

Abstract
The Escherichia coli single stranded DNA binding protein (SSB) is crucial for DNA replication, recombination and repair. Within each process, it has two seemingly disparate roles: it stabilizes single‐stranded DNA (ssDNA) intermediates generated during DNA processing and, forms complexes with a group of proteins known as the SSB‐interactome. Key to both roles is the C‐terminal, one‐third of the protein, in particular the intrinsically disordered linker (IDL). Previously, they have shown using a series of linker deletion mutants that the IDL links both ssDNA and target protein binding by mediating interactions with the oligosaccharide/oligonucleotide binding fold in the target. In this study, they examine the role of the linker region in SSB function in a variety of DNA metabolic processes in vitro. Using the same linker mutants, the results show that in addition to association reactions (either DNA or protein), the IDL is critical for the release of SSB from DNA. This release can be under conditions of ssDNA competition or active displacement by a DNA helicase or recombinase. Consistent with their previous work these results indicate that SSB linker mutants are defective for SSB–SSB interactions, and when the IDL is removed a terminal SSB–DNA complex results. Formation of this complex inhibits downstream processing of DNA by helicases such as RecG or PriA as well as recombination, mediated by RecA. A model, based on the evidence herein, is presented to explain how the IDL acts in SSB function.
Keywords: SSB, RecG, OB‐fold, SH3 domain, PXXP motif, ssDNA binding
Abbreviations
- IDL
intrinsically disordered linker
- SSB
single stranded DNA binding protein
- ssDNA
single stranded DNA
Introduction
The Escherichia coli single stranded DNA binding protein (SSB) plays a central role in DNA metabolism.1, 2, 3 It binds to single stranded DNA (ssDNA) non‐specifically and with high affinity, destabilizing DNA secondary structure and hence stabilizing ssDNA intermediates produced during DNA processing.4 SSB also interacts with an array of at least fourteen proteins that has been termed the SSB‐interactome.5, 6 These two seemingly disparate roles are intimately linked via a previously under‐appreciated region of the protein known as the intrinsically disordered linker or IDL.7
SSB exists as a homo‐tetramer consisting of four, 18, 843 Dalton monomers.8 Each subunit is composed of two domains: an N‐terminal domain of approximately 116 residues that consists of an oligonucleotide/oligosaccharide binding fold (OB fold) and a C‐terminal domain composed of the last 62 residues that extends outward from the core [Fig. 1(A)].9 SSB employs its OB fold to bind to ssDNA via interactions with the phosphodiester backbone and base stacking with nucleotide bases.11, 12, 13
Figure 1.
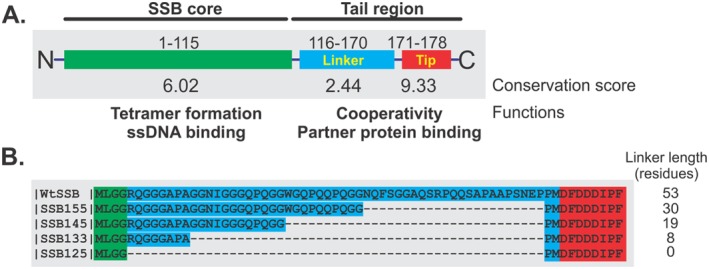
Organization of the SSB proteins used in this study. (A) Wild type SSB protein is shown in schematic form divided into the N‐terminal core (green) and C‐terminal tail regions by proteolytic cleavage. The tail can be further sub‐divided into the IDL and acidic tip, labeled as linker (blue) and tip (red), respectively. The conservation scores for each region were calculated from alignments using Praline.10 (B) Sequence alignment of wild type and linker domain mutants. The colored boxes behind the sequence correspond to the domain organization in panel A. The alignment was done using Praline.
The C‐terminal domain of SSB has not been modeled successfully due to its highly disordered structure, even when it is bound to ssDNA.11, 14, 15 It consists of a linker region of approximately 54 amino acids that has been termed the intrinsically disordered linker (IDL) and a highly conserved, C‐terminal acidic tip that is formed by the last 8–10 residues. The acidic tip is thought to play a major role in interacting with SSB‐interactome partners as peptides comprising this sequence bind to interactome partners with high affinity and a mutant lacking the last eight residues cannot mediate binding.6, 16, 17, 18, 19, 20, 21, 22 In contrast, the IDL is the least conserved region among prokaryotic SSB proteins.3, 4 Initially, it was proposed to be non‐essential, as a mutant SSB with residues 116–167 deleted was able to complement an ssb deletion.9 Very little work has focused on this region until recently. Lohman's group reported that the IDL is involved in cooperative ssDNA binding by SSB in its (SSB)35 mode, which is established at low salt concentrations and high protein‐to‐DNA ratios.23 They showed using a series of linker deletion mutants that removal of the C‐terminal tip or, shortening of the linker region, reduces cooperative binding. Further, they demonstrated that removal of the entire linker region or replacing it with that from Plasmodium falciparum SSB, eliminated cooperative ssDNA binding completely.23 Recently, and using a series of IDL deletion and hybrid mutants, we found that the IDL is critical in mediating protein‐protein interactions.7 A model was proposed whereby the linker(s) of an SSB tetramer bind to the OB‐folds present in either a neighboring SSB or a member of the interactome. When binding to another SSB occurs, this results in rapid and cooperative ssDNA binding, producing a stable complex that can resist displacement by a variety of enzymes that might otherwise damage the exposed portion of the genome. When the IDL binds to a protein such as RecG, the helicase is loaded onto the DNA, leading to the rescue of stalled replication forks.21, 24, 25
A key component to the IDL–OB‐fold interactions was the identification of PXXP motifs in the C‐terminal half of the linker.7 This is significant as these motifs bind Src homology (SH3) domains in eukaryotic cells. These domains are approximately 50 residue modules that often occur in signaling and cytoskeletal proteins in eukaryotes.26, 27, 28, 29 They have a characteristic fold consisting of five to six beta‐strands arranged as two tightly packed anti‐parallel beta sheets. Furthermore, and germane to this work, SH3 domains are very similar in structure to the OB‐fold, which is found in SSB and in each interactome partner for which a structure exists30 (P. Bianco, unpublished). We proposed that the PXXP‐OB fold interaction involving SSB is analogous to the SH3‐PXXP binding that occurs in eukaryotic systems and have taken advantage of in RecG loading.31, 32
If the IDL‐OB‐fold model is correct, and if SSB tetramers are linked to one another on a single stranded DNA molecule, then it is likely that changes in the IDL would impact several DNA metabolic processes in which SSB is involved. To test this, we examined the activities of various enzymes in the presence of WtSSB and the linker deletion mutants used previously.7 Results show that an SSB without a linker fails to stimulate the repair helicases RecG and PriA and actually inhibited their ssDNA‐dependent ATPase activity. Furthermore, SSB mutants with short or no linkers inhibited RecA‐dependent DNA strand exchange. Finally, single molecule experiments using a hairpin substrate and magnetic tweezers show that the elimination of the IDL produces an SSB that is a greater obstacle to displacement by RecG. Collectively, these results suggest that in addition to the already assigned roles in association with DNA and proteins, the linker is necessary to facilitate dissociation of SSB from ssDNA. This finding is consistent with our model that protein–protein interactions, mediated by the IDL of an SSB monomer within one tetramer and the OB‐fold of an adjacent partner protein are essential to SSB function.
Results
Site size of SSB linker mutants
To understand the role of the IDL in SSB function, a series of linker deletions were constructed and are shown schematically in Figure 1(B). These were studied previously using an in vivo binding assay.7 The results showed that the IDL was critical to target protein binding. A separate study from the Lohman group using a similar set of linker deletion mutants showed that changes in the IDL produced defects in cooperative binding to ssDNA.23
To further understand how changes in the IDL affect ssDNA binding, we purified several SSB proteins (wild type and linker deletions) as N‐terminally histidine‐tagged versions. Tagging was necessary to minimize the level of incorporation of wild type subunits which are known to form heterotetramers with mutant SSB proteins when expressed in the same cell.33 Following purification to near homogeneity, we evaluated the ability of each protein to bind DNA by monitoring the fluorescence quenching of the intrinsic fluorescence of SSB that occurs upon binding to ssDNA.34 For the studies described below, poly d(T) with an average length 359 nt was used as the DNA cofactor.
The results show that wild type untagged SSB bound to poly d(T) with a site size of 9.2 ± 0.5 nt/monomer [Fig. 2(A), Table 1]. Under identical conditions, his‐wtSSB bound to the ssDNA cofactor with a site size of 9.4 ± 0.3 nt/monomer. These values correspond to 36 nt/tetramer, consistent with previous reports for the wild type protein.34 Therefore, the addition of a histidine tag does not alter binding of the protein to ssDNA, consistent with prior work.33
Figure 2.
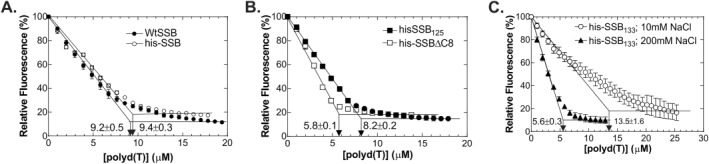
The histidine tag does not alter the intrinsic site size. (A) The site size of wild type is unaffected by the presence of a histidine tag. (B) Removal of the C‐terminal eight residues decreases the site size, whereas elimination of the linker does not. (C) The site size of hisSSB133 is inversely related to the monovalent cation concentration. Experiments were done as described in the Materials and Methods. Following a 4 min thermal equilibration of 1 μM protein, aliquots of poly d(T), that is, 1 μM increments, were added. Subsequent additions only occurred once the fluorescence signal had stabilized. Thereafter, the signal at each DNA concentration was expressed as a fraction of the initial fluorescence of SSB.
Table 1.
Site Size of Wild Type SSB and Linker Mutants a
| Site size (nucleotides) | |
|---|---|
| wtSSB | 9.2 ± 0.5 |
| His‐wtSSB | 9.4± 0.3 |
| His‐SSB155 | 8.5 ± 0.4 |
| His‐SSB145 | 7.2 ± 0.5 |
| His‐SSB133 | 13.5 ± 1.6; 5.6 ± 0.3b |
| His‐SSB125 | 8.1 ± 0.2 |
| His‐SSBΔC8 | 4.3 ± 0.2; 5.8 ± 0.1 |
Site size of SSBs were determined as described in Materials and Methods. For wild type and his‐SSB, duplicate experiments were done on separate days, with two separate titrations in each. For each of the mutants, duplicate titrations were done on the same day.
For this mutant, experiments were done in 10 and 200 mM NaCl. The larger site size was obtained at low salt.
Next, the binding of each linker mutant was evaluated. For SSB155, SSB145, and SSB125, a small but detectable decrease in site size by an average of 1 nt was observed [Table 1, Fig. 2(B) for SSB125]. Surprisingly, a significant reduction in the site size for SSBΔC8 (a mutant lacking the acidic tip) was observed with values ranging from 4 to 6 nt/monomer. The range in values came from two separate purifications of the protein. Finally, the site size for SSB133 was 13.5 ± 1.6 nt/monomer [Table 1, Fig. 2(C)]. This is an estimation at best, as a clear plateau in the titration could not be reached. Surprisingly, when the monovalent cation concentration was increased from 10 to 200 mM, the site size for SSB133 decreased 2.4‐fold to 5.6 ± 0.3 nt/monomer. The altered site size suggests that this mutant linker is somehow interfering with binding to DNA. This behavior is the opposite of wild type SSB which occludes 35 nt/tetramer at low salt and 65 nt/tetramer at higher salt concentrations.34 For SSB133 the values are 54 nt/tetramer at 10 mM NaCl and 22 nt/tetramer at 200 mM NaCl.
SSB linker region affords protection from trypsin cleavage
Previous studies with chymotrypsin and trypsin have demonstrated that SSB is susceptible to cleavage, predominantly after arginine 115.35 These studies showed that the core of SSB is stable and the C‐terminal linker is mobile (labile). To determine whether the changes in the linker by altering its length have affected protein stability, we used protease cleavage with a limiting amount of trypsin. Reactions were done as described in Materials and Methods with aliquots removed at various time points and subjected to gel electrophoresis.
The resulting gels show that, under the conditions used, his‐wtSSB was cleaved at a rate of 1.4%/min in the absence of ssDNA [Figs. 3(A) and 4(A)]. When the linker mutants were examined, cleavage occurred even more rapidly. The rates for hisSSB155 and hisSSB125 were 3.6%/min and 2.2%/min, respectively [Figs. 3(B,C) and 4(B,C)]. Surprisingly, hisSSB133 was resistant to cleavage under these conditions, with only 30% of the protein being cleaved during the 2 hr assay (Table 2). This suggests that for this mutant, the linker is sequestered into a configuration that is inaccessible to trypsin. This is consistent with the aberrant migration in SDS‐PAGE gels where the apparent molecular weight is 16k Da and predicted molecular weight is 18k Da [Fig. 3(D)].7
Figure 3.
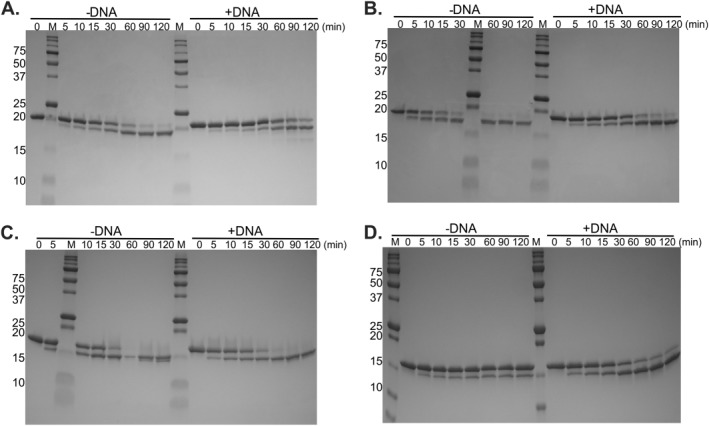
The SSB linker affords protection from cleavage by trypsin. About 10 µg of protein with or without M13 ssDNA was incubated in Binding Buffer (10 mM Tris‐HCl, pH 8.0, 200 mM NaCl, 5 mM MgCl2, 1 mM DTT, and 1 mM ADP), on ice for 10 min. The reaction was then adjusted to a final concentration of 50 mM Tris‐HCl, pH 8.0, and 20 mM CaCl2. About 0.1 µg of Trypsin was added to each reaction and the trypsin digestion was carried out at 37°C. At each time point, aliquots were removed and incubated on ice with 2 μL of 100 mM PMSF for at least 10 min. SDS‐PAGE loading dye was added to each sample which was then boiled for 5 min and subjected to gel electrophoresis. The resulting Coomassie stained, 18% Criterion TGX gels are presented. (A) hisSSB; (B) hisSSB155; (C) hisSSB125; (D) hisSSB133. Time points are indicated at the top of each lane. M, Molecular weight marker.
Figure 4.
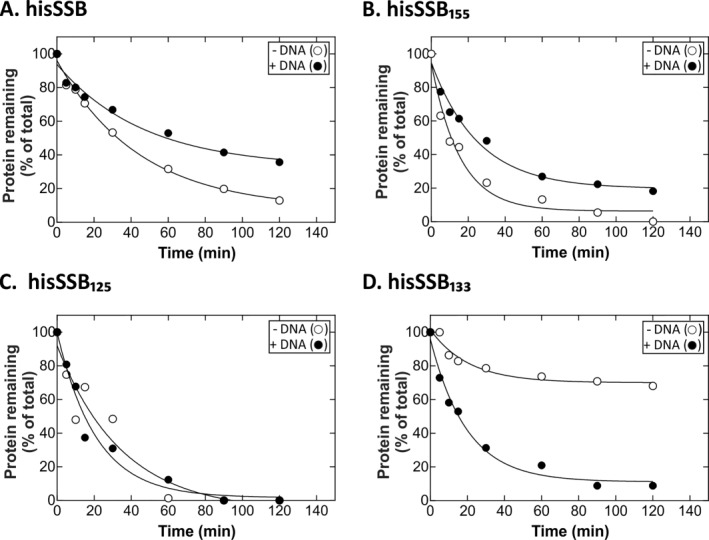
The IDL affords enhanced protection in the presence of ssDNA. Analysis of gels presented in Figure 2. Coomassie stained gels were photographed and analyzed using ImageQuant 5.2. Graphs of substrate disappearance over time are shown. Data points were obtained by expressing amount of protein present in each lane as a fraction of t = 0. (A) hisSSB; (B) hisSSB155; (C) hisSSB125; (D) hisSSB133. The data in each graph were approximated by a single exponential producing the parameters in Table 2.
Table 2.
Binding to ssDNA Alters the Sensitivity to Cleavage by Trypsin
| Half‐life (minutes)a | Extent (%)b | ||||
|---|---|---|---|---|---|
| −ssDNA | +ssDNA | −ssDNA | +ssDNA | Extent ratioc | |
| His‐WtSSB | 30.71 | 32.59 | 7.95 | 32.57 | 4.09 |
| His‐SSB155 | 10.67 | 17.54 | 6.29 | 19.66 | 3.13 |
| His‐SSB145 | 12.60 | 14.81 | 15.25 | 26.36 | 1.73 |
| His‐SSB133 | 14.23 | 13.59 | 70.01 | 11.16 | 0.16 |
| His‐SSB125 | 24.22 | 14.57 | 0 | 1.7 | none |
The half‐life disappearance of SSB due to trypsin cleavage was determined by approximating the time course data with a single exponential (Fig. 4).
Extent of reaction was calculated form the plateau value in the fit in (a).
Extent ratio is defined as (extent +ssDNA)/(extent −ssDNA).
To determine whether ssDNA binding affords protection from tryptic digestion, cleavage assays were done using stoichiometric ratios of each SSB bound to M13 ssDNA. The results show that, hisSSB, hisSSB155 and hisSSB145 were more resistant to cleavage by trypsin in the presence of DNA (Figs. 3 and 4; and data not shown for hisSSB145). This is evident in the increase in half‐life of each protein resulting in a several fold decrease in the amount of protein cleaved (Table 2). Once the length of the linker was decreased further, intriguing behavior was seen. For hisSSB125, which has essentially no linker, DNA does not afford any protection from trypsin cleavage [Figs. 3 and 4(C); Table 2]. Surprisingly, for SSB133, binding to ssDNA that is cleaved seven fold readily than when it is in solution [Figs. 3 and 4(D); Table 2). Further, when the ratios of the extents of cleavage in the absence and presence of ssDNA are analyzed, this ratio decreases in linear fashion from 4 for full length linker (53 residues) to 0 for SSB125 (0 residues; Supporting Information Fig. S1).
Alterations in the IDL affect fork rescue helicases differently
Previous work has shown that SSB binds to PriA and separately, to RecG both in vivo and in vitro.6, 18, 21 In addition, SSB loads RecG onto model fork substrates.24 Bulk phase analysis revealed that while wild type SSB stabilizes RecG on DNA, SSBΔC8 destabilizes the DNA helicase.21 To determine whether the linker mutants stabilize these helicases, we evaluated the effect of SSB linker mutants on the ATPase activity of RecG on M13 ssDNA and PriA on ϕx174 ssDNA (Fig. 5).
Figure 5.
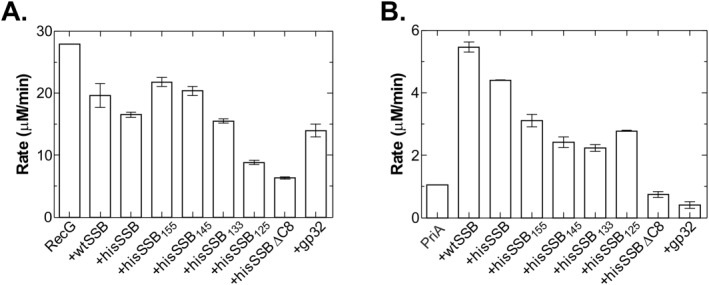
SSB linker mutants affect RecG and PriA differently. ATPase assays were carried out as described in Materials and methods. For RecG, the DNA substrate was M13 ssDNA and for PriA, it was ϕx174 ssDNA. For each helicase, DNA substrate (10 μM nucleotides) was incubated separately with each single strand binding protein at 37°C for 2 min, prior to the addition of RecG (100 nM, final) or separately, PriA (10 nM, final) to initiate reactions. In RecG assays, SSB proteins were present at 125 nM tetramer and gp32 at 500 nM monomer. For PriA, SSB proteins were present at 200 nM tetramer and gp32 was 800 nM monomer. (A) Linker deletion mutants inhibit RecG more than wtSSB. (B) Linker mutants are not effective stimulators of PriA. The analysis in each panel represents two assays done on the same day. Error bars represent the standard deviation.
The results show that in the presence of wtSSB, the rate of ATP hydrolysis by RecG decreased 1.4‐fold, down from 28 to 19.6 μM/min [Fig. 5(A)]. Addition of the histidine tag to SSB reduced the rate to 16.8 μM/min, but this is within experimental error, the same as untagged wild type. When the effects of linker mutants were compared, there is an almost inverse linear relationship between linker length and inhibition of RecG, reaching a maximum when the linker is completely removed. In fact, for hisSSB125, the level of inhibition observed was within experimental error, the same, as that for hisSSBΔC8 (8.8 and 10.8 μM/min, respectively). Gene 32 protein, which binds to ssDNA with a polarity opposite to that of SSB and which does not bind to RecG, also inhibits the helicase two‐fold.21, 36
In contrast to RecG, the ATPase activity of PriA on ϕx174 ssDNA is stimulated 5‐fold by SSB, from 1.1 to 5.5 μM/min [Fig. 5(B)]. The level of stimulation for hisSSB (4.4 μM/min) was comparable to wtSSB. On the other hand, the linker mutants only stimulated the ATPase activity two‐ to three‐fold compared with wild type. The level of stimulation was affected in almost linear fashion by the length of the IDL. That is, as linker length decreased, the level of stimulation of the ATPase activity of PriA, decreased accordingly. When the acidic tip is removed, as in hisSSBΔC8 which does not interact appreciably with PriA, no stimulation was observed. Finally, gp32 protein inhibits PriA 2‐fold.
Previous work has shown that SSB stabilizes RecG on ssDNA producing a two‐ to five‐fold increase in the salt titration midpoint.21, 37 To determine whether changes in the IDL affect the ability of SSB to stabilize RecG on ssDNA, the effects of mutants on the salt titration midpoint (STMP) of RecG, and separately of PriA, were assessed. The results show that wt and hisSSB increased the STMP approximately three‐fold (Table 3). When the mutants were studied, they separated into two groups: hisSSB155 and hisSSB145 increased the STMP and the level of stimulation was comparable to that of weight. Once 48–53 residues were deleted as in the hisSSB133 and hisSSB125 proteins, only a two‐fold enhancement in the STMP was observed. The last eight residues at the C‐terminus of SSB are required for the stabilization of RecG on ssDNA, as previously reported.21 As expected, the presence of hisSSBΔC8 eliminated the enhancement in the STMP.
Table 3.
The SSB Linker is Required for Stabilization of RecG But Not PriA a
| Salt titration mid‐pointb (mM) | ||
|---|---|---|
| RecG | PriA | |
| Helicase only | 34 ± 0 | 41 ± 1 |
| +wtSSB | 95± 1 | 159 ± 9 |
| +hisSSB | 103 ± 1 | 168 ± 4 |
| +hisSSB155 | 96 ± 6 | 179 ± 9 |
| +hisSSB145 | 92 ± 7 | 168 ± 3 |
| +hisSSB133 | 81 ± 1 | 166 ± 7 |
| +hisSSB125 | 59 ± 2 | 138 ± 8 |
| +hisSSBΔC8 | 40 ± 2 | 71 ± 1 |
| +gp32 | NDc | 40 ± 2 |
Assays were carried out with the pre‐incubation of DNA substrate with or without SSB in reaction buffer at 37°C and initiated with addition of helicase.
Calculation of the salt titration midpoint is described in the Materials and Methods.
ND, not done.
In contrast to RecG, alterations in the linker had little to no effect on the increase in the STMP for PriA (Table 3). However, the STMP in the presence of hisSSBΔC8 decreased 2.4‐fold compared with wild type SSB, down from 168 to 71 mM. T4 gene 32 protein which does not bind to PriA, had no effect on the STMP of the helicase. Thus, changes the IDL affect each helicase differently.
RecBCD unwinding is unaffected by SSB linker length
In contrast to RecG and PriA which both bind to SSB, RecBCD is a DNA helicase that utilizes SSB to bind to unwound strands of the duplex produced in the wake of the advancing enzyme.38, 39 While no physical interaction between this helicase and SSB is known, SSB does control the nuclease activity of RecBCD.40 To determine whether the linker region of SSB plays a role in binding of ssDNA generated by RecBCD, we used a fluorescence‐based assay to monitor linear dsDNA unwinding by RecBCD.38 Here, the intrinsic fluorescence of SSB, which is quenched upon binding to ssDNA, reflects DNA unwinding.
First, we compared the effects of wild type and his‐wtSSB on DNA helicase activity. Results show that the observed unwinding rate is within experimental error, the same as wild type consistent with previous work (Table 4 and Ref. 32). Then, we assayed hisSSBΔC8 and T4 gene 32 protein, the results show that helicase activity was unaffected. Finally, we assayed each of the linker mutants separately. The results show that RecBCD unwinds dsDNA in the presence of hisSSB133 at a 12% higher rate than in the presence of wild type. In contrast, for the remaining mutants, an 18%–29% decrease in unwinding rate was observed. Collectively, the results show that, changes in the length of the IDL have minimal effects on the helicase activity of RecBCD, and these are not consistent.
Table 4.
SSB Linker Length Does Not Affect the Helicase Activity of RecBCD a
| Rate (nM bps s−1)b | Relative ratec | |
|---|---|---|
| wtSSB | 124 ± 1 | 1.00 |
| hisSSB | 114 ± 9 | 0.92 |
| hisSSBΔC8 | 163 ± 10 | 1.31 |
| hisSSB155 | 89 ± 22 | 0.71 |
| hisSSB145 | 101 ± 6 | 0.82 |
| hisSSB133 | 139 ± 5 | 1.12 |
| hisSSB125 | 99 ± 1 | 0.80 |
| gp32 | 148 ± 20 | 1.19 |
Assays were carried out as described in Materials and Methods. Three assays were done on the same day.
The rate of DNA unwinding is calculated as described.39
Rate of DNA unwinding are normalized to the control, wild type SSB.
Elimination of the IDL inhibits DNA strand exchange by RecA
The DNA strand exchange reaction consists of three stages: presynapsis, synapsis, and branch migration (Supporting Information Fig. S2 and Ref. 40). During presynapsis, RecA protein binds to ssDNA and ATP to form a nucleoprotein filament. Residual DNA secondary structure is removed by SSB which is then displaced by additional RecA polymerization, facilitating complete filament formation.42 Even though they may co‐exist on the same ssDNA, SSB and RecA do not interact.43 During synapsis, the RecA nucleoprotein filament binds to duplex DNA and initiates the homology search. Once homology is found, strands are exchanged and branch migration ensues, with SSB binding to the displaced single strand of DNA.44 As SSB has key roles in this reaction, we wanted to ascertain the effects of alterations in the IDL on each stage of the reaction. Reactions were done as described in the Materials and Methods and the effects on substrate disappearance, intermediate formation and loss and, product formation were assessed.
In the presence of wtSSB, RecA catalyzes an efficient DNA strand exchange reaction. During synapsis, 80% of the dsDNA substrate is rapidly taken up into presynaptic filaments at a rate of 4%/min [Fig. 6(A)]. Accordingly, the dsDNA substrate is converted to joint molecule intermediates at a rate of 4.88% ± 0.1%/min, reaching a maximum at 10 min following reaction initiation [Fig. 6(B)]. Thereafter, the level of intermediates declines to 3.4% ± 1.1%, as joint molecules are converted to nicked circle product. This phase of the reaction is biphasic and occurs at a rate of 0.95%/min in the first phase, followed by a slower second phase (0.24%/min). The disappearance of intermediates is coupled to the corresponding appearance of product which forms at a rate of 1.55%/min, reaching a maximum level of 70% at 80 min [Fig. 6(C)].
Figure 6.
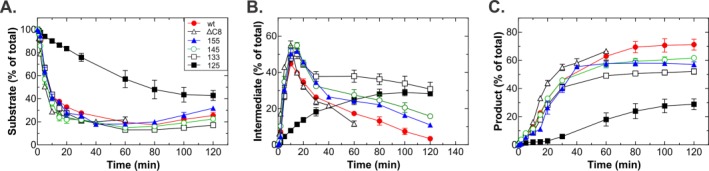
Elimination of the SSB linker significantly impairs DNA strand exchange by RecA. (A) Deletion of the linker inhibits substrate disappearance. (B) hisSSB125 inhibits both intermediate formation and disappearance. (C) Efficient product formation requires the SSB IDL. Data points were obtained by expressing the amount of each band at each time point as a fraction of the total count of each lane. Each data point represents two assays done on the same day, with error bars represent the standard deviation. wt, untagged wild type SSB; ΔC8, hisSSBΔC8; 125, hisSSB125; 133, hisSSB133; 145, hisSSB145; 155, hisSSB155. Assays were carried out as described in Materials and methods and were initiated by the addition of using 5′‐end labeled dsDNA. At various time points, aliquots were removed and subjected to electrophoresis in 1% agarose gels. Following electrophoresis, gels were dried and exposed to PhosphorImager screens. Analysis of gels was performed using ImageQuant software.
When the reaction in the presence of his‐SSBΔC8 was examined, the half‐life of substrate disappearance decreased 1.6‐fold, from 5.87 ± 0.6 to 3.63 (Table 5). No effect on intermediate formation was observed, whereas the rates of intermediate disappearance were 1.6‐fold more rapid (both phases faster), but the extent was reduced three‐fold. Product formation was slightly more rapid (1.5‐fold faster) and the yield of product was comparable to wild type, even though this reaction was only done for 60 min as opposed to 120 min (all other proteins).
Table 5.
Changes in the IDL Affect DNA Strand Exchange at Stages Where SSB Dissociation is Required a
| Stage of DNA strand exchange | ||||||
|---|---|---|---|---|---|---|
|
SSB protein present |
Half‐life of substrate disappearanceb (min) | Rate of intermediate formationc (%/min) | Rate I of intermediate disappearancec (%/min) | Rate II of intermediate disappearancec (%/min) | Extent of intermediate disappearance (%)d | Rate of product formationc (%/min) |
| wtSSB | 5.87 ± 0.6 | 4.88 ± 0.1 | 0.92 ± 0.07 | 0.26 ± 0.02 | 3.4 ± 1.1 | 1.55 ± 0.0 |
| hisSSBΔC8e | 3.63 ± 0.0 | 5.33 ± 0.1 | 1.51 ± 0.18 | 0.41 ± 0.09 | 11.7 ± 1.5 | 2.27 ± 0.1 |
| hisSSB155 | 6.11 ± 0.6 | 3.89 ± 0.0 | 0.82 ± 0.13 | 0.25 ± 0.02 | 11.0 ± 0.0 | 1.07 ± 0.1 |
| hisSSB145 | 4.35 ± 0.4 | 5.96 ± 0.2 | 1.05 ± 0.16 | 0.18 ± 0.02 | 15.8 ± 1.4 | 1.60 ± 0.0 |
| hisSSB133 | 6.47 ± 0.8 | 3.84 ± 0.1 | 0.55 ± 0.15 | 0.07 ± 0.04 | 30.8 ± 3.8 | 1.46 ± 0.1 |
| hisSSB125 | 49.61 ± 7.0 | 0.41 ± 0.0 | – | – | – | 0.34 ± 0.0 |
RecA strand exchange assays were performed as described in Materials and Methods. Two separate assays for each SSB protein were carried out on the same day.
The half‐life for disappearance of dsDNA substrate was calculated by using approximating the data in Figure 5(A) by a single exponential decay.
The rates were calculated by fitting a straight line to the linear portion of time courses in Figure 5(B,C). Rate of intermediate formation is calculated from 0 to 10 min, rate I of intermediate disappearance is calculated from the initial phase (10–30 min) and rate II of intermediate disappearance is calculated from 30 min onward.
The extent of the reaction was determined by the amount of intermediate remaining at the end of assay at 2 h from data in Figure 5(B).
Reactions with hisSSBΔC8 were carried out for 1 h.
Reactions with the linker mutants can be divided into three groups. In the first group, hisSSB155 and hisSSB145 had only modest effects on each stage of the reaction (Fig. 6, Table 5). For these proteins intermediates formed as rapidly as wild type and for the rates of intermediate disappearance were similar. However, the extent of intermediate disappearance was reduced three‐ to five‐fold compared with wild type and was accompanied by a 10% reduction in the amount of nicked circle product formed.
Group two contains the next longest IDL mutant, hisSSB133 and which enables near wild type levels of the rates of substrate disappearance and intermediate and product formation. However, the extent of intermediate disappearance is reduced 10‐fold, occurring at a two‐fold reduced rate in phase I and four‐fold slower rate in phase II (Table 5). This is accompanied by a 1.6‐fold reduction in the amount of product formed [Fig. 6(C)]. Thus, SSB135 is defective at the branch migration stage of the reaction where joint molecule intermediates are converted to product. This correlate with less than 20% of the intermediates being converted to product, whereas wild type converts more than 95%.
Finally, when the linker is eliminated as in the hisSSB125 protein (group three), each stage of the reaction is dramatically affected. First, the rate of substrate disappearance during synapsis decreases 8.5‐fold and only 60% is converted to joint molecules versus 80% for wtSSB. Accordingly, rate of intermediate formation is reduced 12‐fold [Table 5, Fig. 6(B)]. Furthermore, while intermediates form and are converted to product in easily discernible phases when each of the other SSB proteins are present, for SSB125 intermediates only accumulate. Regardless, some of these intermediates do get converted to nicked circle product, but their appearance is delayed 20–30 min, form at a five‐fold reduced rate and reach a maximum extent of 30%.
Collectively, the results show that changes in the IDL affect stages of DNA strand exchange where dissociation of SSB from the DNA is required. Small, but noticeable defects are observed for 155 and 145 mutants, a larger defect for 133 and once the linker is eliminated, the defects are severe.
SSB125 has difficulty releasing from DNA
Previously, we used a single molecule approach to understand the biochemical mechanism of fork regression by the RecG helicase.45 In this study, we showed that RecG was able to catalyze regression while working against more than 35pN of opposing force and that it could use this power to easily displace SSB from the arms of forks. Recently, we proposed that the IDLs of one tetramer of SSB bind to the OB‐folds present in the adjacent tetramer forming a stable protein‐DNA complex.7 It is conceivable that in addition to facilitating the forward reaction of SSB the IDL may also facilitate the reverse, that is, the release from ssDNA. A defect in release may explain the inhibition of RecA and of the helicases, RecG and PriA.
To test this, we used the same magnetic tweezers approach employed before and shown schematically in Figure 7(A,B). The DNA substrate is a 480 bp hairpin that is tethered to a glass coverslip surface using DIG/α‐DIG chemistry on one end, and to a magnetic bead on the opposite end using avidin/biotin technology (Supporting Information Fig. S3). This base hairpin was mechanically unzipped by capturing the bead in a magnetic trap (the tweezers). Next, a 30 base oligonucleotide complementary to one arm was introduced and allowed to bind [Supporting Information Fig. S3(A), step 2].46 Once the force is decreased, the hairpin reforms to reveal a fork with a gap in the nascent leading strand (step 3). Thereafter, SSB is added coating the remaining ssDNA regions of the fork. When RecG and ATP are injected, the helicase binds to the fork and during the process of regression, it displaces both the annealed oligonucleotide and the bound SSB protein [Fig. 7(B)].
Figure 7.
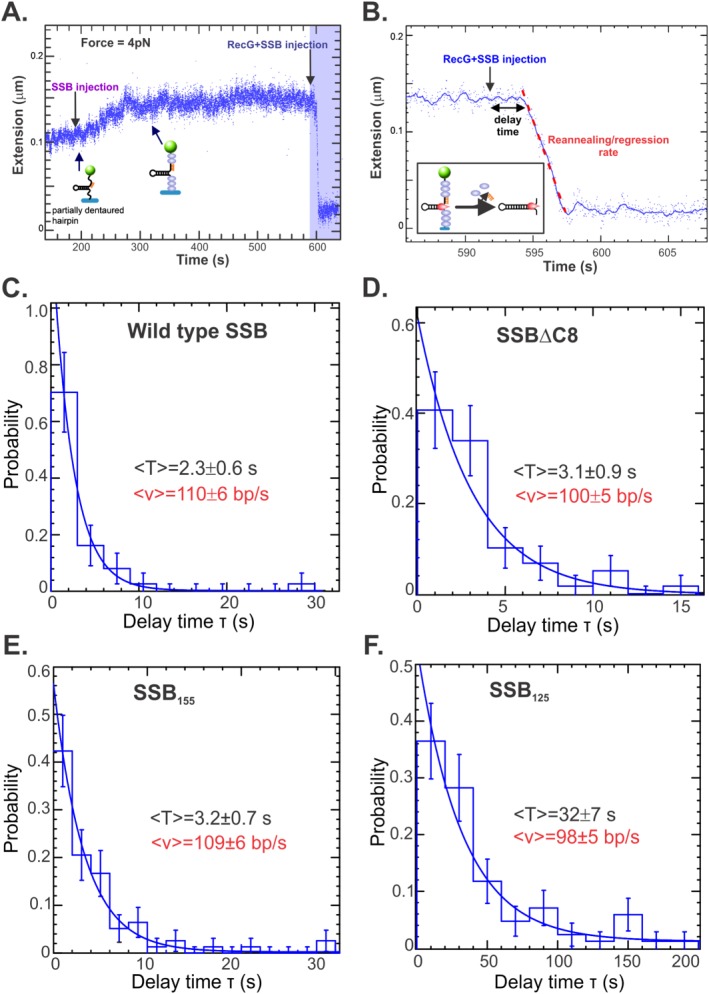
The IDL is required for displacement by RecG. (A). Time course of single molecule reaction progress. Assembly of the fork substrate is described in Supporting Information Figure S2. Once assembled, excess oligonucleotide is washed out, the substrate is held in place in the tweezers using a low force of 4pN, and at t = 200 s, SSB is introduced. Binding causes DNA extension to increase from 0.1 to 0.14 μm (t = 300 s). The length remains constant and at t = 592 s, RecG/SSB is injected. Following a brief delay, extension rapidly decreases to 0.02 μm as RecG unwinds the oligonucleotide, displaces bound SSB and reanneals the hairpin duplex in its wake (blue shaded region). (B). Close inspection of the blue shaded region from panel A. In this assay, RecG/SSB was injected and is followed by a 3‐s delay prior to the onset of the reaction, which is shown in the inset. The rate of the reaction is determined by linear regression as indicated by the red dashed line. (C–F) Analysis of reactions done in the presence of wild type, SSBΔC8, SSB155 or SSB125. The delay time (T) is shown for each protein as well as the resulting reaction rate (v). Rates of reaction were calculated in Supporting Information Figure S3.
By maintaining the DNA fork at 4pN of applied force we observed an increase in the molecular extension once we injected SSB protein [Fig. 7(A); time 200–340 s]. This length increase is a signature for SSB binding since ssDNA elongates when is coated by SSB at low forces.46 Once RecG + SSB and ATP were injected, regression, concomitant with oligo and SSB displacement is detected as a decrease in the DNA molecular extension until the hairpin is completely reformed [Fig. 7(A), blue shaded region, 7(B)]. In these experiments, there is a delay lasting a few seconds between the time RecG is injected and regression initiates [Fig. 7(B)]. If an SSB were defective in the release from ssDNA, this would result in a longer delay before DNA molecule extension ensues.
To test this, we examined the effects of wild type, his‐SSBΔC8, his‐SSB155, and his‐SSB125 on the delay time prior to regression initiation. Each protein was studied separately and by recording many events in parallel, it was possible to measure the distribution in delay times and determined that it follows an exponential behavior (Fig. 7). Most importantly, the results show that the delay time for wild type, SSBΔC8 and SSB155 were found to be within error, the same [2.3 ± 0.6; 3.1 ± 0.9; and 3.2 ± 0.7 s, respectively; Fig. 7(A–C)]. In contrast, when the IDL was deleted, as in SSB125, the delay time was 14 times longer at 32 ± 7 s [Fig. 7(D)]. Thus this mutant inhibits the onset of the RecG reannealing activity. However, once the helicase enters the extension phase of the reannealing reaction there is no effect of the C‐terminal domain of SSB, as the reaction rates are identical [red values, Fig. 7(C–F), Supporting Information Fig. S4].
The association of SSB125 with ssDNA is impaired
One possible explanation for the inhibition of RecG induced by SSB125 could be that this mutant has an enhanced affinity for ssDNA. To determine whether changes in the linker alter affinity for DNA, a fluorescence‐based competition assay was employed.47 This assay was selected as it can provide insight into both the association and dissociation phases of the DNA binding reaction. Here, SSB is bound to a fluorescent oligomer of d(T), resulting in a change in the fluorescence signal. Binding is done in the presence of 1M NaCl so that SSB can transfer between DNA types when challenged. When a large excess of unlabeled competitor ssDNA is added, SSB transfers from the fluorescent oligo d(T)65 to the competitor poly d(T)359, resulting in restoration of the fluorescent signal.47
First, it was necessary to determine whether each SSB linker mutant could bind to oligo d(T)65 in the presence of 1M NaCl. Here unmodified oligo d(T)65 was used and the intrinsic fluorescence of SSB monitored. Results show that each protein bound, with the exception of SSB125, where the ΔF upon the addition of DNA was only 14–28 units, in different experiments and this was 10‐fold lower than wtSSB (data not shown). Consequently, the concentration of NaCl was lowered, in 100 mM increments in separate experiments, until binding by SSB125 could be observed. At 600 mM NaCl, this mutant bound to oligo d(T)65. At 700 mM NaCl, binding still occurred, but it was significantly reduced (data not shown).
Next, and to compare proteins, experiments were done at 600 mM NaCl. Each SSB was bound to oligo d(T)65 labeled with ATTO488 at the 5′‐end. Similar to what was seen with Cy3/5 labeled cofactors,47 the fluorescence of ATTO488 is quenched upon binding to protein [Fig. 8(A)]. When a large excess of unlabeled competitor poly d(T) is added, each SSB transferred from the ATTO488 labeled oligo to the competitor, resulting in recovery of the fluorescence signal [Fig. 8(A)]. At this concentration of NaCl, transfer occurs slowly and can be approximated by a single exponential. This occurs because each protein retains measurable affinity for the 65mer at this concentration of salt, but still transfers as affinity for poly d(T) is greater.
Figure 8.
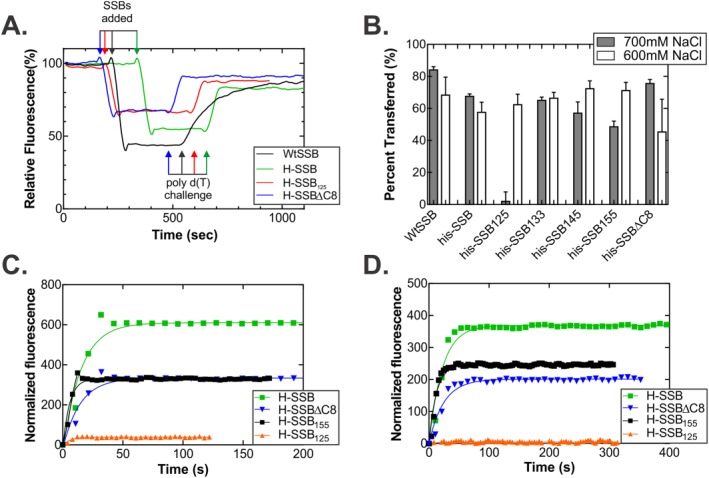
The IDL is required for association with and dissociation from ssDNA. (A) Time course of the association/dissociation reaction. Four representative traces, normalized to the starting fluorescence signal of from the ATTO‐488 dye, are shown from reactions performed in 600 mM NaCl. For clarity, the raw data were smoothed using a 2nd order polynomial with 10 neighbors on each side. For each reaction, 50 nM 5′‐ATTO‐488(dT)65 oligonucleotide was pre‐incubated in reaction buffer for 4 min. Next, 50 nM SSB tetramer was added and once a stable, lower fluorescence signal was observed, poly d(T) was added to 15 μM nucleotides, final. (B) Analysis of the amount of each SSB protein that transfers from the oligo d(T)65 to the poly d(T). The %transferred = (ΔF 2/ΔF 1) * 100, where ΔF 1 is the magnitude of the change in fluorescence upon binding to oligo d(T)65, and ΔF 2 is the magnitude of the change in signal when the SSB transfers from the 65mer to the poly d(T). For each protein, two separate assays were done at each concentration of NaCl. (C and D) Representative time courses done in 700 mM NaCl. In C, the forward reaction or binding to the 5′‐ATTO‐488‐d(T)65 oligonucleotide is shown, and in D, the transfer reaction is shown. Each curve represents the average fluorescence signal from two separate reactions for each protein. The signal is normalized to the starting fluorescence signal for each stage of each reaction.
There are some notable differences between the proteins. The forward reactions occur with similar half‐lives and with the exception of SSB125, the ΔF values are comparable, ranging from 225 to 285 fluorescence units. For this mutant, the ΔF for both the forward and reverse reactions is reduced two‐fold compared with other proteins [Fig. 8(A), Supporting Information Table SI]. Second, for the reverse reaction where transfer occurs, the half‐life for wtSSB is eight‐fold longer than that of the other proteins, suggesting that the histidine tag affects this component of SSB function. This is consistent with the two‐fold reduction in the STMP when each of the subunits of the tetramer is histidine tagged.33 Further, the half‐life for the SSB133 and SSB125 proteins is marginally shorter than SSB145, SSB155 and his‐SSB. When the amount of each SSB transferred to competitor at 600 mM NaCl was analyzed, this value averaged 65% and was independent of IDL length [Fig. 8(B)].
When reactions were done at 700 mM NaCl, the difference between SSB125 and the remaining SSB proteins was magnified [Fig. 8(B–D)]. At this concentration of salt, binding was reduced 20‐fold and transfer was virtually non‐existent with less than 4% transferring. In contrast, SSB, SSBΔC8 and SSB155 bound to the ATTO488 oligonucleotide and 50%–80% transferred to the competitor DNA.
Discussion
The primary conclusion of this article is that the intrinsically disordered linker is critical to the release of SSB from ssDNA. This region of the protein is critical to protein‐protein interactions mediated by the PXXP motifs in the IDL of one SSB tetramer and the OB‐fold in another.7 These interactions are critical to DNA binding, which is rapid and highly cooperative and results in an IDL/OB‐fold network of interactions. When this network of interactions is disrupted, each SSB tetramer binds to DNA as an independent entity. Consequently, each tetramer will release from the DNA also as a separate entity. The data herein do not point to protein–protein interactions between SSB and another protein being required for dissociation since both RecA (does not bind SSB) and RecG (does bind) were inhibited. Instead, it is SSB–SSB communication via the IDL/OB‐fold network that is key.
Previous work has demonstrated that when the IDL is removed, cooperative binding to ssDNA is eliminated.23 We have shown that removal and or mutation of the IDL impairs target protein binding.7 We proposed that the common element is the binding of the IDL in SSB to the OB‐fold of a target protein. Further, this interaction involves the PXXP motif in the linker binding to the OB‐fold in a manner similar to that observed for the Src homology 3 domains binding to these motifs in eukaryotic systems.48, 49 When an SSB tetramer is bound to ssDNA, binding to a second tetramer in solution could facilitate loading, resulting in the immediate juxtaposition of two tetramers, with the process being repeated until the DNA is fully covered [Fig. 9(A)]. This forms an extensive network of IDL/OB‐fold interactions through which SSB tetramers communicate to form a stable complex that requires 2M NaCl for displacement.7
Figure 9.
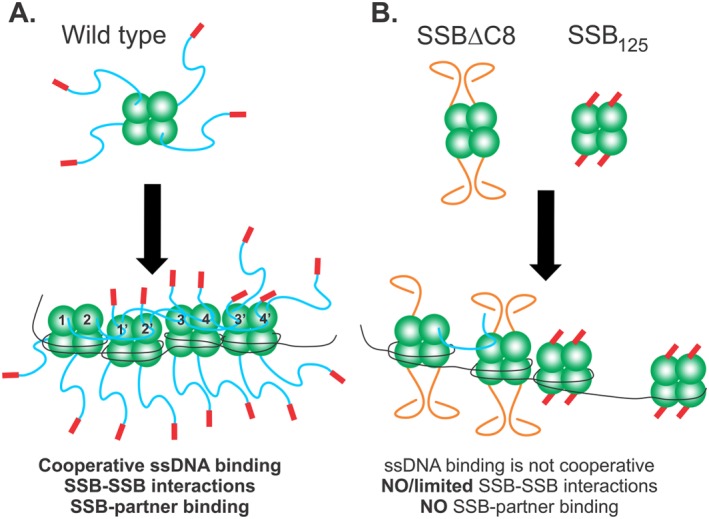
The IDL coordinates protein‐protein interactions to facilitate efficient SSB function. Schematics of wild type and mutant SSB proteins binding to ssDNA. The coloring of SSB monomers follows that of Figure 1, with the core domain in green, linker in blue and acidic tip in red. (A) Wild type protein establishes an IDL/OB‐fold network of interactions. Here tetramers have their functional C‐terminal domains exposed in solution. Upon binding to ssDNA in SSB35 mode for example, the IDLs of monomers 1 and 2, bind to the OB‐folds of monomers 1′ and 2′, respectively. Concurrently, the linkers of monomers 1′ and 2′ bind to the OB‐folds of monomers 3 and 4, and their IDLs bind to monomers 3′ and 4′, respectively. The C‐termini of subunits 3′ and 4′ are available to bind to an incoming tetramer. On the opposite side of each tetramer, C‐termini are available for binding to interactome partners. For simplicity, SSB‐SSB interactions are shown in the top subunits only. (B) SSBΔC8 and SSB125 mutant proteins have defective C‐termini and cannot establish an IDL/OB‐fold network of interactions. For SSBΔC8, the acidic tip is absent and as a result, linkers adopt a configuration that is incompatible with OB‐fold binding. Consequently, and even though this protein can still bind to ssDNA, co‐operativity is diminished, some IDL/OB‐fold interactions occur but binding to target proteins is eliminated. For SSB125 the linker is absent, but the acidic tip is retained. These tetramers cannot interact with other proteins but still retain the ability to bind ssDNA as the N‐terminal domains containing the OB‐folds are unaffected.
However, once the DNA template is bound and protected, SSB must be displaced so that subsequent reactions can occur. Again, the role of the IDL/OB‐old network of interactions is critical. When the linker is absent, binding to DNA still occurs as the OB‐fold remains intact. This idea is supported by measurements of occluded site size and stability in gradients, which are similar to wild type (Fig. 2, Table 1 and Ref. 23). However, and distinct from wild type, binding to DNA by these mutant proteins results in a “terminal complex” that fails to dissociate appropriately. This follows because each tetramer binds to DNA independently forming discrete complexes and which are not linked via the IDL/OB‐fold network. This results in inhibition of RecG and PriA translocation on ssDNA and, inhibition of RecA in DNA strand exchange. The terminal complex does not imply that the association of the SSB mutant with ssDNA is so strong that it cannot be reversed as shown by the competition assays (Fig. 8). Instead, it refers to a complex that is terminal with respect to downstream proteins, that is, RecA or RecG as explained below.
We have shown that SSB binds to RecG both in vivo and in vitro.6, 21 This interaction is important for stalled replication fork rescue where SSB loads RecG onto the DNA and in the process, remodels the wedge domain enabling disengagement of the helicase from the fork coupled to thermal diffusion on the parental duplex.24 We proposed a model for this that involves interactions between the SSB IDL and the OB‐fold present in the wedge domain of RecG.31 In this model, IDL and fork arm binding are mutually exclusive since the binding sites have significant overlap. As a result, that binding of the SSB IDL to RecG prevents fork‐wedge association, dictating that the loaded helicase slides on the duplex DNA average, 36 bp, using thermal diffusion. Once RecG returns to the fork, it displaces SSB protein quite readily.46 This occurs because (i) the helicase can work against opposing forces as high as 35pN, generating sufficient force to displace many tightly bound proteins and (ii), because of the IDL/OB‐fold network, displacement of the first SSB propagates changes to the remaining DNA‐bound tetramers enabling their facile displacement, likely in cooperative fashion. When this network of interactions is unavailable, as in SSB125 which has no linker, displacement takes 10 times longer as shown in the single molecule experiments (Fig. 7). This occurs because when RecG approaches the first SSB tetramer, it advances and applies force to dislodge that protein from the DNA. This process has to be repeated multiple times as each tetramer is encountered and because these tetramers are no longer are able to communicate via the OB‐fold/IDL network. This results in an extended lag time (10‐fold longer) prior to the onset of fork regression.
It is likely that interactions with the RecG via the IDL/wedge are not involved in SSB displacement, since DNA and IDL binding likely to be competitive. Additional evidence against an IDL‐target protein interaction being involved in dissociation comes from the analysis of DNA strand exchange. As RecA and SSB interact functionally but not physically, the displacement of SSB is likely DNA mediated.42, 43, 50 This occurs during all stages of this complex reaction. During presynapsis, RecA binds to the ssDNA template forming nucleoprotein filaments. However, it fails to completely coat this DNA due to residual secondary structure. The addition of SSB results in removal of this impediment, but for complete filament formation to occur, additional binding of RecA leading to the displacement of SSB must take place. This is accompanied by ATP hydrolysis by RecA resulting in redistribution leading to formation of complete nucleoprotein filaments. When a functional IDL is present in SSB, displacement by RecA readily occurs and efficient strand exchange reaction ensues. However, when it is deleted as in SSB125, the bound tetramers cannot be displaced and inhibit the reaction at the synapsis phase where incomplete filaments are attempting to bind dsDNA and locate homologous targets. As ATP hydrolysis occurs throughout strand exchange and RecA filaments are dynamic, this means that ssDNA regions will become exposed during the course of the reaction providing additional targets for SSB to bind. As this is likely to occur during branch migration when heterologous sites are encountered, and if continued polymerization by RecA cannot displace the SSB, additional inhibition will be observed when intermediates are being converted into products. Reactions done in the presence of SSB125 are severely inhibited at this stage (Fig. 6).
We note that significant inhibition of RecG and PriA is also observed in ATPase assays the presence of SSBΔC8 (Fig. 5). We attribute this to two separate but linked effects resulting from defects in the conformation of the IDL. First, the IDL conformation is altered in SSBΔC8 so it cannot bind to these helicases.6 Second, once SSBΔC8 binds to DNA, tetramers bind as separate entities so it cannot establish a fully competent IDL/OB‐fold network as cooperative binding to ssDNA is impaired [Fig. 9(B)].23 This situation is comparable to SSB125, but the defect is not quite as severe, since RecG displaces this protein quite readily (Fig. 7). This defect in the IDL conformation comes about because the acidic tip plays a critical regulatory role in protein function likely by regulating the IDL conformation as suggested previously.51 When it is present, the pI of the last 69 residues of SSB is 4.04. In contrast in its absence as in the SSBΔC8 mutant, the pI of the C‐terminal 61 residues is 9.35. Thus we propose that the acidic tip controls the association of the intrinsically disordered linker with OB‐folds, both within SSB tetramers and with target proteins. When it is absent, this results in an inability to associate with proteins of the interactome and a failure to bind ssDNA cooperatively since SSB‐SSB interactions leading to cooperative binding are negatively impacted.23
The conformation of the linker domain is critical to SSB function. This conformation is maintained by the acidic tip and is formed by a combination the proposed polyproline II helix and the multiple repetitive elements within the IDL containing G, Q and P residues.7 When this conformation is perturbed, there are dramatic changes in the overall structure of the protein as indicated by the tryptic digests in the presence and absence of DNA (Figs. 3 and 4). This was most evident in SSB133 which behaved in a manner opposite to that of full length SSB, becoming more sensitive to cleavage in the presence of ssDNA. In addition, the site size for this protein was affected by the monovalent cation concentration, but again, this was the opposite of wild type SSB (Fig. 2). Therefore, it is likely that changes in the amino acid composition of the length by mutation or deletion, as well as single residue changes in the last eight residues of SSB contribute to the interplay between the IDL and acidic tip, as suggested by preliminary data (LW and PB, unpublished). Consequently, the results in this and our recent work bring into question the role of the acidic tip of SSB in protein function.7 In each linker mutant, the acidic tip has been retained but yet activities involving SSB–SSB or SSB–partner protein interactions are severely impaired. If the acidic tip was responsible for mediating protein–protein interactions as has been widely thought, then each SSB mutant should retain full levels of binding and in addition, cooperative binding to ssDNA should be unaffected. This is not what is observed. Instead, and consistent with a previous report, our results suggest that the acidic tip has a regulatory function and may not be directly involved in protein binding.51, 52, 53
In conclusion, we provide the first evidence for the mechanism of release of SSB from ssDNA. Intrinsic to these interactions is the presence of a fully functional intrinsically disordered linker. When the IDL is perturbed all activities of SSB are affected. This follows because binding of the IDL of one SSB tetramer to the OB‐fold in another protein is critical for efficient SSB function. When the other protein is SSB both the association with and dissociation from ssDNA are controlled via the IDL/OB‐fold network of interactions. This is observed as loss of cooperative ssDNA binding and impaired ability to release from DNA. When the other protein is a member of the interactome such as RecG or PriA, loading of that protein onto the DNA cannot occur. Both the inherent flexibility imparted by the repetitive elements in the IDL as well as the specificity imparted by the PXXP motifs are essential. Flexibility is needed to facilitate cooperative ssDNA binding and dissociation, as well as providing the ability of an SSB tetramer to bind to target proteins of various sizes and likely, in different structural configurations, that is, bound to ssDNA, in solution or within multi‐subunit complexes. The specificity provided by the PXXP motifs in the donor linker and the OB‐folds in the target protein, enables SSB to control the function of all proteins of the SSB interactome, including itself, in spatial and temporal fashion.
Materials and Methods
Chemicals and reagents
All chemicals were reagent grade and were made up in Nanopure water and passed through 0.2 µm pore size filters. Isopropyl β‐D‐1‐thiogalactopyranoside (IPTG), sodium chloride (NaCl), calcium chloride (CaCl2) and sodium phosphate dibasic (Na2HPO4) were from Fisher Scientific (NJ, USA). Coomassie Brilliant Blue R‐250, Tris‐base, potassium chloride (KCl), sodium dodecyl sulfate (SDS), and acetic acid were from Amresco (OH, USA). Ethylenediaminetetraacetic acid (EDTA), potassium acetate, magnesium chloride (MgCl2), glycerol, potassium phosphate monobasic (KH2PO4), sodium phosphate monobasic (NaH2PO4), and nickel sulfate (NiSO4(H2O)6) were from J.T. Baker (NJ, USA). Imidazole, adenosine diphosphate (ADP), dithiothreitol (DTT) and magnesium acetate (MgOAc) were from Acros Organics (NJ, USA). The infusion kit was from Clontech (CA, USA). Phenylmethylsulfonyl fluoride (PMSF) was from Omnipur (NJ, USA). HisTrap FF crude columns, HiPrep DEAE FF 16/10, Q Sepharose Fast Flow, Mono Q HR 10/10, HiPrep SP Sepharose FF 16/10, HiPrep Heparin FF 16/10, MonoS 5/50 GL, and adenosine triphosphate (ATP) were from GE Healthcare Life Sciences (NJ, USA).
Trypsin‐ultra (Mass Spectrometry Grade), Φx174 Virion DNA, Φx174 RFI DNA and restriction enzymes were purchased from New England BioLab (MA, USA). Primers and HPLC purified 5′ATTO488(dT)65 were purchased from Integrated DNA Technologies (IA, USA). Phosphoenol pyruvate (PEP), Polymin P, nicotinamide adenine dinucleotide (NADH), pyruvate kinase (PK), deoxyribonucleic acid−cellulose single‐stranded from calf thymus DNA (ssDNA cellulose resin), and lactate dehydrogenase (LDH) were from Sigma Aldrich (MO, USA). Protenaise K was purchased from Roche (IN, USA). About 18% Criterion TGX Precast gel and Bio‐Gel HTP Hydroxyapatite were from Bio‐Rad Laboratories, Inc. (CA, USA). Hydrochloric acid (HCl) was from Macron Chemical (PA, USA). Potassium phosphate dibasic (K2HPO4) was from BDH Chemicals (PA, USA). Ammonium sulfate was purchased from MP Biomedicals, LLC (OH, USA). ATP Gamma P32 was from Perkin Elmer (MA, USA).
DNA cofactors
M13 mp18 single stranded DNA (ssDNA) was prepared as described.37 The concentration of DNA was determined spectrophotometrically using an extinction coefficient of 8780 M−1cm−1 (nucleotides). Purified DNA was stored in small aliquots at −80°C. Covalently closed circular DNA (pPB248) used in RecBCD unwinding assay was purified from pPB248 using procedures as described previously.37, 54 The method utilized alkaline lysis followed by two successive isopycnic centrifugations in CsCl gradients. The concentration of double stranded DNA (dsDNA) was determined spectrophotometrically using an extinction coefficient of 6500 M−1 cm−1 (nucleotides). Poly(dT) (average length 359 nt) was purchased from Amersham Biosciences, dissolved in 1× TE and stored in small aliquots at −80°C. The concentration was determined spectrophotometrically using an extinction coefficient of 8520 M−1cm−1 (nucleotides).
Proteins
RecG protein was purified as described previously,21, 37 with the following modifications: the first column was a 100 mL Q‐Sepharose column equilibrated in Buffer A (20 mM Tris‐HCl (pH 8.0), 1 mM EDTA, 1 mM DTT, 10 mM NaCl). The protein was eluted using a linear gradient (10–1000 mM NaCl) with RecG eluting between 270 and 370 mM NaCl. The pooled fractions were subjected to ssDNA cellulose column equilibrated in Buffer C (20 mM Tris HCl, pH 7.5, 1 mM EDTA, 1 mM DTT) and eluted with a 100–1000 mM NaCl gradient. Next column used was hydroxylapatite chromatography equilibrated with 10 mM KPO4, pH 6.8, 1 mM DTT, and 150 mM KCl. RecG was eluted off the hydroxylapatite column with a linear gradient of 10–600 mM KPO4. Pooled fractions from the hydroxylapatite column were dialyzed overnight into S Buffer (10 mM KPO4 (pH 6.8), 1 mM DTT, 1 mM EDTA, and 100 mM KCl). The protein was applied to a 1 mL MonoS column and eluted using a linear KCl gradient (100–1000 mM) with RecG eluting at 500 mM KCl. The fractions containing RecG were pooled and dialyzed overnight against storage buffer (20 mM Tris‐HCl (pH 7.5), 1 mM EDTA, 1 mM DTT, 100 mM NaCl, and 50% (v/v) glycerol). The protein concentration was determined spectrophotometrically using an extinction coefficient of 49,500 M−1 cm−1.55
The his‐PriA protein was purified by ammonium sulfate precipitation followed by affinity chromatography using HisTrap FF crude column, SP Sepharose column (Equilibrated with 20 mM potassium phosphate, pH 7.6, 150 mM KCl, 0.1 mM EDTA, and 1 mM DTT; Eluted with a linear 150–500 mM KCl gradient) and Heparin column (Equilibrated with 20 mM Tris‐OAc, pH 7.5, 0.1 mM EDTA, 1 mM DTT, 10% (v/v) Glycerol, and 100 mM KCl; Eluted with a linear KCl gradient of 100–600 mM). Fractions containing PriA were pooled and dialyzed overnight against storage buffer (20 mM Tris‐HCl (pH 7.5), 1 mM DTT, 400 mM KCl, and 50% (v/v) glycerol). The PriA concentration was determined using extinction coefficient of 104,850 M−1 cm−1.55
Wild type SSB protein was purified from strain K12ΔH1Δtrp as described.56 Histidine tagged SSBΔC8 was purified as described previously.33 The concentration of the purified protein was determined at 280 nm using ε = 30,000 M−1 cm−1. Gene 32 protein was purified as described previously.21
Histidine‐tagged SSB proteins
Cultures were lysed and the cleared cell lysate was subjected to affinity chromatography using HisTrap FF crude columns, equilibrated in Binding Buffer (20 mM sodium phosphate, pH 7.4, 600 mM NaCl, 30 mM Imidazole). The column was sequentially washed with 400 mL Binding Buffer, 350 mL Binding Buffer with 0.2% NP40 and 250 mL Binding Buffer only. A linear gradient of 30–500 mM of Imidazole was used to elute bound proteins from the column. Proteins were assessed by SDS‐PAGE. Following electrophoresis, gels were stained with Coomassie Brilliant Blue, de‐stained, and photographed. Protein fractions were pooled, precipitated in 70% ammonium sulfate, resuspended, and dialyzed overnight against storage buffer (20 mM Tris‐HCl pH 8.0, 1 mM EDTA, 500 mM NaCl, and 50% (v/v) Glycerol). The concentration of various histidine‐tagged SSBs were determined using extinction coefficients as calculated from amino acid composition using ProtParam (ExPASy): his‐SSB, 27,960 M−1 cm−1; his‐SSB125, 22,460 M−1 cm−1; hisSSB130, 22,460 M−1 cm−1; his‐SSB145, 22,460 M−1 cm−1; and his‐SSB155, 27,960 M−1 cm−1.55
RecA was purified from E. coli strain GE1171.57 The procedure used to purify RecA is based on the procedures of Weinstock et al.58 and Griffith and Shores59 with the modification of using DEAE and HAP instead of phosphocellulose chromatography. The procedure involves a selective extraction from Polymin P followed by DEAE chromatography (equilibrated with 20 mM NaPO4, pH 6.8, 1 mM DTT, 10% glycerol, and 200 mM NaCl) using salt step elution (0.5M and 2M), HAP column (equilibrated with 20 mM NaPO4, pH 6.8, 1 mM DTT, and 10% glycerol), eluting using linear phosphate gradient (20–300 mM phosphate), spermidine acetate precipitation of RecA, followed by MonoQ chromatography using salt elution (100–1000 mM NaCl; linear gradient). The concentration of purified RecA was determined at 280 nm using ε = 27,000 M−1 cm−1.
RecBCD holoenzyme
Growth of cells, induction of expression of genes and protein purification was done as described previously.39 The concentration of purified enzyme was determined at 280 nm using ε = 40,000 M−1 cm−1. The amount of active RecBCD enzyme (100%) was determined using a fluorescence based helicase assay as described.38
Site size determination of SSB
The binding of SSB to poly d(T) was determined by monitoring the quenching of the intrinsic fluorescence of SSB that occurs on binding to ssDNA, as described.34 Here, reactions were done at 25°C in a 500 μL volume. Reactions contained 10 mM Tris‐HCl (pH 8.0), 1 mM DTT and 10 mM NaCl, and 1 μM SSB, in monomer. In these assays, SSB was titrated with increasing amounts of poly d(T)359 until the maximum amount of intrinsic fluorescence was quenched.
Trypsin cleavage assay
In all experiments, the standard SSB:Trypsin ratio was 1:112 in moles. About 10 µg of SSB protein with or without M13 ssDNA was incubated in Binding Buffer (10 mM Tris‐HCl, (pH8.0), 200 mM NaCl, 5 mM MgCl2, 1 mM DTT, and 1 mM ADP), on ice for 10 min. M13 single‐stranded DNA was used in excess to saturate all SSB binding sites (1 protein monomer: 10 DNA nucleotides). Trypsin buffer was then added to each sample to achieve a final concentration of 50 mM Tris‐HCl, pH 8.0, 20 mM CaCl2. The samples were incubated at 37°C for 2 min. Samples for Time = 0 were collected and incubated on ice with 2 μL of 10 mM PMSF. About 0.1 µg of Trypsin was added to all samples and the trypsin digestion was carried out at 37°C. At each time point (5, 10, 15, 30, 60, 90, and 120 min), samples were collected and incubated on ice with 2 μL of 10 mM PMSF for at least 10 min. About 5 µL of SDS‐PAGE Loading Dye was added to each time point samples, boiled at 100°C for 2 min, and subjected to SDS‐PAGE using Bio‐Rad 18% Criterion TGX Pre‐cast gels. Gels were then stained with Coomassie Brilliant Blue and quantitated using ImageQuant 5.2 software, GE healthcare Bioscience. Data points for graphs of percent relative to total were obtained by expressing the percentage of intensity of substrate band at each time point to total intensity of each lane. Data were analyzed using non‐linear curve fitting in Prism v 5.04 (GraphPad Software, Inc.).
ATP hydrolysis assay
The hydrolysis of ATP was monitored using a coupled spectrophotometric assay carried at 37°C as described previously.21, 37 The standard reaction buffer contained 20 mM Tris‐OAc (pH 7.5), 1 mM DTT, 0.3 mM NADH, 7.5 mM PEP, 20 U/mL PK, 20 U/mL LDH, 100 nM RecG or 10 nM PriA, 1 mM ATP, and 10 mM MgOAc. In assays with RecG, 125 nM of SSB tetramer and 10 μM of M13 ssDNA were present. Assays with PriA contained 200 nM of SSB tetramer, 10 μM of Φx174 ssDNA for ATPase activity assay and 100 μM of Φx174 ssDNA for salt titration.
Assays were performed in a reaction volume of 150 μL, and were initiated by the addition of RecG or separately PriA, following a 2 min pre‐incubation at 37°C of all other components. The rate of ATP hydrolysis was calculated by multiplying the slope of a tangent drawn to linear portions of time courses by 159. In a typical reaction, close to 200 data points were used to draw a linear fit to the data to calculate reaction rates. To obtain kinetic parameters, data were analyzed using non‐linear curve fitting in Prism v 5.04 (GraphPad Software, Inc.).60
In salt‐titration experiments, the same reaction buffers were used (see above). Once a steady‐state rate of ATP hydrolysis was achieved, NaCl was added in increments. This was repeated until all ATP hydrolysis ceased. The resulting hydrolysis rate in each steady‐state region was calculated and expressed as a percent of the steady‐state rate in the absence of NaCl. The total volume used to calculate final concentration of NaCl was adjusted after each addition in order to correct for the additions themselves. A line of best fit was drawn for data points between each addition to obtain the ATP hydrolysis rate after each salt increment. These rates were subsequently graphed to determine the concentration of NaCl resulting in a 50% reduction in the rate of ATP hydrolysis which corresponds to the salt‐titration mid‐point.
RecBCD helicase assay
The unwinding of dsDNA by RecBCD was monitored by observing the quenching of the intrinsic fluorescence of SSB that occurs when the protein binds to ssDNA.38 The dsDNA substrate used was pPB248, linearized with XhoI and incubated at 65°C to inactivate the restriction endonuclease. Reactions contained 10 μM nucleotides of linearized dsDNA, 1 μM SSB monomer, and 2 nM RecBCD. Reactions were carried out at 37°C in a 500 μL volume of buffer, containing 20 mM Tris‐OAc, pH 7.5, 1 mM ATP, 1 mM DTT, and 2 mM MgOAc. Following thermal equilibration at 37°C for 3 min, RecBCD was added to initiate the reaction. The intrinsic fluorescence of SSB is excited at 290 nm and monitored at emission wavelength of 340 nm. The unwinding rates were calculated by multiplying DNA concentration in base pairs by the ratio of total change in fluorescence to the slope of initial drop in fluorescence signal. In a typical reaction, close to 200 data points were used to draw a linear fit to the data to calculate slope of initial drop.
DNA strand exchange
DNA strand exchange was assayed as described.61 Unless otherwise specified, reaction conditions were as follows: reaction mixtures (160 μL, 37°C) contained 20 mM Tris‐OAc, (pH 7.5), 1 mM DTT, 8 mM magnesium acetate, 1 mM ATP, 6.0 μM RecA, 1 μM SSB monomer, 100 μM Φx174 ssDNA, 20 μM linear Φx174 dsDNA, and an ATP‐regenerating system consisting of 7.5 mM phosphoenolpyruvate and 0.02 U/μL pyruvate kinase. In these reactions, linearized Φx174 dsDNA was 5′‐end labeled. Reactions which contained ssDNA, ATP and the regenerating system were pre‐incubated at 37°C for 2 min, RecA was added; 2 min later, SSB was added and finally, radioactively labeled linear dsDNA was added to start the reaction. At various time points, aliquots were removed and the reaction was quenched by the addition of gel‐loading solution to final concentrations of 1% SDS, 50 mM EDTA and 0.4 mg/mL proteinase K. Samples were incubated at 37°C for 5 min right before loading. About 15 μL of sample was loaded onto a 1% agarose gel and subjected to electrophoresis in TAE buffer (40 mM Tris‐OAc and 2 mM EDTA) at 1 V/cm for 16.5 h. Following electrophoresis, gels were dried and exposed to PhosphorImager screens. Analysis of gels was performed using ImageQuant software. Data was analyzed using non‐linear curve fitting or linear regression in Prism v 5.04 (GraphPad Software, Inc.).
Dissociation of SSB from ATTO(dT)65
The assay used has been described previously except that CY3 or Cy5 labeled oligonucleotides were used.47 Dissociation of SSB from 5′‐ATTO488(dT)65 was studied by monitoring the recovery of fluorescence after addition of the competitor DNA, poly d(T) (average length = 359 nt). ATTO488 fluorescence was being monitored with excitation wavelength of 509 nm and emission wavelength of 521 nm. In each assay, 50 nM 5′‐ATTO488(dT)65 oligonucleotide was pre‐incubated in buffer containing NaCl (1; 0.7; or 0.6M), 10 mM Tris‐HCl, pH 8.0, 0.1 mM EDTA for 4 min. Next, 50 nM SSB tetramer was added. When SSB binds to 5′‐ATTO488(dT)65, the fluorescence of the ATTO dye is quenched. Once a stable, lower fluorescence signal was observed (typically after 2–4 min), poly d(T) was added to 15 μM nucleotides, finally. The fluorescence signal increases when SSB dissociates from 5′‐ATTO488(dT)65 and binds to poly d(T). The percent of fluorescence decrease was calculated by expressing the change in fluorescence after addition of SSB relative to the initial fluorescence. The percent recovery was calculated by expressing the fluorescence signal after addition of poly d(T) as a percentage to change in fluorescence after addition of SSB.
Magnetic tweezers experiments
Magnetic tweezers experiments are performed with a PicoTwist magnetic tweezers instrument (www.picotwist.com). DNA hairpins (480 bp) were prepared as described elsewhere62 and tethered between a glass surface treated with anti‐digoxigenin antibody (Roche) and a 1‐μm streptavidin‐coated Dynal magnetic bead (Invitrogen). The DNA hairpins were mechanically stretched by capturing the bead in a magnetic trap generated by a pair of permanent magnets. By using video‐microscopy, the extension of tethered DNA the molecule can be recorded in real time and the value of the applied force is deduced from the measurement of the transverse fluctuations in the bead position.63, 64 DNA forks with long ssDNA tails where generated as described elsewhere4 and use to follow the RecG DNA reannealing activity in real time in presence of different SSB variants. The conversion from measured molecular extension in nm to number of base pairs rewound or base pairs migrated by UvsW or RecG was done by using the previously measured elasticity of ssDNA.64 Conditions used in these experiments were 3 µM for each SSB variant, 5 nM RecG, and 1 mM ATP.
Magnetic tweezers data analysis
Raw data, corresponding to the real‐time changes of the DNA extension in nm, was converted into the number of base pairs reannealed using a calibration factor determined from the elastic properties of ssDNA and dsDNA as described previously.46 Instantaneous enzymatic rates were obtained from a linear fit to the traces filtered with a third‐order Savitzky–Golay filter over a sliding time window of varying size depending on the applied force.65 Delay times were calculated by visual inspection of each reaction trace and histograms were approximated by a single exponential. The error bars shown in the histograms are proportional to the inverse of the square root of the number of points for each individual bin.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting information
Supporting Information
The authors declare that they have no conflicts of interest.
References
- 1. Meyer RR, Laine PS (1990) The single‐stranded DNA‐binding protein of Escherichia coli. Microbiol Rev 54:342–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kowalczykowski SC, Dixon DA, Eggleston AK, Lauder SD, Rehrauer WM (1994) Biochemistry of homologous recombination in Escherichia coli . Microbiol Rev 58:401–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shereda RD, Kozlov AG, Lohman TM, Cox MM, Keck JL (2008) SSB as an organizer/mobilizer of genome maintenance complexes. Crit Rev Biochem Mol Biol 43:289–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lohman T, Ferrari M (1994) Escherichia coli single‐stranded DNA‐binding protein: multiple DNA‐binding modes and cooperativities. Annu Rev Biochem 63:527–570. [DOI] [PubMed] [Google Scholar]
- 5. Costes A, Lecointe F, McGovern S, Quevillon‐Cheruel S, Polard P (2010) The C‐terminal domain of the bacterial SSB protein acts as a DNA maintenance hub at active chromosome replication forks. PLoS Genet 6:e1001238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yu C, Tan HY, Choi M, Stanenas AJ, Byrd AK, Cohan CS, Bianco PR (2016) SSB binds to the RecG and PriA helicases in vivo in the absence of DNA. Genes Cells 21:163–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bianco PR, Pottinger S, Tan HY, Nguyenduc T, Rex K, Varshney U (2016) The IDL of E. coli SSB links ssDNA and protein binding by mediating protein‐protein interactions. Protein Sci (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sancar A, Williams KR, Chase JW, Rupp WD (1981) Sequences of the ssb gene and protein. Proc Natl Acad Sci USA 78:4274–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Curth U, Genschel J, Urbanke C, Greipel J (1996) In vitro and in vivo function of the C‐terminus of Escherichia coli single‐stranded DNA binding protein. Nucleic Acids Res 24:2706–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Simossis VA, Heringa J (2005) PRALINE: a multiple sequence alignment toolbox that integrates homology‐extended and secondary structure information. Nucleic Acids Res 33:W289–W294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Raghunathan S, Kozlov A, Lohman T, Waksman G (2000) Structure of the DNA binding domain of E. coli SSB bound to ssDNA. Nat Struct Biol 7:648–652. [DOI] [PubMed] [Google Scholar]
- 12. Chrysogelos S, Griffith J (1982) Escherichia coli single‐strand binding protein organizes single‐stranded DNA in nucleosome‐like units. Proc Natl Acad Sci USA 79:5803–5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kuznetsov S, Kozlov A, Lohman T, Ansari A (2006) Microsecond dynamics of protein‐DNA interactions: direct observation of the wrapping/unwrapping kinetics of single‐stranded DNA around the E. coli SSB tetramer. J Mol Biol 359:55–65. [DOI] [PubMed] [Google Scholar]
- 14. Savvides SN, Raghunathan S, Futterer K, Kozlov AG, Lohman TM, Waksman G (2004) The C‐terminal domain of full‐length E. coli SSB is disordered even when bound to DNA. Protein Sci 13:1942–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Raghunathan S, Ricard C, Lohman T, Waksman G (1997) Crystal structure of the homo‐tetrameric DNA binding domain of Escherichia coli single‐stranded DNA‐binding protein determined by multiwavelength x‐ray diffraction on the selenomethionyl protein at 2.9‐A resolution. Proc Natl Acad Sci USA 94:6652–6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lu D, Keck JL (2008) Structural basis of Escherichia coli single‐stranded DNA‐binding protein stimulation of exonuclease I. Proc Natl Acad Sci USA 105:9169–9174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lu D, Windsor MA, Gellman SH, Keck JL (2009) Peptide inhibitors identify roles for SSB C‐terminal residues in SSB/exonuclease I complex formation. Biochemistry 48:6764–6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cadman CJ, McGlynn P (2004) PriA helicase and SSB interact physically and functionally. Nucleic Acids Res 32:6378–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shereda RD, Bernstein DA, Keck JL (2007) A central role for SSB in Escherichia coli RecQ DNA helicase function. J Biol Chem 282:19247–19258. [DOI] [PubMed] [Google Scholar]
- 20. Suski C, Marians KJ (2008) Resolution of converging replication forks by RecQ and topoisomerase III. Mol Cell 30:779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Buss J, Kimura Y, Bianco P (2008) RecG interacts directly with SSB: implications for stalled replication fork regression. Nucleic Acids Res 36:7029–7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kozlov AG, Jezewska MJ, Bujalowski W, Lohman TM (2010) Binding specificity of Escherichia coli single‐stranded DNA binding protein for the chi subunit of DNA pol III holoenzyme and PriA helicase. Biochemistry 49:3555–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kozlov AG, Weiland E, Mittal A, Waldman V, Antony E, Fazio N, Pappu RV, Lohman TM (2015) Intrinsically disordered C‐terminal tails of E. coli single‐stranded DNA binding protein regulate cooperative binding to single‐stranded DNA. J Mol Biol 427:763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sun Z, Tan HY, Bianco PR, Lyubchenko YL (2015) Remodeling of RecG helicase at the DNA replication fork by SSB protein. Sci Rep 5:9625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bianco PR (2015) I came to a fork in the DNA and there was RecG. Prog Biophys Mol Biol 117:166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dalgarno DC, Botfield MC, Rickles RJ (1997) SH3 domains and drug design: ligands, structure, and biological function. Biopolymers 43:383–400. [DOI] [PubMed] [Google Scholar]
- 27. Sudol M (1998) From Src Homology domains to other signaling modules: proposal of the 'protein recognition code'. Oncogene 17:1469–1474. [DOI] [PubMed] [Google Scholar]
- 28. Kay BK, Williamson MP, Sudol M (2000) The importance of being proline: the interaction of proline‐rich motifs in signaling proteins with their cognate domains. faseb J 14:231–241. [PubMed] [Google Scholar]
- 29. Ponting CP, Aravind L, Schultz J, Bork P, Koonin EV (1999) Eukaryotic signalling domain homologues in archaea and bacteria. Ancient ancestry and horizontal gene transfer. J Mol Biol 289:729–745. [DOI] [PubMed] [Google Scholar]
- 30. Agrawal V, Kishan RK (2001) Functional evolution of two subtly different (similar) folds. BMC Struct Biol 1:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bianco PR (2016) The tale of SSB. Prog Biophys Mol Biol. DOI:10.1016/j.pbiomolbio.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bianco PR, YL Lyubchenko (2017) SSB and the RecG DNA helicase: An intimate association to rescue a stalled replication fork. Protein Sci. DOI:10.1002/pro.3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu J, Choi M, Stanenas AG, Byrd AK, Raney KD, Cohan C, Bianco PR (2011) Novel, fluorescent, SSB protein chimeras with broad utility. Protein Sci 20:1005–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lohman TM, Overman LB (1985) Two binding modes in Escherichia coli single strand binding protein‐single stranded DNA complexes. Modulation by NaCl concentration. J Biol Chem 260:3594–3603. [PubMed] [Google Scholar]
- 35. Williams KR, Spicer EK, LoPresti MB, Guggenheimer RA, Chase JW (1983) Limited proteolysis studies on the Escherichia coli single‐stranded DNA binding protein: evidence for a functionally homologous domain in both the Escherichia coli and T4 DNA binding proteins. J Biol Chem 258:3346–3355. [PubMed] [Google Scholar]
- 36. Delagoutte E, Heneman‐Masurel A, Baldacci G (2011) Single‐stranded DNA binding proteins unwind the newly synthesized double‐stranded DNA of model miniforks. Biochemistry 50:932–944. [DOI] [PubMed] [Google Scholar]
- 37. Slocum SL, Buss JA, Kimura Y, Bianco PR (2007) Characterization of the ATPase activity of the Escherichia coli RecG protein reveals that the preferred cofactor is negatively supercoiled DNA. J Mol Biol 367:647–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roman LJ, Kowalczykowski SC (1989) Characterization of the helicase activity of the Escherichia coli RecBCD enzyme using a novel helicase assay. Biochemistry 28:2863–2873. [DOI] [PubMed] [Google Scholar]
- 39. Dziegielewska B, Beerman TA, Bianco PR (2006) Inhibition of RecBCD enzyme by antineoplastic DNA alkylating agents. J Mol Biol 361:898–919. [DOI] [PubMed] [Google Scholar]
- 40. Anderson DG, Kowalczykowski SC (1998) SSB protein controls RecBCD enzyme nuclease activity during unwinding: a new role for looped intermediates. J Mol Biol 282:275–285. [DOI] [PubMed] [Google Scholar]
- 41. Bianco PR, Tracy RB, Kowalczykowski SC (1998) DNA strand exchange proteins: a biochemical and physical comparison. Front Biosci 3:D570–D603. [DOI] [PubMed] [Google Scholar]
- 42. Kowalczykowski SC, Krupp RA (1987) Effects of Escherichia coli SSB protein on the single‐stranded DNA‐dependent ATPase activity of Escherichia coli RecA protein. Evidence that SSB protein facilitates the binding of RecA protein to regions of secondary structure within single‐stranded DNA. J Mol Biol 193:97–113. [DOI] [PubMed] [Google Scholar]
- 43. Kowalczykowski SC, Clow J, Somani R, Varghese A (1987) Effects of the Escherichia coli SSB protein on the binding of Escherichia coli RecA protein to single‐stranded DNA. Demonstration of competitive binding and the lack of a specific protein‐protein interaction [Erratum. J Mol Biol 198:81–95. (1987) J Mol Biol 193: [DOI] [PubMed] [Google Scholar]
- 44. Lavery PE, Kowalczykowski SC (1992) A postsynaptic role for single‐stranded DNA‐binding protein in recA protein‐promoted DNA strand exchange. J Biol Chem 267:9315–9320. [PubMed] [Google Scholar]
- 45. Manosas M, Perumal SK, Bianco PR, Ritort F, Benkovic SJ, Croquette V (2014) Corrigendum: RecG and UvsW catalyse robust DNA rewinding critical for stalled DNA replication fork rescue. Nat Commun 5:4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Manosas M, Perumal SK, Bianco PR, Ritort F, Benkovic SJ, Croquette V (2013) RecG and UvsW catalyse robust DNA rewinding critical for stalled DNA replication fork rescue. Nat Commun 4:2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kozlov A, Lohman T (2002) Kinetic mechanism of direct transfer of Escherichia coli SSB tetramers between single‐stranded DNA molecules. Biochemistry 41:11611–11627. [DOI] [PubMed] [Google Scholar]
- 48. Mayer BJ (2001) SH3 domains: complexity in moderation. J Cell Sci 114:1253–1263. [DOI] [PubMed] [Google Scholar]
- 49. Saksela K, Permi P (2012) SH3 domain ligand binding: what's the consensus and where's the specificity?. FEBS Lett 586:2609–2614. [DOI] [PubMed] [Google Scholar]
- 50. Kowalczykowski SC, Clow JC, Somani R, Varghese A (1987) Effects of the Escherichia coli SSB protein on the binding of Escherichia coli RecA protein to single‐stranded DNA: demonstration of competitive binding and the lack of a specific protein‐protein interaction. J Mol Biol 193:81–95. [DOI] [PubMed] [Google Scholar]
- 51. Su XC, Wang Y, Yagi H, Shishmarev D, Mason CE, Smith PJ, Vandevenne M, Dixon NE, Otting G (2014) Bound or free: interaction of the C‐terminal domain of Escherichia coli single‐stranded DNA‐binding protein (SSB) with the tetrameric core of SSB. Biochemistry 53:1925–1934. [DOI] [PubMed] [Google Scholar]
- 52. Shishmarev D, Wang Y, Mason CE, Su XC, Oakley AJ, Graham B, Huber T, Dixon NE, Otting G (2014) Intramolecular binding mode of the C‐terminus of Escherichia coli single‐stranded DNA binding protein determined by nuclear magnetic resonance spectroscopy. Nucleic Acids Res 42:2750–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mason CE, Jergic S, Lo AT, Wang Y, Dixon NE, Beck JL (2013) Escherichia coli single‐stranded DNA‐binding protein: nanoESI‐MS studies of salt‐modulated subunit exchange and DNA binding transactions. J Am Soc Mass Spectrom 24:274–285. [DOI] [PubMed] [Google Scholar]
- 54. Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 55. Gill SC, von Hippel PH (1989) Calculation of protein extinction coefficients from amino acid sequence data [Erratum. Anal Biochem (1990) 189:283]. Anal Biochem 182:319–326. [DOI] [PubMed] [Google Scholar]
- 56. Lohman TM, Green JM, Beyer RS (1986) Large‐scale overproduction and rapid purification of the Escherichia coli ssb gene product. Expression of the ssb gene under lambda PL control. Biochemistry 25:21–25. [DOI] [PubMed] [Google Scholar]
- 57. Bianco PR, Weinstock GM (1996) Interaction of the RecA protein of Escherichia coli with single‐stranded oligodeoxyribonucleotides. Nucleic Acids Res 24:4933–4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Weinstock GM, McEntee K, Lehman IR (1981) Hydrolysis of nucleoside triphosphates catalyzed by the recA protein of Escherichia coli. Characterization of ATP hydrolysis. J Biol Chem 256:8829–8834. [PubMed] [Google Scholar]
- 59. Griffith J, Shores CG (1985) RecA protein rapidly crystallizes in the presence of spermidine: a valuable step in its purification and physical characterization. Biochemistry 24:158–162. [DOI] [PubMed] [Google Scholar]
- 60. Segel IH (1976) Biochemical calculations, 2nd ed. Hoboken, NJ: John Wiley and Sons. [Google Scholar]
- 61. Roman LJ, Kowalczykowski SC (1986) Relationship of the physical and enzymatic properties of Escherichia coli recA protein to its strand exchange activity. Biochemistry 25:7375–7385. [DOI] [PubMed] [Google Scholar]
- 62. Camunas‐Soler J, Manosas M, Frutos S, Tulla‐Puche J, Albericio F, Ritort F (2015) Single‐molecule kinetics and footprinting of DNA bis‐intercalation: the paradigmatic case of Thiocoraline. Nucleic Acids Res 43:2767–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Strick TR, Allemand JF, Bensimon D, Bensimon A, Croquette V (1996) The elasticity of a single supercoiled DNA molecule. Science 271:1835–1837. [DOI] [PubMed] [Google Scholar]
- 64. Gosse C, Croquette V (2002) Magnetic tweezers: micromanipulation and force measurement at the molecular level. Biophys J 82:3314–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Manosas M, Meglio A, Spiering MM, Ding F, Benkovic SJ, Barre FX, Saleh OA, Allemand JF, Bensimon D, Croquette V (2010) Magnetic tweezers for the study of DNA tracking motors. Methods Enzymol 475:297–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information