel J, Thibault S, Walker N, Wang X, Wang Z, Wanska M, Wehn PM, Weidner MF, Zhang AJ, Zhao X, Kamb A, Wickramasinghe D, Dai K, McGee LR, Medina JC (2014) Discovery of AMG 925, a FLT3 and CDK4 dual kinase inhibitor with preferential affinity for the activated state of FLT3. J Med Chem 57:3430–3449. [DOI] [PubMed] [Google Scholar]
- 9. Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, Toogood PL (2004) Specific inhibition of cyclin‐dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 3:1427–1438. [PubMed] [Google Scholar]
- 10. Durrant JD, McCammon JA (2011) Molecular dynamics simulations and drug discovery. BMC Biol 9:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Friedman R, Boye K, Flatmark K (2013) Molecular modelling and simulations in cancer research. Biochim Biophys Acta 1836:1–14. [DOI] [PubMed] [Google Scholar]
- 12. Bártová I, Otyepka M, Kríz Z, Koca J (2004) Activation and inhibition of cyclin‐dependent kinase‐2 by phosphorylation: a molecular dynamics study reveals the functional importance of the glycine‐rich loop. Protein Sci 13:1449–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bártová I, Otyepka M, Kríz Z, Koca J (2005) The mechanism of inhibition of the cyclin‐dependent kinase‐2 as revealed by the molecular dynamics study on the complex CDK2 with the peptide substrate HHASPRK. Protein Sci 14:445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Iveta B, Jaroslav K, Michal O (2008) Regulatory phosphorylation of cyclin‐dependent kinase 2: insights from molecular dynamics simulations. J Mol Model 14:761–768. [DOI] [PubMed] [Google Scholar]
- 15. Otyepka M, Bártová I, Kríz Z, Koca J (2006) Different mechanisms of CDK5 and CDK2 activation as revealed by CDK5/p25 and CDK2/cyclin A dynamics. J Biol Chem 281:7271–7281. [DOI] [PubMed] [Google Scholar]
- 16. Iveta B, Jaroslav K, Michal O (2008) Functional flexibility of human cyclin‐dependent kinase‐2 and its evolutionary conservation. Protein Sci 17:22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Andrews MJ, McInnes C, Kontopidis G, Innes L, Cowan A, Plater A, Fischer PM (2004) Design, synthesis, biological activity and structural analysis of cyclic peptide inhibitors targeting the substrate recruitment site of cyclin‐dependent kinase complexes. Org Biomol Chem 2:2735–2741. [DOI] [PubMed] [Google Scholar]
- 18. Park H, Yeom MS, Lee S (2004) Loop flexibility and solvent dynamics as determinants for the selective inhibition of cyclin‐dependent kinase 4: comparative molecular dynamics simulation studies of CDK2 and CDK4. Chembiochem 5:1662–1672. [DOI] [PubMed] [Google Scholar]
- 19. Khuntawee W, Rungrotmongkol T, Hannongbua S (2012) Molecular dynamic behavior and binding affinity of flavonoid analogues to the cyclin dependent kinase 6/cyclin D complex. J Chem Inf Model 52:76–83. [DOI] [PubMed] [Google Scholar]
- 20. Patel JS, Berteotti A, Ronsisvalle S, Rocchia W, Cavalli A (2014) Steered molecular dynamics simulations for studying protein–ligand interaction in cyclin‐dependent kinase 5. J Chem Inf Model 54:470–480. [DOI] [PubMed] [Google Scholar]
- 21. Friedman R (2013) Drug resistance missense mutations in cancer are subject to evolutionary constraints. PLoS One 8:e82059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mukherjee K, Sharma M, Urlaub H, Bourenkov GP, Jahn R, Sudhof TC, Wahl MC (2008) CASK Functions as a Mg2+‐independent neurexin kinase. Cell 133:328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Placke T, Faber K, Nonami A, Putwain SL, Salih HR, Heidel FH, Krämer A, Root DE, Barbie DA, Krivtsov AV, Armstrong SA, Hahn WC, Huntly BJ, Sykes SM, Milsom MD, Scholl C, Fröhling S (2014) Requirement for CDK6 in MLL‐rearranged acute myeloid leukemia. Blood 124:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Awad MM, Katayama R, McTigue M, Liu W, Deng YL, Brooun A, Friboulet L, Huang D, Falk MD, Timofeevski S, Wilner KD, Lockerman EL, Khan TM, Mahmood S, Gainor JF, Digumarthy SR, Stone JR, Mino‐Kenudson M, Christensen JG, Iafrate AJ, Engelman JA, Shaw AT (2013) Acquired resistance to crizotinib from a mutation in CD74‐ROS1. N Engl J Med 368:2395–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tamura K, Mukai H, Naito Y, Yonemori K, Kodaira M, Tanabe Y, Yamamoto N, Osera S, Sasaki M, Mori Y, Hashigaki S, Nagasawa T, Umeyama Y, Yoshino T (2016) A phase 1 study of palbociclib, a cyclin‐dependent kinase 4/6 inhibitor, in Japanese patients Cancer Sci 107:755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guex N, Peitsch MC (1997) SWISS‐MODEL and the Swiss‐PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723. [DOI] [PubMed] [Google Scholar]
- 27. Zoete V, Cuendet MA, Grosdidier A, Michielin O (2011) SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem 32:2359–2368. [DOI] [PubMed] [Google Scholar]
- 28. Berendsen HJC, van der Spoel D, Vandrunen R (1995) Gromacs—a message‐passing parallel molecular‐dynamics implementation. Comput Phys Commun 91:43–56. [Google Scholar]
- 29. van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC (2005) Gromacs: fast, flexible, and free. J Comput Chem 26:1701–1718. [DOI] [PubMed] [Google Scholar]
- 30. MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph‐McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz‐Kuczera J, Yin D, Karplus M (1998) All‐atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 31. Bjälkmar P, Per L, Cuendet MA, Berk H, Erik L (2010) Implementation of the CHARMM force field in GROMACS: analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J Chem Theor Comput 6:459–466. [DOI] [PubMed] [Google Scholar]
- 32. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935. [Google Scholar]
- 33. Hess B, Bekker H, Berendsen HJC, Fraaije JGEM (1997) LINCS: a linear constraint solver for molecular simulations. J Comp Chem 18:1463–1472. [Google Scholar]
- 34. Miyamoto S, Kollman PA (1992) SETTLE: an analytical version of the SHAKE and RATTLE algorithms for rigid water models. J Comp Chem 13:952–962. [Google Scholar]
- 35. Bussi G, Donadio D, Parrinello M (2007) Canonical sampling through velocity rescaling. J Chem Phys 126:014101. [DOI] [PubMed] [Google Scholar]
- 36. Berendsen HJC, Postma JPM, DiNola A, Haak JR (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81:3684–3690. [Google Scholar]
- 37. Parrinello M, Rahman A (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys 52:7182–7190. [Google Scholar]
- 38. Darden T, York D, Pedersen L (1993) Particle mesh Ewald: an N‐log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092. [Google Scholar]
- 39. Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 40. Daura X, Gademann K, Jaun B, Seebach D, van Gunsteren WF, Mark AE (1999) Peptide folding: when simulation meets experiment. Angew Chem Int Ed 38:236–240. [Google Scholar]
- 41. Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38. [DOI] [PubMed] [Google Scholar]
- 42. Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hartley JL, Temple GF, Brasch MA (2000) DNA cloning using in vitro site‐specific recombination. Genome Res 10:1788–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Faber K, Bullinger L, Ragu C, Garding A, Mertens D, Miller C, Martin D, Walcher D, Döhner K, Döhner H, Claus R, Plass C, Sykes SM, Lane SW, Scholl C, Fröhling S (2013) CDX2‐driven leukemogenesis involves KLF4 repression and deregulated PPARγ signaling. J Clin Invest 123:299–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information 1
Supporting Information 2