

Abstract
In this study, we report two high‐resolution structures of the pyridoxal 5′ phosphate (PLP)‐dependent enzyme kynurenine aminotransferase‐I (KAT‐I). One is the native structure with the cofactor in the PLP form bound to Lys247 with the highest resolution yet available for KAT‐I at 1.28 Å resolution, and the other with the general PLP‐dependent aminotransferase inhibitor, aminooxyacetate (AOAA) covalently bound to the cofactor at 1.54 Å. Only small conformational differences are observed in the vicinity of the aldimine (oxime) linkage with which the PLP forms the Schiff base with Lys247 in the 1.28 Å resolution native structure, in comparison to other native PLP‐bound structures. We also report the inhibition of KAT‐1 by AOAA and aminooxy‐phenylpropionic acid (AOPP), with IC50s of 13.1 and 5.7 μM, respectively. The crystal structure of the enzyme in complex with the inhibitor AOAA revealed that the cofactor is the PLP form with the external aldimine linkage. The location of this oxime with the PLP, which forms in place of the native internal aldimine linkage of PLP of the native KAT‐I, is away from the position of the native internal aldimine, with the free Lys247 substantially retaining the orientation of the native structure. Tyr101, at the active site, was observed in two conformations in both structures.
Keywords: aminotransferase, kynurenine, pyridoxyl phosphate, kynurenic acid, transferase
Short abstract
Introduction
Kynurenine aminotransferases (KATs) catalyze the synthesis of kynurenic acid (KYNA), an intermediate in the tryptophan catabolic pathway, by irreversible transamination of l‐kynurenine (KYN). Tryptophan metabolism occurs in most cells and is important in liver, leucocytes, heart, lungs, and astrocytes and microglia in the brain. Both indoleamine 2,3‐dioxygenases (IDO‐1 and IDO‐2)1, 2, 3 and tryptophan 2,3‐dioxygenase (TDO)4 are also key enzymes that initiate the pathway and degrade tryptophan into KYN.
KYNA is a broad spectrum antagonist of the three ionotropic excitatory amino acid receptors.5, 6 At low concentrations, KYNA preferentially blocks the glycine coagonist site of the N‐methyl‐d‐aspartate (NMDA) receptor (NMDA‐R) [IC50 ∼15 μM].7, 8 It has neuroprotective and anticonvulsant properties. It was discovered over a century ago, as a tryptophan metabolite,9 and a considerable gap occurred until it was also identified as a constituent of the mammalian brain.7, 10 The main arm of the tryptophan catabolism pathway produces 3‐hydroxykynurenine and then quinolinic acid. KYN can pass through the blood–brain‐barrier and 60% of the brain KYN comes from the periphery.11
Imbalance in the metabolites of tryptophan degradation has been implicated in a number of psychoses. The role of KYNA as an endogenous NMDA‐R antagonist supports the hypothesis of NMDA hypofunctioning in schizophrenia,12, 13, 14 where deficient glutaminergic neurotransmission has been linked to the disease, especially after the original observation of reduced glutamate levels in the cerebrospinal fluid (CSF) of schizophrenic patients.15 Glutaminergic hypofunction provided an improved explanation of neurochemical disturbances in schizophrenia based on a role for KYNA. KYNA is the only known endogenous antagonist of the NMDA subtype of a glutamate receptor in brain.16 High concentrations of KYNA in postmortem prefrontal cortex17 and in the CSF18 occur in people experiencing schizophrenia when compared with control samples. As KYNA is also a α7‐nicotinic acetylcholine receptor antagonist, this represents another potential mechanism by which psychotic symptoms could be induced.19
In the cerebrum it seems the synthesis of KYNA is catalyzed almost exclusively by two kynurenine aminotransferases (KAT‐I and KAT‐II). These enzymes, KAT‐I [glutamine transaminase K (GTK; EC 2.6.1.64)] and KAT‐II [α‐aminoadipate aminotransferase (α‐ADA; EC 2.6.1.7)], are distinguished by substrate specificity and other discrete biochemical and biophysical characteristics.20, 21, 22 The amino acid sequence identity between KAT‐I and KAT‐II is 11% for the two human enzymes. The KAT‐I structure has been previously reported at 2.0 Å resolution23 (PDB ID 1W7L) in which the homodimeric structure was reported with the fold‐type I of pyridoxal 5‐phosphate (PLP)‐dependent enzymes. In its PLP form, KAT‐I carries the PLP molecule covalently bound in the active site by a Schiff‐base linkage to the catalytic Lys247, and the substrate binding site of KAT‐I consists of a region of the aromatic residues Trp18, Tyr101, and Phe125 of one monomer, and Tyr63*, Phe278*, and His279* from the second monomer in which the substrate side chain inserts. The hydrophobicity at the binding site generally explains the efficiency of KAT‐I transamination of relatively hydrophobic amino acids such as leucine and tryptophan. The native human KAT‐II crystal structure has also been solved at 2.30 Å resolution24 (PDB ID 2VGZ), and at 1.83 Å resolution25 (PDB ID: 5EUN). The protein architecture of KAT‐II revealed that it, like KAT‐I, belongs to the fold‐type I PLP‐dependent enzymes, but it was considered a novel subclass because of the different folding at its 65 N‐terminal residues. The chain shift of residues 16–31 affects binding and catalysis in KAT‐II and is consistent with a shift in substrate profile. The aromaticity of the active site is more pronounced in KAT‐I than in KAT‐II and is also larger in KAT‐II in part due to not having an amino acid equivalent to Trp18. Two other KAT molecules have been reported in mammalian brain. These are KAT‐III/cysteine conjugate beta‐lyase 2 (EC 4.4.1.13) and KAT‐IV/glutamic‐oxaloacetic transaminase 2/mitochondrial aspartate aminotransferase2 (EC 2.6.1.1, EC 2.6.1.7).
Analyses of transgenic mice in which KAT‐II was knocked out were consistent with a relatively modest decrease in brain KYNA levels and normalisation of behavior and KYNA levels occurred as the mice approached maturity.26, 27, 28 From this, an expectation has emerged that KAT‐II, rather than KAT‐I, is the enzyme producing the majority of the kynurenic acid in the rodent brain.29 The differences suggest the possibility of important and different roles for each of the KATs.
The structures exist for KAT‐I in complex with several important inhibitors, such as indole‐3‐acetic acid at 1.55 Å resolution30 (PDB ID 3FVU) and with phenylalanine at 2.7 Å resolution,23(PDB ID 1W7M) and KAT‐II inhibitor complexes were subsequently more actively pursued and exist for BFF‐12231 (PDB ID 2XH1), PF‐04859989 and analogues derived from the scaffold32, 33 (PDB ID 3UE8, 4GE4, 4GE7, 4GE9, and 4GEB), and with alpha‐ketoglutarate alone34 (PDB ID 3DC1). Along with BF‐122, PF‐04859989, and analogues form covalent adducts to the PLP cofactor. Aminooxyacetic acid (AOAA) has long been known as an inhibitor of aminotransferases,35, 36 and inhibition by aminooxy‐phenylpropionic acid (AOPP) had been reported previously but specifically for tyrosine aminotransferase alone.37 We sought a description of the structure of AOAA inhibited KAT‐I, to determine the mode of action, and also to extend the understanding of the modes of inhibition of this enzyme.
Results
Inhibition assays
Enzyme inhibition assays have identified AOPP and AOAA as potent inhibitors of PLP‐dependent KAT‐I. In addition, the inhibition assays have included tryptophan and phenylalanine (Fig. 1) because inhibition of KAT‐I by these amino acids has been reported previously, and our assays for Phe (IC50 = 3.0 mM) and Trp (IC50 = 1.0 mM) are consistent with these reports.38 In contrast, inhibition by AOPP is more potent, with IC50 = 5.7 μM. The AOAA IC50 = 13.1 μM.
Figure 1.
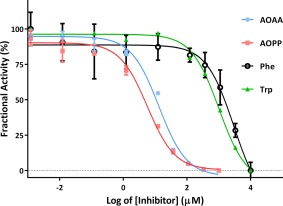
Inhibition assays for aminooxyacetic acid (AOAA), aminooxy‐phenylpropionic acid (AOPP), phenylalanine (Phe), and tryptophan (Trp).
Structural studies
The crystallographic experiments have provided two high‐resolution KAT‐I structures. One is of the native protein with the cofactor in the PLP form bound to Lys247, with the highest resolution yet available for KAT‐I at 1.28 Å resolution, and another with the general PLP‐dependent aminotransferase inhibitor, AOAA, covalently bound with the cofactor at 1.54 Å (Fig. 2). In the AOAA inhibited structure the native Schiff base covalent linkage to the cofactor is not observed and rather the linkage is an oxime covalent bond to the AOAA. The structure of the KAT‐I:AOPP complex could not be achieved. Although there were physical indications that the KAT‐I:AOPP complex existed in the crystals in which we had AOPP, based on an obvious change in color of the yellow native crystals to colorless, the measured potency of AOPP, and the observation of discontinuous electron density consistent with the presence of the free end of Lys247, the inhibitor could not be unambiguously placed within the active site.
Figure 2.
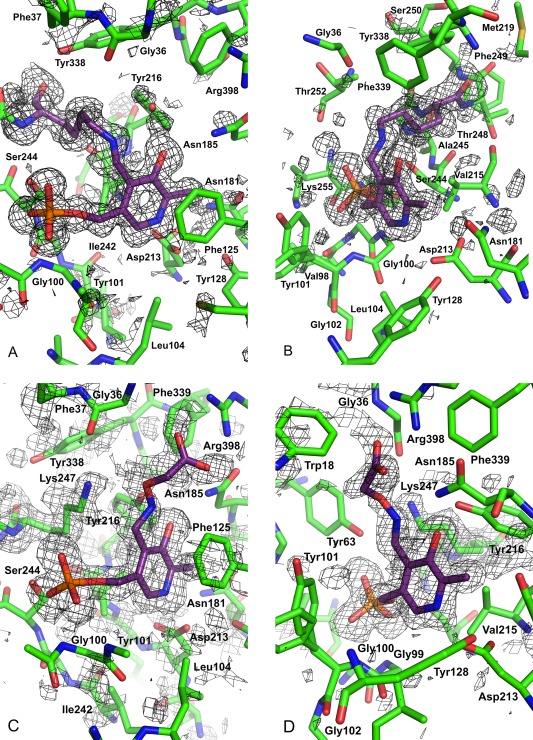
OMIT maps and structure in the vicinity of the cofactor and ligands. 2mFo – DFc OMIT maps are shown, and the structure has been clipped from the front. The maps are after refinement, in the native and inhibitor structures, in which the components omitted are, respectively, the cofactor and the inhibitor with the cofactor bound. In addition to these omissions from the refinements, Lys247 is omitted in both. The carbons of the cofactor and Lys247 in the native structure are colored mauve. (A) and (B) The native structure showing the contiguous PLP and Lys247 density with no inhibitor. The map is contoured at 1σ. In addition to the cofactor the carbons of Lys247 are colored mauve. (C) and (D) The aminooxyacetate bound structure in the vicinity of the ligand. The map is contoured at 1σ. In addition to the cofactor the carbons of the inhibitor are colored mauve. The views have been chosen to be approximately a 90° rotation and relatively similar, with each to best depict the structure in its locale. Figure was created with PyMOL.39
Both the structures exhibit the homodimeric structure with its two active sites located at the domain interface in each subunit, and at the subunit interface in the dimer. This commentary applies to both monomers. The interface of the monomers provides the active site. The cavity at the interface mainly derives from loops (Tyr63*, Phe125, Phe278*, His279*) and some amino acids that are close to the N‐termini of α‐helices (Trp18, Tyr101). Another important contribution to the stability of the KAT‐I dimer is provided by the first residues of the N‐termini. The KAT‐I active site was originally revealed by Rossi et al.,23 and descriptions in that work hold true in our high resolution structure, most fundamentally with the PLP bound to Lys247. The active site is formed of amino acid residues from both monomers, and conformations of both active sites were observed to be similar in both structures. The cofactor ring is part of a stack with the Phe125 aromatic ring, the nitrogen of the ring hydrogen bonded to a carboxylate Oδ of Asp213, and the phosphate group of the cofactor is engaged in interactions with residues contributing to the “PLP‐phosphate binding cup” as it was originally referred to in PLP‐dependent enzymes.40 Structures of the active site with corresponding electron density maps are presented (Fig. 2) as well as superposed (Fig. 3). The PLP and the Lys247 to which it is linked through the aldimine occupies the same general region as observed in other structures, although small conformational differences were observed in the vicinity of the aldimine linkage itself.
Figure 3.
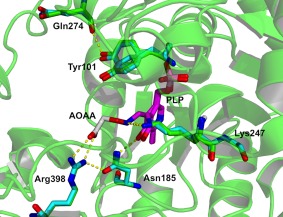
View of the active site and interaction of the AOAA with hKAT‐1 superimposed on the native structure. Carbon atoms are colored according to their structural origins: on the PLP colored mauve, native structure blue and AOAA inhibited structure green. Figure was created with PyMOL.39
In the AOAA‐bound structure the cofactor is the PLP form linked by the external aldimine where the Lys247 is no longer bonded, and essentially the Lys247 keeps the conformation of the native protein, except for the Nζ, which is a viable location as the aldimine bond securing the AOAA is impervious to the actions of Lys247, and the oxime linkage is swivelled away to the other side of the face of the pyridine ring. The only significant overall change to the AOAA‐bound structure of KAT‐I is at the Val19‐Glu26 α‐helix and the Trp18 preceding it, in which the main chain is approximately 2.0 Å further away from the active site in the AOAA‐bound structure. Of the residues with significant overall changes it is the Trp18 side chain indole that is closest to the AOAA and the active site and may be responsible for promulgating the changes.
In the native structure, hydrogen bonded solvent is displaced from the location that the oxygens of the carboxylate of the AOAA occupy. In the native structure, the carboxylates of the AOAA form salt bridges to both guanidine nitrogens of Arg398. The network of hydrogen bonds here is more extensive, with one of the carboxylate oxygens of the AOAA in a hydrogen bond to Nδ 2 of Asn185, which is also hydrogen bonded to the hydroxyl of the cofactor. The network is completed by a hydrogen bond of the Oδ 1 of Asn185 back to the adjacent guanidine nitrogen of Arg398.
Tyr101 is quite near the active site, and has the side chain closest to the site at which the aldimine bond is formed with the cofactor. It was observed similarly in both native and AOAA‐bound structures with two conformations, and is the only observation of alternate positions in these structures. The Tyr101 conformations are separated by 0.8 Å at the Oη in a comparison of these two structures, which may be consistent with the presence of the small inhibitor AOAA. In a comparison of the alternate conformations of Tyr101 in both the native and the AOAA‐bound structure, the centroid of the aromatic ring of Tyr101 and the Oη is approximately 1.5 Å and 3.1 Å distant, respectively. An extensive Tyr101 shift was first observed in the KAT‐I‐indole acetic acid complex structure,30 (PBD ID 3FVU) representing a situation in which the indole acetic acid was considered sizeable to induce the Tyr101 Oη shift of 3.7 Å, moving the Tyr101 phenol away from the cofactor, and also with the Oη then achieving a hydrogen bond to O(Gln274). Although the two similar conformations of Tyr101 are observed in the vicinity of the inhibitor binding location, there is also evidence for hydrogen bonding disruption at Tyr101 on the attachment of the AOAA inhibitor, due to its proximity to the external aldimine bond. Additionally, the region occupied cannot support a hydrogen bond network of two solvent molecules and Oη (Tyr101), as observed in the native structure.
AOAA inhibited PLP‐dependant aminotransferase structures have been reported for five other proteins: chicken aspartate aminotransferase (open and closed conformations)41 (PDB IDs 1OXO and 1OXP), E. coli gamma aminobutyrate aminotransferase42 (PDB ID 1SFF), bacterial (Mesorhizobium sp.)(Mes), S‐selective aminotransferase43 (PDB ID 2YKV), and a novel Variovorax paradoxus (Vp) β‐phenylalanine aminotransferase44 (PDB ID 4AOA). The hydrogen bonding of the carboxylate of the AOAA to an arginine is a common theme in these structures except for the case of the aspartate aminotransferase. Hydrogen bonding with the O of the oxime linkage of the Schiff base lysine is also observed in most cases, but while the Nζ is close by, no such hydrogen bond is observed in the case of KAT‐I. For the gamma aminobutyrate aminotransferase structure the displacement of the Nζ from the O of the oxime is observed to be more distant again than that observed for KAT‐I.
Discussion
This work has reported the inhibition behavior of KAT‐1 in which AOAA and AOPP were studied, with the AOPP suggested from the addition of the phenyl ring to AOAA as a rational bioisosteric modification and a hybrid derived using structural features of AOAA and l‐Phe. The IC50 of AOPP is 5.7 μM, which is half that of AOAA (13.1 μM). We also performed our inhibition assay to determine the IC50 of Phe and Trp (1.0 mM and 3.0 mM, respectively) as comparators that have been reported previously. AOAA is a nonselective inhibitor of PLP‐dependent aminotransferases, as both AOAA and AOPP contain the O‐alkylhydroxylamine moiety that irreversibly forms an oxime with PLP present in these enzymes as a co‐factor covalently bound to the catalytic lysine by a Schiff‐base linkage. This bonding prevents the regeneration of enzyme‐bound PLP and thus ablates the enzyme activity. AOPP, however, represents an inhibitor which is more potent than AOAA, indicating a synergistic effect resulting from the addition of the phenyl ring to the AOAA. The involvement of the hydrogen bonding of the carboxylate of the AOAA to Arg398 is observed, but no hydrogen bonding is observed at the oxime linkage. The observations of the Tyr101 conformations suggest that it is sensitive to inhibitor binding. The most systematic effect on the protein on binding AOAA is a movement of the main chain of approximately 2 Å at Trp18 and the first residues of the helix beyond.
Inhibitor studies have previously been performed on tryptophan and indole‐3‐pyruvic acid, along with four similar indole derivatives selected because the α‐amino and α‐keto groups were absent.30 In that study, indole‐3‐propionic acid (IC50 = 0.14 mM) and DL‐indole‐3‐lactic acid (IC50 = 0.22 mM) were the more potent of the six compounds studied and provided potential lead candidate inhibitors. The crystal structure of KAT‐I bound to DL‐indole‐3‐acetic acid shows that the compound inserts into the hydrophobic pocket with its carboxylic moiety forming hydrogen bonds with Arg398,30 with both of the oxygens satisfying hydrogen bonds with the guanidinium hydrogens, and also analogous to the binding of the carboxylate of phenylalanine23 (PDB ID 1W7M). More recent work has shown other compounds of series of indole and phenylhydrazone to be approximately sixfold–sevenfold more potent KAT‐I inhibitors.45 The most potent indole based molecule was provided by a bromo‐substitution with an IC50 of 271 μM, while the 3‐ and 4‐chlorophenylhdrazone hexanoic acid derivatives were significantly more potent (both with IC50 = 20 μM).
This work has demonstrated an approach toward the design of improved irreversible inhibitors for KAT‐I, and the anticipated covalent bonding created by the inhibitor was observed in the inhibitor‐enzyme crystal structure. Although the high resolution crystal structures reported here are generally consistent with the other AOAA‐bound structures of PLP‐dependent aminotransferases, this work supports the notion of the possibility of the design of further more potent and specific inhibitors of KAT‐I using the O‐alkylhydroxylamine moiety with substituents on the phenyl ring of AOPP, or with other substitutions.
Materials and Methods
Macromolecule production
The KAT‐I ORF, cloned into the transfer vector pBlueBac4.5 (pBlueBac4.5–utKAT‐I), was kindly provided by the Jianyong Li group at the University of Illinois at Urbana‐Champaign, Urbana, IL. Recombinant baculovirus containing the sequence‐verified KAT‐I ORF was generated and purified by plaque assay using the Bac‐N‐Blue transfection kit (Invitrogen; Carlsbad, CA) according to the manufacturer's instructions. A number of blue putative recombinant plaques were propagated and high titer baculovirus stocks from individual viral strains with the largest potency were selected for protein expression in Spodoptera frugiperda (Sf9) insect cells (Invitrogen; Carlsbad, CA) according to manufacturer's instructions.
Large‐scale protein expression was carried out in Sf9 cells grown in SF900II serum free media as described previously.46 Suspension cultures used 250 mL culture per 1 L Schott laboratory bottles (Aunet Pty Ltd, Perth, WA) and shaking at 140 rpm at 27°C. High titer KAT‐I baculovirus stock was added to cultures when cell density reached 2 × 106 cells/mL at an approximate multiplicity of infection of two viral particles per cell. Infected cells were harvested 96 h postinfection by centrifugation at 800 rcf for 15 min at 4°C and stored at −80°C until required.
Protein purification
Harvested Sf9 cell pellets containing intracellularly expressed KAT‐I were resuspended in Buffer A (1 mM phenylmethylsulfonyl fluoride, 10 mM sodium phosphate, pH 7.0; 100 μL per 2 × 106 cells). The cells were lysed by sonication for 5 min on an ice/water slurry. The cell debris was removed by centrifugation at 18000 rcf for 20 min at 4°C and the resulting clarified total cellular lysate was precipitated with 35% ammonium sulphate to retain the supernatant fraction, and again at 60% ammonium sulphate to retain the precipitated protein fraction. The precipitated protein, containing KAT‐I, was resuspended in Buffer L (10% AS, 10 mM sodium phosphate, pH 7.0) and applied onto 4 × 5 mL Phenyl Sepharose columns using Buffer L at 1 mL/min. KAT‐I was eluted from the media by a single step gradient at 0% AS with Buffer A. The eluted KAT‐I fraction was dialyzed against 4 L Buffer A and the resulting dialysate was loaded onto 3 × 1 mL DEAE Sepharose FF columns using Buffer A at 1 mL min−1. The protein‐bound media were washed at 50 mM, 75 mM, 125 mM, 150 mM, 175 mM, 200 mM, and 225 mM NaCl with Buffer C (500 mM NaCl, 10 mM sodium phosphate, pH 7.0) at 4 mL/min. A distinct KAT‐I elution peak was observed at 125 mM NaCl. The KAT‐I enzymatically active fractions were desalted in Buffer A and loaded onto a 5 mL Blue Sepharose FF column using Buffer A at 1 mL/min. The KAT‐I enzymatically active fraction was present only in the flow through. The resulting Blue Sepharose flow through fraction containing KAT‐I was reapplied onto 3 × 1 mL DEAE Sepharose FF columns using Buffer A at 1 mL min−1. The protein‐bound columns were washed at 75 mM, 125 mM, and 500 mM NaCl with Buffer C.
Enzymatically active KAT‐I that was present only in the 125 mM NaCl fraction was concentrated to 1.8 mL and the resulting sample was loaded on a Superdex200 prep grade column pre‐equilibrated with Buffer K (100 mM NaCl in 10 mM Tris, pH 7.6). The sample was passed through the column isocratically using Buffer K and KAT‐I enzyme activity was confirmed in the retention peak corresponding to the enzyme dimer. The fractions within this peak were pooled and was stored at 4°C, protected from light for subsequent enzymology, biochemical characterization assays and crystallization studies.
Protein purification was monitored and controlled using an ÄKTA PrimePlus or ÄKTA Purifier system (GE Amersham). Pre‐packed columns containing up to 5 mL chromatographic media were used for every 20 mL sample up to a maximum of 4–5 columns to avoid system back‐pressure increasing above 0.3 MPa. The elution of KAT‐I fractions during the purification steps was identified by KAT‐I enzyme assay.
KAT‐I enzyme inhibition activity assay
All chemicals were purchased from Sigma (St Louis, MO) unless otherwise stated. AOPP was from Wako (Osaka, Japan). Methodologies for the determination of KAT‐I enzyme activity, and inhibition analysis by HPLC‐UV was based on our previous report,45 and was originally described by Okuno et al. and Li and Li.47, 48 A typical reaction mixture of 30 µL containing 15 µL of substrate mixture (5 mM KYN, 2 mM α‐ketobutyrate, 40 mM PLP), 2 µg purified KAT‐1 in 5 µL solution and 10 µL of inhibitor at different concentrations was prepared in 200 mM phosphate buffer at pH 8. Each inhibitor was incorporated into the reaction mixture at final concentrations of 0.0004, 0.004, 0.037, 0.37, 3.7, 37, 111, 333, 1000, and 3000 µM.
KAT‐1 was pre‐incubated with each inhibitor (or buffer) for 10 minutes at room temperature before adding the substrate mixture. The mixture was incubated at 45°C for 10 minutes and the reaction was stopped by adding 30 µL of 1 M trichloroacetic acid. The resulting mixture was centrifuged at 15,000 rcf at 4°C for 5 min, and the supernatant used as the analyte on the HPLC. A positive control reaction (inhibitor substituted with buffer) and negative control reaction (KAT‐1 solution and inhibitor substituted with buffer) were simultaneously performed with all assays. All reactions were performed in triplicates.
HPLC detection of KYNA
KYN and KYNA were measured using reverse‐phase CLASS‐VP (Shimadzu) at 330 nm. The sample was isocratically passed through a 5 mm C18 column (150 mm × 2 mm; PerkinElmer Life and Analytical Sciences) using 50 mM sodium acetate and 2.5% (v/v) acetonitrile, pH 6.2. Samples were injected in a volume of 20 µL at a flow rate of 0.4 mL min−1 for 10 min. The amount of KYNA observed in each reaction was deduced from standards using authentic reagents. The reduction in observed activities (as determined by the amount of KYNA produced) were expressed as fractional activities and plotted against the corresponding inhibitor concentrations. The software GraphPad Prism (v5.04 for Windows; GraphPad Software, San Diego, CA) was used for graphing, statistical and regression analysis.
Crystallization
Purified KAT‐I sample were buffer‐exchanged into 10 mM potassium phosphate, pH 6.8 and concentrated to 10 mg/mL. The optimized crystallization conditions yielding diffraction grade KAT‐I crystals were obtained using hanging drop vapor diffusion technique at 20°C. Four 1 μL drops with 0.5 μL protein and 0.5 μL reservoir (1:1 ratio) were suspended from thin siliconized coverslips over a reservoir of 500 μL. The reservoir solution contained 200 mM sodium acetate, 29% PEG4000 in 100 mM Tris, pH 7.3. Crystals were obtained within 2–5 days.
Data collection, processing, structure solution, and refinement
The experiments were performed on the MX1 beamline of the Australian Synchrotron, using the ADSC quantum 210r area detector. The wavelength used was 0.9537 Å and crystals cooled to 100 K. The cell data were refined with MOSFLM.49 The programs MOSFLM and SCALA 49, 50 were used for data reduction. A lower than desirable completeness does not arise from low intensity, but rather losses due to ice rings as data in the highest resolution bin was well represented, and diffraction beyond the reported resolution was clear.
Initial phases for the KAT‐I crystals were solved by molecular replacement using the software PHASER51 from the CCP4 suite.52 The previously published 3FVX structure30 (1.5 Å resolution) was used as the search model to initially solve the crystal structure of the KAT‐I (PDB ID 4WLH; 1.28 Å). Subsequent structure determination was performed using this higher resolution structure as the search model for the aminooxyacetate (AOAA)‐bound KAT‐I complex (PDB ID 4WLJ). Iterative cycles of crystallographic refinements were performed with the software PHENIX53 alternated with manual model rebuilding using the program COOT.54 Solvent molecules were subsequently added in PHENIX and the validity of the water positions were manually checked for valid interactions to account for hydrogen bonding with protein or other solvent atoms. These procedures converged to an acceptable R‐factor and free R‐factor values, with ideal geometry. The model was refined using isotropic temperature factors, data collection, and refinement statistics are presented in Table 1.
Table 1.
Summary of X‐Ray Data Collection, Processing, and Refinement Statistics
| KAT‐I (PLP‐form) | KAT‐I in complex with aminooxyacetate | |
|---|---|---|
| Wavelength (Å) | 0.9537 | 0.9537 |
| Oscillation range (°) | 0.5 | 0.5 |
| Number of images | 380 | 720 |
| Detector distance (mm) | 110 | 150 |
| Space group | C2 | C2 |
| a, b, c (Å) | 102.4, 107.6, 81.5 | 102.7, 107.7, 81.8 |
| α, β, γ (°) | 90, 112.6, 90 | 90, 113.0, 90 |
| Resolution range (Å) | 20.1–1.28 (1.30–1.28)a | 26.9–1.54 (1.58–1.54) |
| Total No. of reflections | 805418 (114267) | 683702 (84844) |
| No. of unique reflections | 208782 (30461) | 108232 (16441) |
| Completeness (%) | 100.0 (100.0) | 89.1 (93.9) |
| Redundancy | 3.9 (3.8) | 6.3 (5.2) |
| 〈 I/σ(I)〉 | 11.4 (2.0) | 13.2 (2.2) |
| R merge | 0.039 (0.40) | 0.035 (0.36) |
| R meas | 0.055 (0.57) | 0.055 (0.50) |
| R p.i.m. | 0.039 (0.40) | 0.035 (0.36) |
| CC1/2 | 0.998 (0.676) | 0.999 (0.699) |
| Overall B factor from Wilson plot (Å2) | 9.79 | 14.48 |
| σ cutoff | F > 1.34σ(F) | F > 1.34σ(F) |
| No. of reflections, working set | 198278 (6575) | 102037 (3332) |
| No. of reflections, test set | 10493 (347) | 5348 (206) |
| Final R cryst | 0.172 (0.259) | 0.197 (0.248) |
| Final R free | 0.187 (0.274) | 0.227 (0.297) |
| No. of non‐H atoms | ||
| Protein | 6601 | 6618 |
| Ligand | 0 | 42 |
| Solvent | 631 | 392 |
| Total | 7313 | 7076 |
| R.m.s. deviations | ||
| Bonds (Å) | 0.009 | 0.011 |
| Angles (°) | 1.347 | 1.361 |
| Average B factors (Å2) | ||
| Protein | 12.7 | 15.7 |
| Ligand | 17.5 | |
| Water | 21.1 | 21.3 |
| Ramachandran plotb | ||
| Favored (%) | 98.1 | 97.2 |
| Allowed (%) | 100.0 | 100.0 |
| PDB code | 4WLH | 4WLJ |
Values for the outer shell are given in parentheses.
Ramachandran analysis was performed using MolProbity.55
Exploratory refinements of alternate side chains for which there was evidence were performed by refining their temperature factors and occupancies. Alternate sidechain conformations were not kept in the final model if in the final refinements they had any atoms with occupancies < 0.2. We ensured the double bond character of the aldimine linkage was modeled in the native structure in the bond from the Nζ of Lys247 to the C4' of PLP.
Acknowledgments
The authors are grateful to Alaina Ammit, Lenka Munoz, Nicholas West, Cy Jeffries, Gayan Jayawickrama, Danny Hua and Christine Gee for their discussions and support, to Tim Werner for assistance with data collection, and to Reza Nematollahi for his reading of the manuscript. Thank you to Ali Achaechi for assistance with the figures. The authors wish to thank members of their respective research groups for a supportive and collegial environment. The Australian Synchrotron Research Fund supported the crystallographic data collection at beamline MX1. Accession Numbers: The atomic coordinates and structure factors corresponding to the structures determined in this study were deposited on 2014‐10‐07 and released on 2014‐12‐03 and are accessible at the Protein Data Bank (http://www.rcsb.org/pdb) under the accession codes 4WLH (KAT‐I) and 4WLJ (KAT‐I with aminooxyacetate). The Authors Declare No Conflict of Interest. Competing interests: The authors have declared that no competing interests exist. Authors contributions: Performed the experiments: NAN, JHP, FNA, WBC. Designed the experiments: NAN, NKS, VK, MDG, WBC. Analyzed the data: NAN, NKS, CAC, WBC. Wrote the article: NAN, MDG, WBC.
References
- 1. Heyes MP, Saito K, Major EO, Milstien S, Markey SP, Vickers JH (1993) A mechanism of quinolinic acid formation by brain in inflammatory neurological disease. Attenuation of synthesis from L‐tryptophan by 6‐chlorotryptophan and 4‐chloro‐3‐hydroxyanthranilate. Brain 116:1425–1450. [DOI] [PubMed] [Google Scholar]
- 2. Mellor AL, Munn DH (1999) Tryptophan catabolism and T‐cell tolerance: immunosuppression by starvation? Immunol Today 20:469–473. [DOI] [PubMed] [Google Scholar]
- 3. Ball HJ, Sanchez‐Perez A, Weiser S, Austin CJ, Astelbauer F, Miu J, McQuillan JA, Stocker R, Jermiin LS, Hunt NH (2007) Characterization of an indoleamine 2,3‐dioxygenase‐like protein found in humans and mice. Gene 396:203–213. [DOI] [PubMed] [Google Scholar]
- 4. Knox WE, Mehler AH (1950) The conversion of tryptophan to kynurenine in liver. I. The coupled tryptophan peroxidase‐oxidase system forming formylkynurenine. J Biol Chem 187:419–430. [PubMed] [Google Scholar]
- 5. Perkins MN, Stone TW (1982) An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res 247:184–187. [DOI] [PubMed] [Google Scholar]
- 6. Ganong AH, Lanthorn TH, Cotman CW (1983) Kynurenic acid inhibits synaptic and acidic amino acid‐induced responses in the rat hippocampus and spinal cord. Brain Res 273:170–174. [DOI] [PubMed] [Google Scholar]
- 7. Moroni F, Russi P, Lombardi Beni M, Carla V (1988) Presence of kynurenic acid in the mammalian brain. J Neurochem 51:177–180. [DOI] [PubMed] [Google Scholar]
- 8. Kessler M, Terramani T, Lynch G, Baudry M (1989) A glycine site associated with N‐methyl‐D‐aspartic acid receptors: characterization and identification of a new class of antagonists. J Neurochem 52:1319–1328. [DOI] [PubMed] [Google Scholar]
- 9. Ellinger A (1904) Die entstehung der Kynurensaure. Z Physiol Chem 43:325–337. [Google Scholar]
- 10. Turski W, Nakamura M, Todd W, Carpenter B, Whetsell WJ, Schwarcz R (1988) Identification and quantification of kynurenic acid in human brain tissue. Brain Res 454:164–169. [DOI] [PubMed] [Google Scholar]
- 11. Gal EM, Sherman AD (1980) L‐kynurenine: its synthesis and possible regulatory function in brain. Neurochem Res 5:223–239. [DOI] [PubMed] [Google Scholar]
- 12. Olney JW, Farber NB (1995) NMDA antagonists as neurotherapeutic drugs, psychotogens, neurotoxins, and research tools for studying schizophrenia. Neuropsychopharmacology 13:335–345. [DOI] [PubMed] [Google Scholar]
- 13. Olney JW, Newcomer JW, Farber NB (1999) NMDA receptor hypofunction model of schizophrenia. J Psychiatric Res 33:523–533. [DOI] [PubMed] [Google Scholar]
- 14. Muller N, Schwarz M (2006) Schizophrenia as an inflammation‐mediated dysbalance of glutamatergic neurotransmission. Neurotoxicity Res 10:131–148. [DOI] [PubMed] [Google Scholar]
- 15. Gattaz WF, Gattaz D, Beckmann H (1982) Glutamate in schizophrenics and healthy controls. Arch Psychiatr Nervenkr 231:221–225. [DOI] [PubMed] [Google Scholar]
- 16. Stone TW (1993) Neuropharmacology of quinolinic and kynurenic acids. Pharmacol Rev 45:309–379. [PubMed] [Google Scholar]
- 17. Schwarcz R, Rassoulpour A, Wu H‐Q, Medoff D, Tamminga CA, Roberts RC (2001) Increased cortical kynurenate content in schizophrenia. Biol Psychiatry 50:521–530. [DOI] [PubMed] [Google Scholar]
- 18. Erhardt S, Blennow K, Nordin C, Skogh E, Lindstrom L, Engberg G (2001) Kynurenic acid levels are elevated in the cerebrospinal fluid of patients with schizophrenia. Neurosci Lett 313:96–98. [DOI] [PubMed] [Google Scholar]
- 19. Schwieler L, Erhardt S (2003) Inhibitory action of clozapine on rat ventral tegmental area dopamine neurons following increased levels of endogenous kynurenic acid. Neuropsychopharmacology 28:1770–1777. [DOI] [PubMed] [Google Scholar]
- 20. Cooper AJ, Gross M (1977) The glutamine transaminase‐omega‐amidase system in rat and human brain. J Neurochem 28:771–778. [DOI] [PubMed] [Google Scholar]
- 21. Buchli R, Alberati‐Giani D, Malherbe P, Kohler C, Broger C, Cesura AM (1995) Cloning and functional expression of a soluble form of kynurenine/alpha‐aminoadipate aminotransferase from rat kidney. J Biol Chem 270:29330–29335. [DOI] [PubMed] [Google Scholar]
- 22. Cooper A (2004) The role of glutamine transaminase K (GTK) in sulfur and alpha‐keto acid metabolism in the brain, and in the possible bioactivation of neurotoxicants. Neurochem Int 44:557–577. [DOI] [PubMed] [Google Scholar]
- 23. Rossi F, Han Q, Li J, Li J, Rizzi M (2004) Crystal structure of human kynurenine aminotransferase I. J Biol Chem 279:50214–50220. [DOI] [PubMed] [Google Scholar]
- 24. Rossi F, Garavaglia S, Montalbano V, Walsh MA, Rizzi M (2008) Crystal structure of human kynurenine aminotransferase II, a drug target for the treatment of Schizophrenia. J Biol Chem 283:3559–3566. [DOI] [PubMed] [Google Scholar]
- 25. Nematollahi A, Sun G, Harrop SJ, Hanrahan JR, Church WB (2016) Structure of the PLP‐form of the human kynurenine aminotransferase II in a novel spacegroup at 1.83 A resolution. Int J Mol Sci 17:446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alkondon M, Pereira E, Yu P, Arruda E, Almeida L, Guidettu P, Fawcett W, Sapko M, Randall W, Schwarcz R, Tangle DA (2004) Targetted deletion of the kynurenine aminotransferase II gene reveals a critical role of endogenous kynurenic acid in the regulation of synaptic transmission via a7 nicotinic receptors in the hippocampus. J Neurosci 24:4635–4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yu P, Di Prospero NA, Sapko MT, Cai T, Chen A, Melendez‐Ferro M, Du F, Whetsell WO, Guidetti P, Schwarcz R, Tagle DA (2004) Biochemical and phenotypic abnormalities in kynurenine aminotransferase II‐deficient mice. Mol Cell Biol 24:6919–6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sapko MT, Guidetti P, Yu P, Tagle DA, Pellicciari R, Schwarcz R (2006) Endogenous kynurenate controls the vulnerability of striatal neurons to quinolinate: implications for Huntington's disease. Exp Neurol 197:31–40. [DOI] [PubMed] [Google Scholar]
- 29. Guidetti P, Okuno E, Schwarcz R (1997) Characterization of rat brain kynurenine aminotransferases I and II. J Neurosci Res 50:457–465. [DOI] [PubMed] [Google Scholar]
- 30. Han Q, Robinson H, Cai T, Tagle DA, Li J (2009) Structural insight into the inhibition of human kynurenine aminotransferase I/glutamine transaminase K. J Med Chem 52:2786–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rossi F, Valentina C, Garavaglia S, Sathyasaikumar KV, Schwarcz R, Kojima S, Okuwaki K, Ono S, Kajii Y, Rizzi M (2010) Crystal structure‐based selective targeting of the pyridoxal 5'‐phosphate dependent enzyme kynurenine aminotransferase II for cognitive enhancement. J Med Chem 53:5684–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dounay AB, Anderson M, Bechle BM, Campbell BM, Claffey MM, Evdokimov A, Evrard E, Fonseca KR, Gan X, Ghosh S, Hayward MM, Horner W, Kim J‐Y, McAllister LA, Pandit J, Paradis V, Parikh VD, Reese MR, Rong S, Salafia MA, Schuyten K, Strick CA, Tuttle JB, Valentine J, Wang H, Zawadzke LE, Verhoest PR (2012) Discovery of brain‐penetrant, rrreversible kynurenine aminotransferase II inhibitors for Schizophrenia. ACS Med Chem Lett 3:187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tuttle JB, Anderson M, Bechle BM, Campbell BM, Chang C, Dounay AB, Evrard E, Fonseca KR, Gan X, Ghosh S, Horner W, James LC, Kim JY, McAllister LA, Pandit J, Parikh VD, Rago BJ, Salafia MA, Strick CA, Zawadzke LE, Verhoest PR (2013) Structure‐based design of irreversible human KAT II inhibitors: discovery of new potency‐enhancing interactions. ACS Med Chem Lett 4:37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Han Q, Cai T, Tagle DA, Robinson H, Li J (2008) Substrate specificity and structure of human aminoadipate aminotransferase/kynurenine aminotransferase II. Biosci Rep 28:205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rej R (1977) Aminooxyacetate is not an adequate differential inhibitor of aspartate aminotransferase isoenzymes. Clinical Chem 23:1508–1509. [PubMed] [Google Scholar]
- 36. John RA, Charteris A, Fowler LJ (1978) The reaction of amino‐oxyacetate with pyridoxal phosphate‐dependent enzymes. Biochem J 171:771–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. De‐Eknamkul W, Ellis BE (1987) Purification and characterization of tyrosine aminotransferase activities from Anchusa officinalis cell cultures. Archiv Biochem Biophys 257:430–438. [DOI] [PubMed] [Google Scholar]
- 38. Baran H, Okuno E, Kido R, Schwarcz R (1994) Purification and characterization of kynurenine aminotransferase I from human brain. J Neurochem 62:730–738. [DOI] [PubMed] [Google Scholar]
- 39.The PyMOL Molecular Graphics System, Version 1.7.6.2 Schrödinger, LLC.
- 40. Fukumoto Y, Tanase S, Nagashima F, Ueda S, Ikegami K, Morino Y (1991) Structural and functional role of the amino‐terminal region of porcine cytosolic aspartate aminotransferase. Catalytic and structural properties of enzyme derivatives truncated on the amino‐terminal side. J Biol Chem 266:4187–4193. [PubMed] [Google Scholar]
- 41. Markovic‐Housley Z, Schirmer T, Hohenester E, Khomutov AR, Khomutov RM, Karpeisky MY, Sandmeier E, Christen P, Jansonius JN (1996) Crystal structures and solution studies of oxime adducts of mitochondrial aspartate aminotransferase. Eur J Biochem 236:1025–1032. [DOI] [PubMed] [Google Scholar]
- 42. Liu W, Peterson PE, Carter RJ, Zhou X, Langston JA, Fisher AJ, Toney MD (2004) Crystal structures of unbound and aminooxyacetate‐bound Escherichia coli gamma‐aminobutyrate aminotransferase. Biochemistry 43:10896–10905. [DOI] [PubMed] [Google Scholar]
- 43. Wybenga GG, Crismaru CG, Janssen DB, Dijkstra BW (2012) Structural determinants of the beta‐selectivity of a bacterial aminotransferase. J Biol Chem 287:28495–28502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Crismaru CG, Wybenga GG, Szymanski W, Wijma HJ, Wu B, Bartsch S, de Wildeman S, Poelarends GJ, Feringa BL, Dijkstra BW, Janssen DB (2013) Biochemical properties and crystal structure of a beta‐phenylalanine aminotransferase from Variovorax paradoxus. Appl Environ Microbiol 79:185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Akladios FN, Nadvi NA, Park J, Hanrahan JR, Kapoor V, Gorrell MD, Church WB (2012) Design and synthesis of novel inhibitors of human kynurenine aminotransferase‐I. Bioorg Med Chem Lett 22:1579–1581. [DOI] [PubMed] [Google Scholar]
- 46. Park J, Knott HM, Nadvi NA, Collyer CA, Wang XM, Church WB, Gorrell MD (2008) Reversible inactivation of human didpepidyl peptidases. Open Enzyme Inhibition J 1:52–60. [Google Scholar]
- 47. Okuno E, Du F, Ishikawa T, Tsujimoto M, Nakamura M, Schwarcz R, Kido R (1990) Purification and characterization of kynurenine‐pyruvate aminotransferase from rat kidney and brain. Brain Res 534:37–44. [DOI] [PubMed] [Google Scholar]
- 48. Li J, Li G (1997) Transamination of 3‐hydroxykynurenine to produce xanthurenic acid: a major branch pathway of tryptophan metabolism in the mosquito, Aedes aegypti, during larval development. Insect Biochem Mol Biol 27:859–867. [DOI] [PubMed] [Google Scholar]
- 49. Leslie AGW, Powell HR (2007) Processing Diffraction Data with Mosflm. In: Read R.J., Sussman J.L. (eds). Evolving methods for macromolecular crystallography. NATO Science Series, vol 245. Springer, Dordrecht.
- 50. Kabsch W (1988) Evaluation of single‐crystal X‐ray diffraction data from a position‐sensitive detector. J Appl Cryst 21:916–924. [Google Scholar]
- 51. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Cryst 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Cryst D 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L‐W, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Cryst D 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Cryst D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 55. Lovell SC, Davis IW, Arendall WB, 3rd de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC (2003) Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins 50:437–450. [DOI] [PubMed] [Google Scholar]