

Abstract
The spirochete Treponema pallidum is the causative agent of syphilis, a sexually transmitted infection of major global importance. Other closely related subspecies of Treponema also are the etiological agents of the endemic treponematoses, such as yaws, pinta, and bejel. The inability of T. pallidum and its close relatives to be cultured in vitro has prompted efforts to characterize T. pallidum's proteins structurally and biophysically, particularly those potentially relevant to treponemal membrane biology, with the goal of possibly revealing the functions of those proteins. This report describes the structure of the treponemal protein Tp0737; this polypeptide has a fold characteristic of a class of periplasmic ligand‐binding proteins associated with ABC‐type transporters. Although no ligand for the protein was observed in electron‐density maps, and thus the nature of the native ligand remains obscure, the structural data described herein provide a foundation for further efforts to elucidate the ligand and thus the function of this protein in T. pallidum.
Keywords: syphilis, X‐ray crystallography, differential scanning fluorimetry, analytical ultracentrifugation, ABC transporter
Short abstract
PDB Code(s): 5U2P
Abbreviations
- ABC
ATP‐binding cassette
- AUC
analytical ultracentrifugation
- DSF
differential scanning fluorimetry
- LBP
ligand‐binding protein
- rTp0737
a recombinant version of gene product tp0737
- PCR
polymerase chain reaction
- PIPE
polymerase‐incomplete primer extension
- SV
sedimentation velocity
Introduction
Despite the efficacy and widespread availability of chemotherapeutic strategies, the sexually transmitted human disease syphilis remains a significant threat to human health on a global scale.1, 2, 3 Worldwide rates and prevalences of the disease have failed to decline,1 and alarming increases in case rates, even in developed countries, have been observed.4, 5, 6, 7 Furthermore, the emergence of macrolide resistance in Treponema pallidum, the etiological agent, has been alarming.8 Finally, decades of intensive efforts have failed to yield a syphilis vaccine.
Treponema pallidum subsp. pallidum is an obligate parasite of humans. This microorganism's lifestyle and its minimized genome (containing only 1041 genes9) necessitate that it acquires the vast majority of its nutrients from its human host. In many cases, nutrient procurement is achieved via ABC‐type transporters.3 In T. pallidum, these systems typically initially secure target small‐molecule compounds through their binding to membrane‐tethered ligand‐binding proteins (LBPs) that protrude into the periplasm.3 These lipoproteins ostensibly facilitate the translocation of the nutrient to the organism's cytoplasm via the action of transmembrane permeases, whose actions are driven by cytoplasmic ATPases.10 Many such systems have been elucidated in T. pallidum, including transporters for transition metal ions,11, 12 L‐methionine,13 D‐glucose,14, 15 purine nucleosides,16 and riboflavin.17
The gene product of tp0737 is an unusual protein in the context of the biology of T. pallidum. The protein bears sequence homology to sugar‐binding proteins of ABC‐type transporters, and is annotated as such in databases.9 However, the protein does not harbor the N‐terminal sequence signatures that are ordinarily necessary for the post‐translational modification of lipoproteins. Rather, an N‐terminal sequence that putatively targets the protein to the periplasm (i.e. to signal peptidase I) is present.18 Whereas this may suggest that Tp0737 can exist free in the treponemal periplasm, it is possible that the signal sequence is not cleaved, thus serving as a “signal anchor” that tethers the protein to the cytoplasmic membrane, with its soluble portion harbored in the periplasm.19, 20, 21 Moreover, the genetic environment of tp0737 suggests that the mRNA derived therefrom is monocistronic. This organization is unusual insofar as T. pallidum commonly transcribes the LBP, permease, and ATPase(s) of ABC transporters on the same mRNA.16, 17, 22
These unique features and the unknown identity of the protein's native ligand prompted us to study the structure of Tp0737 recombinantly expressed without its N‐terminal signal sequence (rTp0737). The crystal structure was determined at a resolution of 1.76 Å. The structure reveals a two‐lobed protein with a deep cleft between the lobes; as predicted, it resembles a LBP for an ABC‐type transporter. Unfortunately, no electron density for a potential ligand could be observed. Although a series of biophysical studies also did not elucidate the identity of any ligand, the rTp0737 structure sets the stage for future efforts in ligand discovery.
Results and Discussion
Structure description
The crystal structure of rTp0737, a recombinant fragment of Tp0737, was determined and refined at a resolution of 1.76 Å (Table 1, Fig. 1). All residues are visible in the electron‐density maps except for 28‐39, 182‐184, 394‐396, and 430‐434 (N.B.: although only 408 residues are present in the protein construct, the numbering scheme reflects the primary structure of the immature, unprocessed protein). Overall, rTp0737 comprises two domains that are connected via three crossover points (Fig. 1). In this report, the two domains are termed the “N domain” (residues 40‐142 and 315‐360) and the “C domain” (residues 146‐312 and 364‐429), as the N‐ and C‐termini are located in those respective domains. The N and C domains have similar structures insofar as they both are composed of a central (mostly parallel) β sheet that is flanked on both sides by helices and regions with no regular secondary structures. The central β‐sheet in the N domain encompasses five β‐strands; these are all parallel except one [10; Fig. 1(A)]. The analogous β‐sheet in the C domain is more complex, as it contains three parallel strands and one antiparallel strand. A secondary, minor sheet is formed by the packing of strands 9 and 11 against one of the strands of the main sheet; both sheets in the C domain share strand 5. There are 14 α‐helices in the structure, and DSSP23 identifies an inordinate number of 310 helices: 12. There is no evidence that the large number of 310 helices has significant functional consequences for the protein. The three crossover regions (residues 143‐145, 313‐314, and 361‐363) do not have regular secondary structures and appear to form a hinge between the two domains.
Table 1.
Diffraction and Structure Solution Statistics
| Data Set | rTp0737 | rTp0737‐SeMet |
|---|---|---|
| PDB accession no. | 5U2P | |
| Data collection | ||
| Wavelength (Å) | 0.97918 | 0.97929 |
| Temperature (K) | 100 | 100 |
| Space group | P3121 | P3121 |
| Unit cell dimensions (Å) | ||
| a | 55.925 | 55.887 |
| b | 55.925 | 55.887 |
| c | 237.632 | 237.426 |
| α = β (°) | 90 | 90 |
| γ (°) | 120 | 120 |
| Resolution (Å) | 37.43–1.76 (1.79–1.76)a | 48.4–1.95 (1.98–1.95) |
| Completeness (%) | 97.4 (92.4) | 98.8 (79.8) |
| Multiplicity | 3.5 (2.5) | 7.1 (3.9) |
| Unique reflections | 43,052 (1,994) | 32,407 (1,274) |
| Rr.i.m. b | 0.052 (0.471) | 0.061 (0.311) |
| <I>/σI | 19.6 (2.9) | 12.7 (2.6) |
| Wilson B (Å2) | 22.8 | 18.8 |
| Phasing | ||
| Sites found | N/A | 4 |
| Overall figure of merit | N/A | 0.250 |
| Automatically built residues | N/A | 368 |
| Refinement | ||
| Resolution (Å) | 37.54–1.76 | |
| No. nonsolvent atoms | 3,235 | |
| No. water atoms | 232 | |
| No. ions | 3 | |
| Average B‐factors | ||
| Overall (Å2) | 36.64 | |
| Protein (Å2) | 36.53 | |
| Solvent (Å2) | 38.20 | |
| Ion (Å2) | 34.86 | |
| R‐values | ||
| R work | 0.182 | |
| R free | 0.222 | |
| Ramachandran Statistics | ||
| Outliers (%) | 0.0 | |
| Most favored region (%) | 98.7 | |
| r.m.s. deviations | ||
| Bonds (Å) | 0.005 | |
| Angles (°) | 0.90 | |
Numbers in the parentheses are reported for the highest‐resolution shell of reflections.
R merge ( where the outer sum (hkl) is over the unique reflections and the inner sum (i) is over the set of independent observations of each unique reflection) was multiplied by the factor (N/(N − 1))1/2 to estimate Rr.i.m.
Figure 1.
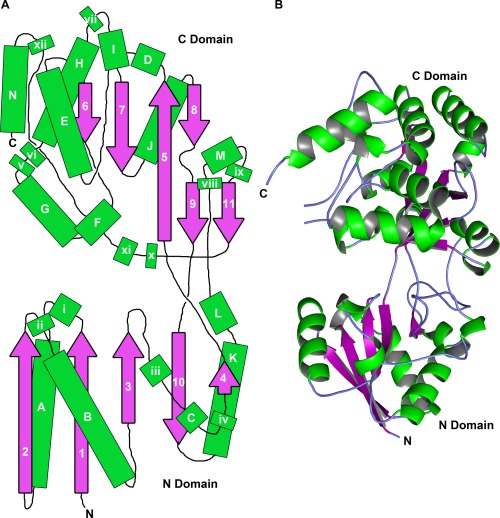
The structure of rTp0737. (A) Schematic of the overall topology of the protein. Helices are depicted as green rectangles; lettered rectangles are α‐helices, while those with lowercase roman numerals are 310 helices. β‐strands are represented by purple, numbered arrows. The relative sizes of these features are approximately to scale. The N and C domains are labeled. (B) Ribbons representation of rTp0737. Secondary structural features are colored as in part (A), with regions of irregular secondary structure colored light blue.
Between the domains is a large, solvent‐filled cleft. There is no electron density for a specifically bound ligand in this cleft nor anywhere else in the structure. Besides bound water molecules, the only other chemical entities bound to the proteins are a bromide anion, two chloride anions, and ethylene glycol, all of which were present in the protein‐storage buffer and/or the cryoprotection buffer.
Comparisons to other structures
A hidden‐Markov‐model‐based strategy24 was used to search the Protein Data Bank for sequences similar to that of rTp0737A. The closest sequence match (16% identity) was to the GacH acarbose/maltose‐binding protein from Streptomyces glaucescens (accession number 3K0125). However, that protein had a significantly different conformation than that of rTp0737, so a secondary‐structure‐matching26 search of the Protein Data Bank was initiated, revealing several X‐ray crystal structures with significant structural homology to that of rTp0737. The first match (accession number 3OO9), with an r.m.s.d. of 2.9 Å over 316 aligned Cα atoms is AcbH from Actinoplanes sp. SE50/110.27 The latter protein is the LBP of a putative ABC transporter for β‐D‐galactose. Such proteins bind ligands that are available extracytoplasmically and deliver them to permeases that traverse the cytoplasmic membrane, where the action of an intracellular ATPase drives the import of the ligand into the cytoplasm, usually against a concentration gradient.28 The second match (accession number 3QUF; r.m.s.d. of 2.8 Å over 302 Cα atoms) was to a protein from Bifidobacterium longum supsp. infantis. Although there is no publication describing the structure and function of this protein, it is presumed to be the LBP of an ABC transporter. Other, weaker matches (i.e. with either less coverage or higher r.m.s.d.'s) include other ABC‐type LBPs, such as those with putative specificities for raffinose (accession number 2HFB) and maltotriose (accession number 2GHB). Despite these evident homologies, we observed no bound ligand in the rTp0737 electron‐density maps.
As intimated from the structural matches enumerated above, rTp0737 is topologically and structurally related to polypeptides that serve as the LBPs for the ATP‐dependent transport of nutrients into bacterial cytoplasms. Specifically, rTp0737's topology conforms to that of “Type II” LBPs in an early nomenclature system29 and to that of “Cluster D” in a more recent classification scheme.30 Indeed, the structural organization of rTp0737 is most closely related to proteins in “Cluster D‐I”, which contains carbohydrate‐binding proteins with specificities for maltose and glucose. Like rTp0737, Cluster D‐I proteins have a five‐stranded β‐sheet in the N domain in the order 2‐1‐3‐n‐4, whereas the C domain has the order 1‐2‐n‐3, where n is respectively the first strand after a crossover from one domain to the other. Further, these proteins have an “extra subdomain” in their respective C domains. In rTp0737, this subdomain comprises α‐helices G, H, and N, along with 310 helices v, vi, and xii.
Significantly, the closest structural matches to rTp0737 are LBPs without any bound ligand. Unliganded LBPs tend to exist in solution in an open‐cleft form, with the two domains not closely apposed.15, 31, 32, 33 When ligand binds, the two domains move closer to one another, effectively closing the cleft and, in many cases, shielding the ligand from solvent.16, 34, 35, 36 Thus, the open form of the rTp0737 observed in the crystal structure is consistent with its lack of bound ligand.
To glean possible hints regarding the cognate ligand of Tp0737, we examined the putative binding pocket (Table 2; Fig. 2). We find that no single class of amino acids predominates in the cleft. Further, we compared the surface features of the cleft to a structurally homologous protein, BxlE of Streptomyces thermoviolaceus OPC‐520 (PDB accession number 3VXC; there is no attendant publication). BxlE binds to a disaccharide called xylobiose, and the binding site is marked on the left side of Figure 2. There are substantial areas of hydrophobicity in the BxlE xylobiose‐binding site, where aromatic residues stack on the surfaces of the linked xylose monomers. The analogous areas of Tp0737 are largely devoid of such features, corresponding to the paucity of aromatic residues in the putative ligand‐binding cleft (Table 2). Moreover, this area on rTp0737 is flatter than the corresponding area in BxlE, and is rich in hydrogen‐bond donors and acceptors. The overall arrangement of arrangement of amino‐acid side chains in the cleft does not immediately suggest the identities of potential ligands beyond the fact that they would likely have to be largely polar.
Table 2.
Residues Whose Side Chains Line the Cleft of rTp0737
| Polar | Charged | Hydrophobic |
|---|---|---|
| H50 | R47 | P55 |
| S52 | K49 | P102 |
| S100 | D77 | L138 |
| S103 | D78 | V145 |
| H105 | D282 | V259 |
| N142 | K286 | I263 |
| S146 | E287 | Y274 |
| N148 | E313 | L279 |
| S191 | D385 | V280 |
| T193 | A283 | |
| S195 | L352 | |
| T199 | V362 | |
| S277 | P364 | |
| H278 | A383 | |
| T309 | A386 | |
| Q315 | A388 | |
| H349 | ||
| Q391 |
Figure 2.
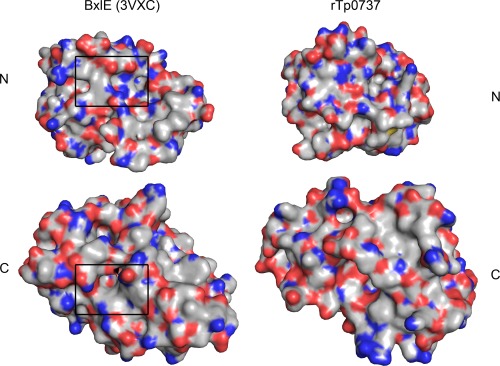
Surface features of rTp0737 and a related LBP. The two LBPs, BxlE (left) and rTp0737 (right) have been split into their respective N and C domains, and these have been individually rotated such that the ligand‐binding surfaces are facing forward; in effect, they have been “opened” along their hinge regions to present the ligand‐binding surfaces. Surfaces contributed by carbon atoms are colored gray, while those representing nitrogens are blue, oxygens are red, and sulfurs are yellow. Importantly, these surfaces are not the result of an electrostatic calculation. Boxes on BxlE represent the xylobiose‐binding site.
Ligand screening of rTp0737
The obvious structural similarities between LBPs and rTp0737 prompted screening experiments to be undertaken for the binding of small molecules to the purified protein. Differential scanning fluorimetry (DSF) was used for this purpose.37 Briefly, the method involves the measurement of the fluorescence of a dye that is included with the protein/ligand solution. The fluorescence intensity of the dye is enhanced when it binds to exposed, hydrophobic regions of the unfolded protein, allowing the observer to estimate an apparent melting temperature, , for the protein in the presence of a temperature gradient. Thus, the protein/ligand/dye solution is subjected to increasing temperature, and the fluorescence of the solution is monitored. Specific binding of a ligand should shift the to a higher value.
DSF was employed in a series of 96‐well plates, each of which contained 95 ligands and a negative control (water; see Materials and Methods). In the negative‐control wells, rTp0737 exhibited a of approximately 48°C (Fig. 3). Overall, 950 ligands/conditions were screened, but none caused a positive excursion from the negative‐control greater than 0.5°C. Thus, 0.5°C probably represents the assay's intrinsic noise. By contrast, in a similar DSF assay performed on a different LBP protein (Tp0684), a compound that bound to Tp0684 with a K D of approximately 30 μM resulted in a +3.5°C change in .15 Given the lack of a response above the noise level and the results of the earlier assay, we concluded that no candidate ligands for rTp0737 were identified. The tested ligands included small molecules that Cluster D‐I members are known or suspected to bind, including D‐glucose, D‐galactose, maltose, maltotriose, raffinose, and xylobiose. Furthermore, not only carbohydrates were tested; amino acids, phosphorus sources, sulfur sources, cofactors, peptides, and osmolytes were among the tested compounds.
Figure 3.
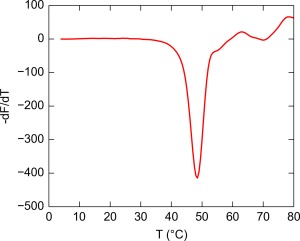
A typical DSF profile for rTp0737. The minimum in this plot was taken as : 48°C.
This negative result could have one of several causes. Most likely, the true ligand (if any) of rTp0737 was not among the tested compounds. However, there are other possibilities. For instance, buffering agents are known to interfere with ligand binding in some LBPs.15, 22, 34 To examine whether the phosphate buffer of the assay was preventing ligand binding, the entire DSF screen was performed again in the presence of Tris as the buffering agent. Still, no significant shifts in could be discerned.
Another potential cause of false negatives in the DSF assay was the solution behavior of the protein. Specifically, rTp0737 could form oligomers or nonspecific aggregates. The interprotein contacts in these assemblies could, in turn, occlude ligand access to the cleft or disfavor the domain motion thought to be necessary for ligand binding.
To address the possibility of oligomers and aggregates, the hydrodynamic properties of rTp0737 were probed using the sedimentation velocity (SV) mode of analytical ultracentrifugation (AUC). At a concentration of 1 mg/mL (roughly twice that employed in the DSF assay), the protein was predominantly monomeric (Fig. 4). More precisely, 95% of the mass present in this rTp0737 preparation could be accounted for by the monomeric form of the protein (3.1 S). A small amount (3%) appears to be either a higher‐mass contaminant or a dimer of rTp0737 (4.2 S). Therefore, it is very unlikely that oligomerization or aggregation was affecting the protein's ability to bind ligands in the DSF assay.
Figure 4.
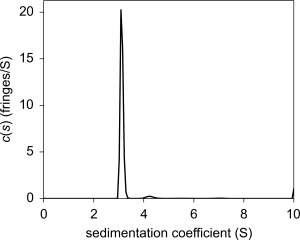
The c(s) distribution for 1 mg/mL rTp0737. The dominant peak corresponds to the monomeric form of the protein, as computed using the Svedberg equation.
Conclusions and implications
The structure of rTp0737 (Fig. 1) conforms closely to other protein structures that serve as LBPs for ABC transporters. Hence, it may be expected that Tp0737 fills this role as well. However, a screen of a large panel of likely nutrients that might be imported by such a transporter revealed no putative ligands for the protein.
That T. pallidum cannot be continuously cultivated in vitro underscores the incomplete understanding of the spirochete's nutritional requirements. It is therefore possible that the true ligand of Tp0737 may not have been included in the chemical panel of known or likely nutrients used in the DSF experiments detailed above. Expanding the chemical space of the screen could reveal the protein's native ligand. In this regard, it is instructive to examine the chemical nature of the amino‐acid residues that line rTp0737's cleft (Fig. 2, Table 2). These residues are a mixture of polar, charged, and hydrophobic, with only one aromatic residue present (Y274). The aromatic portion of this residue is mostly buried, with only the side chain's hydroxyl group presented to the cleft. The lack of aromatic amino acids in the cleft contrasts with most carbohydrate‐binding LBPs, which have a mix of polar and aromatic residues to bind the sugar moiety(ies).38, 39 Again, the evidence favors an atypical ligand.
The bilobed fold of the LBPs is also used for other purposes by microorganisms, e.g. as chemotaxis receptors40 and transcription factors.41 Although there is no evidence for either of these functions for Tp0737, these examples underscore the functional versatility of the fold. Further investigations along these lines are complicated by the aforementioned intractability of culturing T. pallidum in vitro. Another fact that hampers future analysis of this protein is that homologues are not found in genetically tractable spirochetes such as T. denticola and Borrelia burgdorferi.
Despite the difficulties and enigmas presented above, the crystal structure of rTp0737 offers some opportunities. The amino‐acid side chains present in the cleft (Table 2) are informative in this regard. The ligand probably has a mixed chemical character, with a number of polar groups poised to interact with the polar side chains arrayed in Tp0737's cleft. Future investigations of the ligand's identity will proceed taking heed of these structural clues.
Materials and methods
Cloning, overexpression, and protein preparation
To produce a recombinant derivative of Tp0737 (rTp0737) in Escherichia coli, the DNA fragment encoding amino acid residues 28‐436 (cloned without the residues comprising the N‐terminal predicted transmembrane helix (residues 1‐26) plus Cys27) of Tp0737 was polymerase chain reaction (PCR) amplified from T. pallidum genomic DNA by the polymerase incomplete primer extension (PIPE) cloning method using ends‐specific primers (PIPE insert). The expression vector, pSpeedET (DNASU, AZ), which encodes an N‐terminal TEV‐protease cleavable expression and purification hexa‐histidine tag (MGSDKIHHHHHHENLYFQG), was PCR amplified with PIPE‐vector primers. The PIPE‐insert and PIPE‐vector were mixed to anneal the amplified DNA fragments together.42 E. coli HK100 competent cells were transformed with the mixtures (PIPE‐vector and insert) and selected for kanamycin resistance on LB agar plates. Cloning junctions/fragments were verified by DNA sequencing. A verified plasmid in HK100 cell was then used for soluble protein expression. Briefly, E. coli HK100 cells were grown at 37°C in LB medium containing 40 µg/mL of kanamycin until the cell density reached an A 600 of 0.5. The culture was then induced for 3 h at 37°C with 0.2% (w/v) L‐arabinose. The procedures for expression and purification of the recombinant proteins were essentially as previously described.43
For the production of selenomethionine labeled protein, tp0737 was recloned into a pProEx HTb vector (Invitrogen) and methionine mutations (L139M and L336M) were introduced into the plasmid using the QuikChange Multi site‐directed mutagenesis kit (Agilent Technologies). The plasmid was then transformed into a methionine auxotroph E. coli B834 (DE3). The recombinant protein was overproduced and purified as described previously.44
Protein concentrations were determined in buffer A (20 mM Hepes, 0.1 M NaCl, pH 7.5, 2 mM n‐Octyl‐β‐D‐glucopyranoside) using spectrophotometry. Extinction coefficients were calculated using the ProtParam tool of ExPASy (www.expasy.org).45
Crystallization and cryoprotection
Crystals of rTp0737 were obtained by mixing 3 μL of Tp0737 (∼24 mg/mL in buffer A) with 3 μL of crystallization buffer (0.2 M NaBr, 0.1 M Bis‐Tris‐Propane, pH 7.5, 20% (w/v) PEG 3,350) and incubating them over 0.4 mL of the reservoir (containing crystallization buffer) for 7 days. The crystals were transferred to the stabilization buffer (0.2 M NaBr, 0.1 M Bis‐Tris‐Propane, pH 7.5, 20% (w/v) PEG 3,350, 20% (v/v) ethylene glycol). After about 1 min in this solution, the crystals were flash‐cooled in liquid nitrogen. Toward solving the phase problem, crystals of a selenomethionyl derivative of Tp0737 (SeTp0737) were grown by mixing 3 μL of SeTp0737 (∼23 mg/mL in buffer A) with 3 μL of crystallization buffer (0.1 M Tris, pH 8.0, 0.2 M NaCl, 20% (w/v) PEG 6,000) and incubating the drops over 0.4 mL of reservoir solution containing the crystallization buffer for 10 days. The crystals were transferred to the stabilization buffer (0.1 M Tris, pH 8.0, 0.2 M NaCl, 20% (w/v) PEG 6,000, 25% (v/v) ethylene glycol) and cryoprotected as above.
Data collection, processing, and structure determination
Native crystals of rTp0737 diffracted X‐rays to a dmin spacing of 1.76 Å and had the symmetry of space group P3121 using radiation of wavelength 0.97918 Å at beamline 19‐ID at the Structural Biology Center of Argonne National Laboratory (Table 1). The data were reduced and scaled using HKL2000.46 Data from the mutant, selenomethionyl derivative of rTp0737 were acquired at the same facility at a wavelength of 0.97929 Å (Table 1). The single‐wavelength anomalous diffraction (SAD) method was used to calculate phases using HKL‐3000.47 In brief, the HKL‐3000 user interface was utilized to reduce, integrate, and scale the data, to find Se sites (four sites were found) using SHELXD,48 to refine the sites and calculate initial phases using MLPHARE,49 to employ density modification using DM50 (no noncrystallographic symmetry is present), and to perform automated model building using ARP/wARP.51 After these steps, the model covered 94% of the input sequence. This model was subsequently rigid‐body refined against the native data (the space groups of the two crystals were sufficiently isomorphous to support this strategy) in PHENIX.52 The model was completed using iterative rounds of model building and adjustment in Coot53 and refinement in PHENIX, including individual B‐factor, TLS, and positional refinement. Riding hydrogen atoms were included in the protein model throughout refinement. The final model comprises 384 amino‐acid residues, 177 water molecules, 2 chloride ions, 1 bromide ion, and a single molecule of ethylene glycol. The model geometry and related statistics are detailed in Table 1. Residues 182‐184 and 394‐396 (given the uncertain knowledge of the protein's processing in T. pallidum, the numbering for the unprocessed protein is used throughout this paper) were not evident in electron‐density maps. The structure has been deposited in the Protein Data Bank with the accession number 5U2P.
Analytical ultracentrifugation
Sedimentation velocity (SV) AUC was conducted in a Beckman‐Coulter Model XL‐I ultracentrifuge. After assembly of the centrifugation cell, which consisted of a dual‐sector, 1.2‐cm path‐length Epon centerpiece positioned between two sapphire windows enclosed in an aluminum housing, 400 μL of sample buffer was introduced into the reference sector, and 400 μL of rTp0737 at an approximate concentration of 1 mg/mL was dispensed into the sample sector. The cell was inserted into an An50‐Ti rotor, which was then placed into the centrifuge. The rotor was temperature‐equilibrated at 20°C for 2.5 h under vacuum, and then accelerated to a speed of 50,000 rpm. Interference optics were used to monitor the concentration profiles. The c(s) methodology was used to analyze the data54, 55 with both radially independent and time‐independent noise being calculated and accounted for in the analysis.56 Partial‐specific volume, solution viscosity, and solution density were calculated using SEDNTERP.57 The molar mass associated with a given c(s) peak was determined using equations presented by Schuck and coworkers.58 GUSSI59 was used to render the c(s) figure.
Differential Scanning Fluorimetry
The thermal stability of rTp0737 in the absence and presence of small molecules was performed in a 96‐well PCR‐plate (Bio‐Rad Laboratories, Inc., Hercules, CA) using a BioRad CFX96 real‐time PCR instrument coupled to a C1000 thermal cycler. Compounds from Biolog's Phenotype MicroAray (PM) supplied in 96‐well microplates (BIOLOG, Inc.) were dissolved in 50 μL of sterile water to obtain a final concentration of around 10–20 mM. Screenings were performed with plates PM1, PM2A, PM3B, PM4A, PM5, PM6, PM7, PM8, and PM9. Each plate contains 95 compounds and a blank (no ligand) control. Complete plate contents are detailed on the BIOLOG website (www.biolog.com).
The assay mixture (20 μL) contained 10 μM purified protein in a buffer containing 10 mM phosphate, 100 mM NaCl, pH 7.4. This mixture also contained SYPRO Orange that had been diluted 1000‐fold from the purchased stock concentration (Thermo Fisher Scientific, Inc., Waltham, MA). Two microliters of the resuspended Biolog compounds were added to each well. Samples were heated from 4°C to 95°C. The fluorescence values were recorded as a function of temperatures, and the negative first derivative values of these curves (−dF/dT) were calculated. The minima of these curves were taken at the apparent melting temperature (T m , app).
Conflicts of Interest
The authors claim no conflicts of interest.
Acknowledgments
The authors thank Dr. Shih‐Chia Tso for technical assistance. Some of the results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE‐AC02‐06CH11357. This research was supported by grant no. AI056305 from the National Institutes of Health to M.V.N
Impact Statement: Treponema pallidum, the causative agent of syphilis, has a minimalistic genome and thus relies on the human host for nutrients. In this paper, we describe the structural clues regarding the function of a protein that putatively imports a nutrient into this microorganism.
References
- 1. World Health Organization (2011) Prevalence and incidence of selected sexually transmitted infections: Chlamydia trachomatis, Neisseria gonorrhoeae, syphilis and Trichomonas vaginalis Methods and results used by WHO to generate 2005 estimates. Geneva: World Health Organization. [Google Scholar]
- 2. Chao JR, Khurana RN, Fawzi AA, Reddy HS, Rao NA (2006) Syphilis: reemergence of an old adversary. Ophthalmology 113:2074–2079. [DOI] [PubMed] [Google Scholar]
- 3. Radolf JD, Deka RK, Anand A, Smajs D, Norgard MV, Yang XF (in press) Treponema pallidum, the syphilis spirochete: making a living as a stealth pathogen. Nat Rev Microbiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Simms I, Fenton KA, Ashton M, Turner KME, Crawley‐Boevey EE, Gorton R, Thomas DR, Lynch A, Winter A, Fisher MJ, Lighton L, Maguire HC, Solomou M (2005) The re‐emergence of Syphilis in the United Kingdom: The new epidemic phases. Sex Transm Dis 32:220–226. [DOI] [PubMed] [Google Scholar]
- 5. Bowen V, Su J, Torrone E, Kidd S, Weinstock H (2015) Increase in incidence of congenital syphilis– United States, 2012‐2014. Morb Mortal Wkly Rep 64:1241–1245. [DOI] [PubMed] [Google Scholar]
- 6. Golden MR, Marra CM, Holmes KK (2003) Update on syphilis: resurgence of an old problem. J Am Med Assoc 290:1510–1514. [DOI] [PubMed] [Google Scholar]
- 7. Kerani RP, Handsfield HH, Stenger MS, Shafii T, Zick E, Brewer D, Golden MR (2007) Rising rates of syphilis in the era of syphilis elimination. Sex Transm Dis 34:154–161. [DOI] [PubMed] [Google Scholar]
- 8. Stamm LV (2010) Global challenge of antibiotic‐resistant Treponema pallidum. Antimicrob Agents Chemother 54:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fraser CM, Norris SJ, Weinstock GM, White O, Sutton GG, Dodson R, Gwinn M, Hickey EK, Clayton R, Ketchum KA, Sodergren E, Hardham JM, McLeod MP, Salzberg S, Peterson J, Kalak H, Richardson D, Howell JK, Chidambaram M, Utterback T, McDonald L, Artiach P, Bowman C, Cotton MD, Fujii C, Garland S, Hatch B, Horst K, Roberts K, Sandusky M, Weidman J, Smith HO, Venter JC (1998) Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science 281:375–388. [DOI] [PubMed] [Google Scholar]
- 10. Davidson AL, Maloney PC (2007) ABC transporters: how small machines do a big job. Trends Microbiol 15:448–455. [DOI] [PubMed] [Google Scholar]
- 11. Desrosiers DC, Sun YC, Zaidi AA, Eggers CH, Cox DL, Radolf JD (2007) The general transition metal (Tro) and Zn2+ (Znu) transporters in Treponema pallidum: analysis of metal specificities and expression profiles. Mol Microbiol 65:137–152. [DOI] [PubMed] [Google Scholar]
- 12. Lee Y‐H, Deka RK, Norgard MV, Radolf JD, Hasemann CA (1999) Treponema pallidum TroA is a periplasmic zinc‐binding protein with a helical backbone. Nat Struct Biol 184:628–633. [DOI] [PubMed] [Google Scholar]
- 13. Deka RK, Neil L, Hagman KE, Machius M, Tomchick DR, Brautigam CA, Norgard MV (2004) Structural evidence that the 32‐kilodalton lipoprotein (Tp32) of Treponema pallidum is an L‐methionine‐binding protein. J Biol Chem 279:55644–55650. [DOI] [PubMed] [Google Scholar]
- 14. Deka RK, Goldberg MS, Hagman KE, Norgard MV (2004) The Tp38 (TpMg1B‐2) lipoprotein binds glucose in a manner consistent with receptor function in Treponema pallidum . J Bacteriol 186:2303–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brautigam CA, Deka RK, Liu WZ, Norgard MV (2016) The Tp0684 (MglB‐2) lipoprotein of Treponema pallidum: a glucose‐binding protein with divergent topology. PLoS One 11:e0161022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Deka RK, Brautigam CA, Yang XF, Blevins JS, Machius M, Tomchick DR, Norgard MV (2006) The PnrA (Tp0319; TmpC) lipoprotein represents a new family of bacterial purine nucleoside receptor encoded within an ATP‐binding cassette (ABC)‐like operon in Treponema pallidum . J Biol Chem 281:8072–8081. [DOI] [PubMed] [Google Scholar]
- 17. Deka RK, Brautigam CA, Biddy BA, Liu WZ, Norgard MV (2013) Evidence for an ABC‐type riboflavin transporter system in pathogenic spirochetes. MBio 4:e00615–e00612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. [DOI] [PubMed] [Google Scholar]
- 19. Deka RK, Lee Y, Hagman KE, Lingwood CA, Hasemann CA, Norgard MV, Radolf JD, Shevchenko D (1999) Physicochemical evidence that Treponema pallidum TroA is a zinc‐containing metalloprotein that lacks porin‐like structure. J Bacteriol 181:4420–4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Akins DR, Robinson E, Shevchenko D, Elkins C, Cox DL, Radolf JD (1997) Tromp1, a putative rare outer membrane protein, is anchored by an uncleaved signal sequence to the Treponema pallidum cytoplasmic membrane. J Bacteriol 197:5076–5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dalbey RE, Wickner W (1985) Leader peptidase catalyzes the release of exported proteins from the outer surface of the Escherichi coli plasma membrane. J Biol Chem 260:15925–15931. [PubMed] [Google Scholar]
- 22. Machius M, Brautigam CA, Tomchick DR, Ward P, Otwinowski Z, Blevins JS, Deka RK, Norgard MV (2007) Structural and biochemical basis for polyamine binding to the Tp0655 lipoprotein of Treponema pallidum: putative role for Tp0655 (TpPotD) as a polyamine receptor. J Mol Biol 373:681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 24. Söding J, Biegert A, Lupas AN (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33:W244–W248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vahedi‐Faridi A, Licht A, Bulut H, Scheffel F, Keller S, Wehmeier UF, Saenger W, Schneider E (2010) Crystal structures of the solute receptor GacH of Streptomyces glaucescens in complex with acarbose and an acarbose homolog: Comparison with the acarbose‐loaded maltose‐binding protein of Salmonella typhimurium. J Mol Biol 397:709–723. [DOI] [PubMed] [Google Scholar]
- 26. Krissinel E, Henrick K (2004) Secondary‐structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Cryst D60:2256–2268. [DOI] [PubMed] [Google Scholar]
- 27. Licht A, Bulut H, Scheffel F, Daumke O, Wehmeier UF, Saenger W, Schneider E, Vahedi‐Faridi A (2011) Crystal structures of the bacterial solute receptor AcbH displaying an exclusive substrate preference for β‐D‐galactopyranose. J Mol Biol 406:92–105. [DOI] [PubMed] [Google Scholar]
- 28. Davidson AL, Dassa E, Orelle C, Chen J (2008) Structure, function, and evolution of bacterial ATP‐Binding cassette systems. Microbiol Mol Biol Rev 72:317–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fukami‐Kobayashi K, Tateno Y, Nishikawa K (1999) Domain dislocation: a change of core structure in periplasmic binding proteins in their evolutionary history. J Mol Biol 286:279–290. [DOI] [PubMed] [Google Scholar]
- 30. Berntsson RP‐A, Smits SHJ, Schmitt L, Slotboom D‐J, Poolman B (2010) A structural classification of substrate‐binding proteins. FEBS Lett 584:2606–2617. [DOI] [PubMed] [Google Scholar]
- 31. Newcomer ME, Lewis BA, Quiocho FA (1981) The radius of gyration of L‐arabinose‐binding protein decreases upon binding of ligand. J Biol Chem 256:13218–13222. [PubMed] [Google Scholar]
- 32. Borrok MJ, Zhu Y, Forest KT, Kiessling LL (2009) Structure‐based design of a periplasmic binding protein antagonist that prevents domain closure. ACS Chem Biol 4:447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shilton BH, Flocco MM, Nilsson M, Mowbray SL (1996) Conformational changes of three periplasmic receptors for bacterial chemotaxis and transport: the maltose‐, glucose/galactose‐ and ribose‐binding proteins. J Mol Biol 264:350–363. [DOI] [PubMed] [Google Scholar]
- 34. Borrok MJ, Kiessling LL, Forest KT (2007) Conformational changes of glucose/galactose‐binding protein illuminated by open, unliganded, and ultra‐high‐resolution ligand‐bound structures. Protein Sci 16:1032–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pflugrath J, Quiocho F (1985) Sulphate sequestered in the sulphate‐binding protein of Salmonella typhimurium is bound solely by hydrogen bonds. Nature 314:257–260. [DOI] [PubMed] [Google Scholar]
- 36. Newcomer ME, Gilliland GL, Quiocho FA (1981) L‐arabinose‐binding protein‐sugar complex at 2.4 Å resolution. J Biol Chem 256:13213–13217. [PubMed] [Google Scholar]
- 37. Niesen FH, Berglund H, Vedadi M (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc 2:2212–2221. [DOI] [PubMed] [Google Scholar]
- 38. Quiocho FA (1986) Carbohydrate‐binding proteins: tertiary structures and protein‐sugar interactions. Annu Rev Biochem 55:287–315. [DOI] [PubMed] [Google Scholar]
- 39. Vyas NK (1991) Atomic features of protein‐carbohydrate interactions. Curr Opin Struct Biol 1:732–740. [Google Scholar]
- 40. Hazelbauer GL, Adler J (1971) Role of the galactose binding protein in chemotaxis of Escherichia coli toward galactose. Nat New Biol 230:101–104. [DOI] [PubMed] [Google Scholar]
- 41. Schumacher MA, Choi KY, Zalkin H, Brennan RG (1994) Crystal structure of LacI member, PurR, bound to DNA: minor groove binding by α helices. Science 266:763–770. [DOI] [PubMed] [Google Scholar]
- 42. Klock HE, Koesema EJ, Knuth MW, Lesley SA (2008) Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins Struct Funct Genet 71:982–994. [DOI] [PubMed] [Google Scholar]
- 43. Deka RK, Brautigam CA, Liu WZ, Tomchick DR, Norgard MV (2015) Molecular insights into the enzymatic diversity of flavin‐trafficking protein (Ftp; formerly ApbE) in flavoprotein biogenesis in the bacterial periplasm. Microbiologyopen 5:21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Deka RK, Brautigam CA, Goldberg M, Schuck P, Tomchick DR, Norgard MV (2012) Structural, bioinformatic, and in vivo analyses of two Treponema pallidum lipoproteins reveal a unique TRAP transporter. J Mol Biol 416:678–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A Protein identification and analysis tools on the ExPASy server In: Walker JM, Ed. (2005) The proteomics protocols handbook. Totowa, NJ: Humana Press, pp 571–607. [Google Scholar]
- 46. Otwinowski Z, Minor W (1997) Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. [DOI] [PubMed] [Google Scholar]
- 47. Minor W, Cymborowski M, Otwinowski Z, Chruszcz M (2006) HKL‐3000: the integration of data reduction and structure solution – from diffraction images to an initial model in minutes. Acta Cryst D62:859–866. [DOI] [PubMed] [Google Scholar]
- 48. Schneider TR, Sheldrick GM (2002) Substructure solution with SHELXD. Acta Cryst D58:1772–1779. [DOI] [PubMed] [Google Scholar]
- 49. Otwinowski Z (1991) Maximum likelihood refinement of heavy atom parameters. Proc CCP4 Daresbury Study Weekend:80–86.
- 50. Cowtan K, Main P (1998) Miscellaneous algorithms for density modification. Acta Cryst D54:487–493. [DOI] [PubMed] [Google Scholar]
- 51. Langer G, Cohen SX, Lamzin VS, Perrakis A (2008) Automated macromolecular model building for X‐ray crystallography using ARP/wARP version 7. Nat Protoc 3:1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung W, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure determination. Acta Cryst D66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Emsley P, Cowtan K (2004) Coot: Model‐building tools for molecular graphics. Acta Cryst D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 54. Schuck P (2000) Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J 78:1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schuck P, Perugini MA, Gonzales NR, Howlett GJ, Schubert D (2002) Size‐distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys J 82:1096–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schuck P, Demeler B (1999) Direct sedimentation analysis of interference optical data in analytical ultracentrifugation. Biophys J 76:2288–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Laue TM, Shah BD, Ridgeway RM, Pelletier SL Computer‐aided interpretation of analytical sedimentation data for proteins In: Harding SE, Rowe AJ, Horton JC, Ed. (1992) Analytical ultracentrifugation in biochemistry and polymer science. Cambridge, UK: The Royal Society of Chemistry, pp. 90–125. [Google Scholar]
- 58. Brown PH, Schuck P (2006) Macromolecular size‐and‐shape distributions by sedimentation velocity analytical ultracentrifugation. Biophys J 90:4651–4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Brautigam CA (2015) Calculations and publication‐quality illustrations for analytical ultracentrifugation data. Methods Enzymol 562:109–134. [DOI] [PubMed] [Google Scholar]