

Abstract
Myosin activation is a viable approach to treat systolic heart failure. We previously demonstrated that striated muscle myosin is a promiscuous ATPase that can use most nucleoside triphosphates as energy substrates for contraction. When 2‐deoxy ATP (dATP) is used, it acts as a myosin activator, enhancing cross‐bridge binding and cycling. In vivo, we have demonstrated that elevated dATP levels increase basal cardiac function and rescues function of infarcted rodent and pig hearts. Here we investigate the molecular mechanism underlying this physiological effect. We show with molecular dynamics simulations that the binding of dADP.Pi (dATP hydrolysis products) to myosin alters the structure and dynamics of the nucleotide binding pocket, myosin cleft conformation, and actin binding sites, which collectively yield a myosin conformation that we predict favors weak, electrostatic binding to actin. In vitro motility assays at high ionic strength were conducted to test this prediction and we found that dATP increased motility. These results highlight alterations to myosin that enhance cross‐bridge formation and reveal a potential mechanism that may underlie dATP‐induced improvements in cardiac function.
Keywords: pre‐powerstroke, allosteric modification, acto‐myosin interaction, molecular dynamics simulations
Introduction
Myosin is a motor protein that normally uses the energy of ATP hydrolysis to produce force and motion. However, myosin is a promiscuous enzyme that can use a variety of nucleotides as substrates, although most are not as effective as ATP.1, 2 2 deoxy‐ATP (dATP), a nucleotide identical to ATP with the exception of an oxygen missing from the 2‐position on the ribose ring, is normally produced in cells for DNA synthesis and repair. We demonstrated previously that dATP increases hydrolytic activity of both cardiac (MYH6, MYH7)3 and skeletal (MYH2) myosin.4 dATP also increases the magnitude and rate of contraction of demembranated cardiac3, 5 and skeletal muscle cells6, 7 in animal models and failing dog and human myocardium.8, 9 Relatively small increases in dATP in cardiomyocytes, achieved by over‐expression of the enzyme ribonucleotide reductase, are effective in increasing left ventricular pressure development and ejection fraction in normal rodent hearts and recovering lost function in infracted rodents and mini‐pigs.10 Thus, elevating dATP to activate myosin represents a promising strategy for treatment of heart failure that has been shown to be effective in multiple preclinical models.11, 12 Numerous studies focused on characterizing the effects of dATP on the chemo‐mechanical cycle have shown that dATP increases both the rate of force development and the number of cross‐bridges bound in a force generating state, but does not increase in the amount of force per crossbridge.3, 7, 13 These studies also demonstrated that there was no difference in the affinity of myosin for dATP vs. ATP, nor was there a difference in the rate of hydrolysis of dATP vs. ATP. There is evidence that, at least in low strain conditions, dATP also increases the rate of crossbridge detachment, however the primary effect of dATP is on crossbridge attachment.13 The findings that dATP increases the rate of force development (posthydrolysis) and increases force via increased numbers of crossbridges bound (and not via increased force per cross‐bridge) suggest enhanced binding of myosin‐dADP.Pi to actin.3, 7 While these biochemical and mechanical data are convincing that posthydrolytic myosin activity is enhanced by dATP, the structural basis for this is not known. Because myosin translates the chemical energy of ATP or dATP to mechanical force through conformational changes associated with alterations to the binding pocket and actin binding surface, it might be expected that the dATP‐induced enhancement of myosin performance is the direct result of changes to protein structure and dynamics. More specifically, we are interested in the conformation of posthydrolysis, pre‐powerstroke myosin state (the major state targeted for actin binding), and the changes induced by the hydrolysis products of dATP (dADP.Pi). As a basis for the current study, we hypothesize that dADP.Pi induces alterations to the myosin conformation that increase its propensity to bind to actin, and that these structural changes to myosin underlie the physiological observation that when dATP is the substrate for contraction there is an 30–40% increase in the rate of force development and the amount of force produced.3
Myosin is composed of functional subdomains that undergo conformational changes in a nucleotide‐dependent manner, including the N‐terminal, upper 50‐kDa, lower 50‐kDa, and the converter subdomains (Fig. 1). The cleft that separates the upper and lower 50‐kDa domains is bordered by the strut and loop 2 that connect the upper and lower 50‐kDa domains near the actin binding surface and three nucleotide binding regions (p‐loop, switch 1, and switch 2) at the apex of the cleft.14, 15 To better understand how dATP alters myosin performance, we were particularly interested in the conformation, contacts, and dynamics of the regions of myosin that are involved in translating alterations from the nucleotide binding pocket to the actin binding surface. We hypothesized that dADP.Pi‐induced alterations to the binding pocket ultimately result in the stabilization of a myosin structure that has a higher propensity for binding actin than that bound to ADP.Pi. Thus, here we provide a detailed exploration of the P‐loop and switch regions, the cleft conformation, and the actin binding surface structure and charge.
Figure 1.
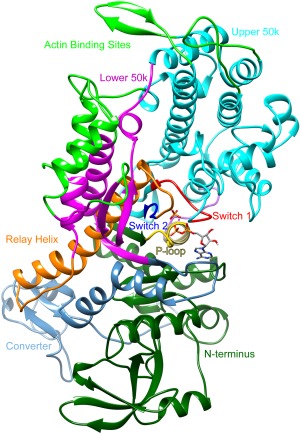
Structure of dictyostelium discoideum myosin S1 head. Functional regions highlighted in different colors: Upper 50k, cyan; Lower 50k, magenta; actin binding sites, bright green; P‐loop, yellow; Switch 1, red; Switch 2, blue; Relay helix, orange; Converter, light blue; N‐terminus, dark green.
We used an in silico approach to assess atom‐level differences in myosin structure and dynamics as a result of dADP.Pi (vs. ADP.Pi) binding to examine the portion of the cross‐bridge cycle that has been demonstrated to be most affected by dADP.Pi in previous in vitro work.3, 7, 13 Multiple, all‐atom molecular dynamics (MD) simulations were performed of the Dictyostelium discoideum myosin S1 head, which was crystallized (PDB ID:1VOM)15 with Mg2+, Pi and either ADP or dADP in the binding pocket for 50 ns (referred to as ADP.Pi simulations and dADP.Pi simulations, respectively, throughout). These simulations provide a better understanding of the structure–function relationship of myosin in the pre‐powerstroke state and how changes in its conformation could directly alter cross‐bridge attachment. Furthermore, the atom‐level assessment of myosin structure, simulated over time, provides unique detail into the dynamics of allosteric regulation that propagates a conformational change from the nucleotide binding pocket to the actin binding surface of myosin. The allosteric mechanism was previously studied primarily through assessment of crystal structures alone. Here we demonstrate how, with all else equal, dADP.Pi binding to myosin in the pre‐powerstroke conformation induces conformational changes within the myosin S1 head that result in an overall shift in the population distribution of energetically favored myosin structures to a conformation that favors actin binding. These changes were precipitated by a change in the contacts made by Phe129, which spends the majority of its time in contact with the O2’ at the 2 position of the ribose ring of ADP; Phe129 reorients and makes altered contacts, however, when the O2’ is absent. The significant conformational alterations in the nucleotide‐binding pocket resulted in an altered contact network and a myosin conformation that is more conducive to actin binding. The MD simulations predict that myosin bound to dADP.Pi should enhance electrostatic interactions between myosin and actin, and actin binding studies in an in vitro motility assay support the MD predictions. These data not only suggest a mechanism for dADP.Pi‐induced changes to the myosin structure and dynamics that favor myosin binding to actin, but they also provide unique insight into the dynamics of pre‐powerstroke myosin bound to ADP.Pi.
Results and Discussion
Three simulations with ADP, Pi, and Mg2+ bound to myosin and three simulations with dADP, Pi, and Mg2+ bound to myosin were performed. These simulations are referred to as ADP.Pi and dADP.Pi simulations throughout this manuscript. Furthermore, while we discuss dADP.Pi binding or dADP.Pi‐induced changes to the binding pocket, all of the simulations also included Mg2+. Each simulation was 50 ns long, and was performed at 37°C and neutral pH. Unless otherwise indicated, values provided below are averages of all three simulations of a given condition over the last 20 ns of simulation time. The last 20 ns were used because the systems had all equilibrated by this time. Error values given are the standard deviations between simulations. All figures showing histograms of data represent the frequency distribution for all six (three ADP.Pi, three dADP.Pi), 50 ns simulations at 100 ps resolution (1503 structures per group).
Effects of dADP.Pi binding on the nucleotide binding pocket of myosin
To investigate the local effects of dADP.Pi in the nucleotide binding pocket of myosin, we specifically assessed the atomic contacts that were altered as a result of removing the O2’ from the starting structure. In the ADP simulations Phe129 Cα, Cδ1, and Cε1 spent over 65% of the simulation time in contact with O2’ (Fig. 2). In the dADP.Pi simulations, however, there were contact changes in the binding pocket because of the absence of the O2’, altering the Cα, Cδ1, and Cε1 contacts. Instead, these atoms primarily contacted the C5 and C6 on the adenine portion of dADP and established new intramolecular contacts with Gln662 and Leu663. Phe129 also lost contact with Arg131 in the dADP.Pi simulations. These immediate changes in the nucleotide‐binding site initiated a cascade of changes in intramolecular interactions that resulted in global changes to myosin's conformation.
Figure 2.
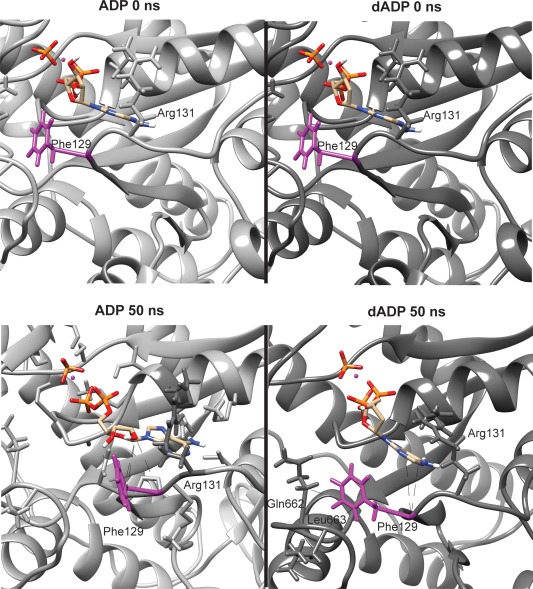
Loss of O2' disrupts contacts in the nucleotide binding pocket. Representative figures showing the conformation of the nucleotide binding pocket at both 0 and 50 ns from ADP.Pi and dADP.Pi simulations. The isolated sphere represents the magnesium ion. Phe129 is highlighted in magenta, and the primary contacts it makes are shown with a dotted black line in the 50 ns figures.
To further assess the local dADP.Pi‐induced conformational changes to the binding pocket, we performed root‐mean‐squared‐deviation (RMSD) analysis of the Cα atoms of the myosin binding pocket residues (considered to be any residue that made contact with the nucleotide throughout the simulation). The binding pocket residues were also used in the alignment for the RMSD calculation, and as such this measurement reflects local changes to the binding pocket conformation. These binding pocket residues are involved in coordinating the nucleotide and experienced larger conformational changes with bound dADP.Pi as compared to ADP.Pi (2.5 ± 0.2 Å vs. 1.9 ± 0.6 Å). While this local shift may appear to be minor, it was associated with a dramatic loss of contacts between myosin and dADP, Pi, and Mg2+, which are critical for maintaining a closed nucleotide‐binding pocket (Figs. 3 and 4). Myosin maintained ∼240 atomic contacts with ADP, Pi, and Mg2+ over the course of simulations. However, ∼3.5 ns into the dADP.Pi simulations, myosin lost ∼12% of its contacts with the nucleotide, Pi, and Mg2+, a change that persisted for the rest of the simulation. The average number of intermolecular contacts was 230 ± 8 for the first 3.5 ns of the dADP.Pi simulations vs. 203 ± 6 for the remainder. The loss of these contacts led to a change in the position of both the binding pocket and the position of the nucleotide within the pocket [Fig. 3(B)]. A detailed description of how dADP was positioned differently (and as a result had altered contacts with myosin) in the binding pocket rearrangement can be found in the supplemental information.
Figure 3.
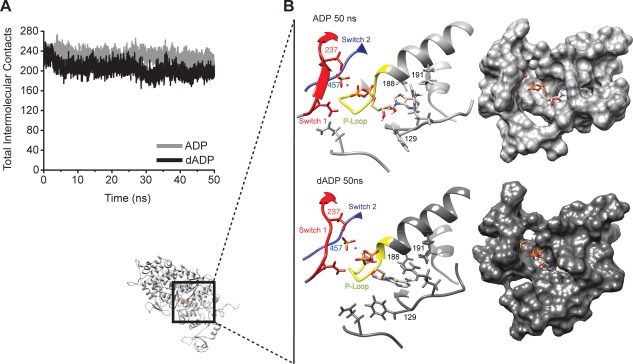
The nucleotide binding pocket conformation is altered in dADP.Pi simulations. (A) Total number of contacts made between myosin and the nucleotide, Pi, and Mg2+ averaged over the three simulations for each condition. (B) Ribbon and surface renderings of representative binding pocket structures highlighting the conformational differences between the binding pocket in the dADP.Pi (dark grey) and ADP.Pi (light grey) simulations. Small myosin structure shows region of focus.
Figure 4.
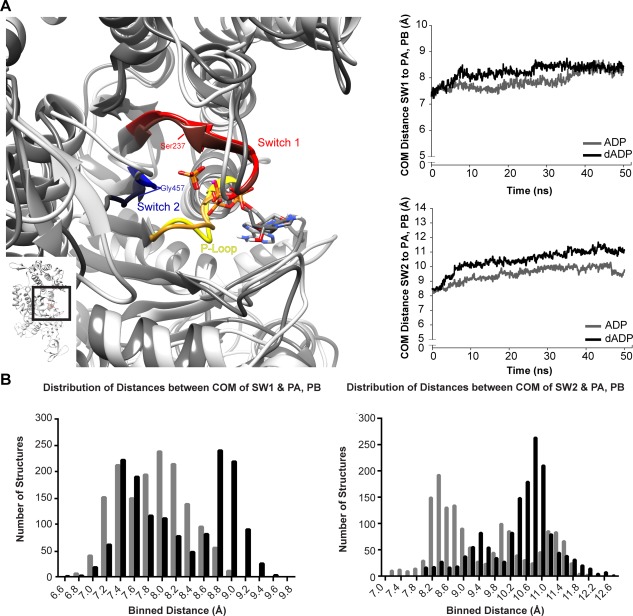
Alterations to the conformation of the switch regions, nucleotide, Pi, and Mg2+ observed in dADP simulations. (A) Representative 50 ns structures of the binding pocket demonstrate minimal differences in the p‐loop (yellow) between ADP.Pi (light grey structure) and dADP.Pi (darker grey structure) simulations in contrast to dramatic alterations to the position of the switch regions (switch 1 (residues 233–240), red; switch 2 (residues 454–459), blue). The entire myosin head is shown on the left corner with the boxed region highlighting the region of interest shown larger to the left. (B) Plots show the distance between the center of mass (COM) of switch I (SW1) and switch II (SW2) and the two phosphates in ADP (labeled PA, PB; see supplement for more information on atom labels). Line graphs show the average of all three simulations within each group. Histograms including structures from all six simulations (three ADP.Pi, three dADP.Pi) at 100 ps granularity (1503 structures in each group) highlight that there are more instances of structures with increased distance between the switch regions and Pi when dADP is bound vs. ADP (all 6 simulations included).
The p‐loop, Switch 1 (SW1), and Switch 2 (SW2) [Fig. 4(A)] coordinate the nucleotide and undergo conformational changes throughout the cross‐bridge cycle to mediate nucleotide binding and release. Switch 1 (SW1) is thought to communicate between the nucleotide binding pocket and the actin binding surface, sensing changes from NTP to NDP, and undergoing conformational shifts that alter affinity for the nucleotide and/or actin. The conformation of SW1 is largely dictated by its interaction with the nucleotide and influences myosin's interaction with actin, where the “open” conformation of SW1 binds actin more tightly than the “closed” conformation of SW1.15, 16, 17 The contact between Ser237 and Mg2+ is critical to maintain the closed conformation SW1.18 The Gly457 (SW2 residue) interaction with Pi is necessary for ATPase activity, and it is lost in the strong actin binding state when SW2 is in the “open” position.15, 18, 19, 20, 21, 22 The loss of contact between Gly457 and Pi was also observed in Targeted Molecular Dynamics (TMD) simulations of myosin binding to actin.20 The conformational change of SW2 and the loss of the essential hydrogen bond with Gly457 are proposed to initiate phosphate release and the powerstroke,20 and they are also thought to be hallmarks of a myosin conformation preferred by actin.19 Unlike the role of the switch regions in allosteric regulation, the role of the p‐loop in communicating between the nucleotide binding pocket and the actin‐binding surface has not been studied extensively. It is known, however, that contacts between the p‐loop and the nucleotide are critical in coordinating the nucleotide in the binding pocket.15, 18, 23
While the p‐loop conformation was similar across all simulations, the conformation and dynamics of the switch regions differed between the dADP.Pi and ADP.Pi simulations. The switch regions and nucleotide, phosphate, and Mg2+ moved away from each other in dADP.Pi simulations, and this increased distance between the switch regions and the ligands destabilized key contacts are required to keep the binging pocket “closed”.15, 16, 17, 18, 19, 20 The “open” binding pocket conformation has been associated with phosphate release (SW2) and higher affinity for actin (SW1, SW2).
Our simulations suggest there are minimal differences between how dADP.Pi and ADP.Pi interact with the p‐loop (residues 179–186). The distance between the center of mass of the two phosphates on ADP and the center of mass of p‐loop residues was stable over the last 20 ns of simulation time in both ADP.Pi and dADP.Pi simulations at 6.9 ± 0.6 Å and 7.0 ± 0.5 Å, respectively. Not only was the p‐loop stable, but it also maintained its conformation throughout the course of all six simulations. Nor did we observe a significant change in the twisting in the central beta sheet between ADP.Pi and dADP.Pi simulations, which supports the conclusion that there is minimal dADP.Pi‐induced movement in the p‐loop.
To investigate the conformation of the binding pocket and evaluate the extent dADP destabilized the closed conformation, we quantified the distance between the center of mass of the switch regions to the ligands as well as the distance between the binding pairs that are hallmarks of the closed binding pocket described above. The distance between the center of mass of SW1 (residues 233–240) and the two phosphates was very slightly increased in the dADP.Pi vs. ADP.Pi simlulations (8.4 ± 0.8 Å vs. 8.2 ± 0.2 Å), and the distance between Ser237 and Mg2+, the contact pair that defines the closed SW1 conformation was increased to a point that contacts were disrupted (5.9 ± 0.7 Å vs. 5.2 ± 0.1 Å for dADP.Pi vs. ADP.Pi). There were more dramatic increases between the center of mass of SW2 (residues 454–459) and the two phosphates (11.0 ± 0.4 Å vs. 9.9 ± 0.9 Å) in dADP.Pi vs. ADP.Pi simulations. Likewise, the distance between Gly457 and Pi was increased in the dADP simulations (7.6 ± 1.2 Å vs. 6.0 ± 1.8 Å). These data suggest that dADP destabilizes the “closed” conformation of the binding pocket while favoring a conformation that is associated with a higher affinity for actin. The distances between the switch regions and the two phosphates on ADP in the crystal structures, 1VOM (ADP.Pi) and 1MMA (ADP) also demonstrate how the switches move toward a more “open” conformation when transitioning toward a strong‐binding form. The distance between the center of mass of SW1 and the two phosphates was 6.9 Å vs. 7.1 Å and between the center of mass of SW2 and the two phosphates was 8.6 Å vs. 11.0 Å for 1VOM vs. 1MMA. The stabilization of pre‐powerstroke myosin in a state with a higher propensity to bind actin in the dADP simulations would increase the rate of attachment of myosin to actin, which supports our previous work studying the impact of dADP on the powerstroke.
Analysis of the conformation of the p‐loop and switch regions as well as their interactions with the nucleotide, Pi, or Mg2+ demonstrated that dADP.Pi increased the propensity of SW1 to adopt an “open” conformation. There was consistently a greater distance between the switch regions and the ligand in the dADP.Pi simulations compared with the ADP.Pi simulations, which is best seen in the histograms in Figure 4(B). The increased distance and associated destabilization of critical intermolecular contacts in the dADP.Pi simulations indicates that dADP.Pi enabled a more “open” conformation of both SW1 and SW2, a conformational state that is associated with weak nucleotide binding and strong actin binding. Importantly, even though there was an increase in the time that switch regions adopted a more “open” conformation in the dADP.Pi simulations (as evidenced by position and loss of contact with the nucleotide, Pi, and Mg2+), there was no evidence suggesting that Pi would be released early with dADP.Pi. Cecchini et al. concluded that contacts between Pi and residues outside of the binding regions are indicative of Pi progressing through the exit tube before release, and that when Pi is stable in the binding pocket, there are only interactions between the p‐loop, switch 1, and switch 2 regions.18 However, other reports using structural and kinetic information have concluded that Pi release may occur earlier.24, 25 In the dADP.Pi simulations, there were no contacts between Pi and regions of myosin outside of the “binding regions” (p‐loop, switch 1, and switch 2). Therefore, this suggests that even though the switch regions may be more open with dADP.Pi, Pi was stable in the binding pocket and did not proceed through the exit tube, which would indicate the potential for early release. The stability of Pi in pre‐powerstroke myosin is important as it can prevent myosin from going through the powerstroke before binding to actin.18 We are limited in our ability to conclude when Pi is released, as our simulations did not include actin, which interacts with myosin during the isomerization and Pi release process, and likely affects the structure of these regions.
The altered position of the switch regions resulted in a more mobile nucleotide that was also more exposed to solvent. dADP had both a higher RMSD than ADP (5.10 ± 0.40 Å vs. 3.31 ± 1.2 Å, respectively) and a higher solvent accessible surface area (SASA) vs. ADP (153.6 ± 6.03 Å2 vs. 88.1 ± 22.6 Å2, respectively) (Supporting Information Fig. S1). An analysis of the binding pocket conformation demonstrated that dADP.Pi initiates a cascade of contact changes beginning with Phe129 that result in an altered binding pocket conformation. Ultimately, dADP was further away from the switch regions (and more exposed to the solvent) than ADP, which prevented contacts between dADP, Pi, and Mg2+, and residues critical for a “closed” nucleotide‐binding pocket. Thus, the most populated states in the dADP.Pi simulations resembled a more “open”‐conformation, as is typically seen in the strong actin binding states.15, 18, 19, 20, 21, 22 The observed alterations to the binding pocket provide insight into the mechanism by which dADP.Pi activates myosin to increase cross‐bridge attachment, as they highlight dADP.Pi‐induced stabilization of a conformation of myosin that is typically observed in strong actin‐binding states.
Effects of dADP.Pi on the cleft orientation
The extent to which the actin binding cleft is open vs. closed is thought to predict whether myosin binding to actin is energetically favorable. Open‐cleft conformations of myosin bind to actin endergonic in contrast to closed‐cleft conformations of myosin, which can bind exergonic.26 We assessed the openness of the actin binding cleft by measuring the angle between the helices bordering the cleft on the lower 50k domain (w‐helix, residues 630–646) and the upper 50k domain (HO helix, residues 411–442). The helices and vectors used to measure this angle are shown from two different views in Figure 5(A). The cleft angle measurements suggest that dADP.Pi stabilized a closed cleft conformation of myosin and specifically highlight the closure of the outer cleft (the portion closest to the actin binding site). Interestingly, in the ADP.Pi simulations, the cleft slowly opened throughout the course of the simulation; at 9 ns into the simulation the cleft angle was ∼28° and by 25 ns the cleft angle reached ∼32° where it remained for the rest of the simulation. The average cleft angle for ADP.Pi simulations was 28° ± 2° for the first 9 ns, 30° ± 4° between 9 ns and 25 ns and reached 32° ± 4° for the last 25 ns of the simulation. The average cleft angle in the dADP.Pi simulations, on the other hand, remained at 26° ± 1° in each time window described above [Fig. 5(B)]. In addition to measuring the angle of the cleft, we also calculated the distances between residues on the inner (inner cleft, residues 277–465; far inner cleft, residues 232–461) and outer (outer cleft, residues 416–589; far outer, residues 365–536) portions of the cleft that have been previously used to assess cleft closure across different species of myosin and in different states of the cross‐bridge cycle.27 These pairs are highlighted by space‐filled residue rendering in Figure 6. Even though some of these atoms reside in flexible loop regions, this analysis demonstrates that dADP.Pi stabilized the closed cleft conformation. Figure 6(B) shows a histogram of structures from dADP.Pi and ADP.Pi simulations for each of the residue pairs listed above. The narrowness of the dADP.Pi distribution relative to the ADP.Pi distribution highlights the stability of the myosin cleft structure with ADP.Pi. The ADP.Pi‐bound myosin sampled a wider range of distances, reflecting fluctuations in the cleft with a propensity for increased distances (in particular in the outer cleft measurements). This analysis also indicates that the outer portion of the cleft was more closed in the dADP.Pi simulations. However, by this measure, the inner cleft was slightly more open in the dADP.Pi vs. ADP.Pi simulations, though it was also more stable than the ADP.Pi simulations. The openness of the inner cleft was because of movement of the residues on the lower 50k domain (Phe461 and Ser465). While residues on the upper 50k domain (Arg232 and Asn277) were in a relatively similar position throughout both ADP.Pi and dADP.Pi simulations, Phe461 and Ser465 moved away from their upper 50k counterparts in the dADP.Pi simulations as a result of the opening of SW2 (residues 454–459) [Fig. 6(C)], as described above. In fact, the mean, median, and mode of the far inner cleft measurement (which contains the residue closest to SW2) of the dADP.Pi simulations were nearly identical to the reported distance in the myosin postrigor crystal structure, 1MMD (12.4 Å),28 where it is expected that SW2 is open.19, 27 Despite the opening of the inner cleft in these simulations, the outer cleft was maintained in a more closed position. This dADP.Pi‐induced stabilization of the cleft conformation with a closed outer region may underlie the increased cross‐bridge attachment and rate of force development observed with dATP in vitro.7
Figure 5.
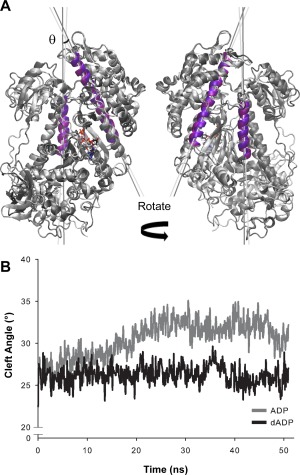
Cleft analysis using helix‐helix measurement highlights dADP.Pi stabilization of the closed‐cleft conformation. (A) Representative structures are shown to highlight the w‐helix (residues 630–646) and HO Helix (residues, 411–442) that are used to measure cleft angle. ADP.Pi structure shown in light grey, and dADP.Pi structure shown in dark grey. Purple helices are those that were fit by vectors used to calculate the cleft angle. Light purple is used on the ADP.Pi structure and dark purple on the dADP.Pi structure. Vectors used to calculate cleft angle are represented by light gray (ADP.Pi) and dark grey (dADP.Pi) lines. (B) Line graphs show the average of all three simulations within each group. Cleft angle was calculated at the point of intersection of these vectors. Vector angle quantification is shown over time and indicates that dADP.Pi stabilizes a closed cleft conformation as compared to ADP.Pi.
Figure 6.
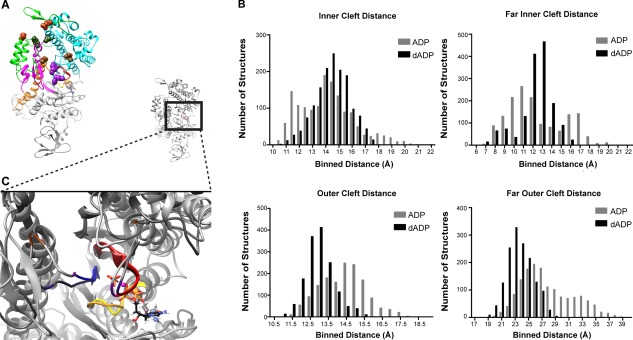
dADP‐induced effects on the outer cleft. (A) Representative structure highlights the residues used in this analysis shown as spheres: inner cleft, residues 277–465 (brown); far inner cleft, residues 232–461 (magenta); outer cleft, residues 416–589 (green); far outer, residues 365–536 (red). (B) Histograms including structures from all six simulations (three ADP.Pi, three dADP.Pi) at 100 ps granularity (1503 structures in each group) highlight the structures from dADP.Pi simulations populate a more closed‐cleft (residues closer together) state. (C) Representative 50 ns structures highlight the movement of residues on the lower 50k region, 461 (purple) and 465 (brown), away from their partner on the upper 50‐kDa region, 232 (purple) and 277 (brown), in dADP.Pi (depicted in darker grey) simulations vs. ADP.Pi (lighter grey) simulations. Reference structure is shown with detail region shown in boxed region.
Effects of dADP.Pi on the actin binding surface
The initial binding interaction between actin and myosin in the pre‐powerstroke state is primarily electrostatic in nature,14, 29, 30, 31 and the rate of attachment and ATPase activity can be dramatically affected by the positive charges presented on myosin's actin binding surface.31, 32, 33 Consequently, we assessed the solvent accessible surface area (SASA) of the positively charged residues on the actin binding surface of myosin, with particular focus on Loop 2, where the charge density has been shown to mediate ATPase activity.31, 32, 33 In the dADP.Pi simulations, there was a modest increase in the SASA of all positively charged residues on the actin binding surface compared with the ADP.Pi simulations (617 Å2 ± 39 Å2 vs. 594 Å2 ± 33 Å2 respectively) (Fig. 7). Further analysis of Loop 2 indicated more of a shift toward increased polar exposure in the dADP.Pi simulations. Not only was there an overall increase in the mean of Loop 2 polar SASA with dADP. Pi (315 ± 21 Å2 vs. 271 ± 26 Å2), but there was also a narrower distribution of dADP.Pi structures with more instances of higher Loop 2 polar SASA [Fig. 7(B)]. This distribution indicates that dADP.Pi stabilized a conformation of myosin with increased polar SASA of Loop 2 residues. Figure 7(C) further highlights this shift in the distribution of structures favoring increased polar SASA in dADP.Pi vs. ADP.Pi simulations. For example, ∼50% of the dADP.Pi structures had polar SASA of 300 Å2 or greater vs. ∼25% of ADP.Pi structures. These results indicate that there was an increase in the occurrence of higher SASA of polar loop 2 residues and positively charged actin binding residues in dADP.Pi simulations and that the distribution was positively skewed as opposed to the negative skew in the ADP.Pi data [Fig. 7(B)]. A discussion relating this increase in polar SASA exposure to experimental increases in charge to loop 2 performed by Joel et al.33 can be found in the supplemental information.
Figure 7.
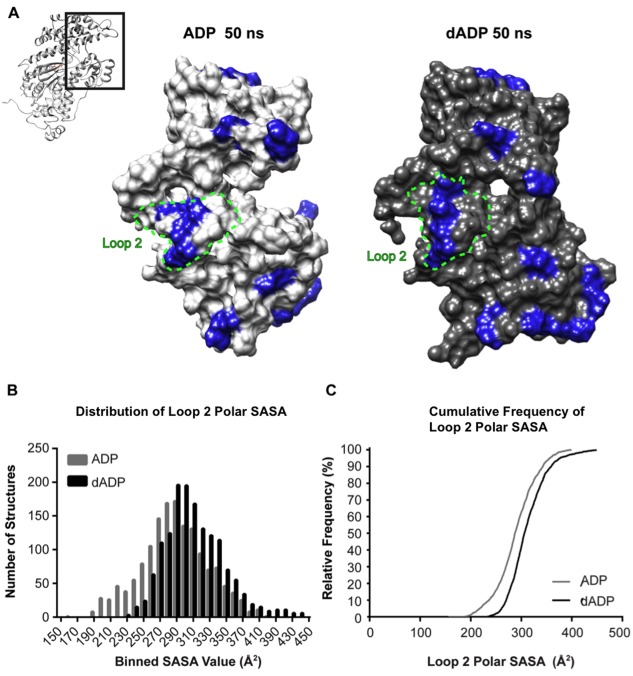
Assessment of dADP.Pi‐induced alteractions to actin binding surface. (A) Representative actin binding surface structures at 50 ns from ADP.Pi and dADP.Pi simulations. For reference, the entire S1 region is shown in context in the ribbon figure and the region in the boxed region is blown up in the surface rendering. Blue highlight indicates positively charged residues. These residues are more exposed to the solvent in the dADP.Pi simulations. (B) Histograms including structures from all six simulations (three ADP.Pi, three dADP.Pi) at 100 ps granularity (1503 structures in each group) highlight the right shift of the distribution of Loop 2 polar SASA values in dADP.Pi simulations. (C) This effect is further evidenced by the cumulative frequency plot that demonstrates at any SASA value observed, there is a higher fraction of dADP.Pi structures that have higher SASA values than ADP.Pi structures.
Together, these analyses suggest that having dADP.Pi in the nucleotide binding pocket results in allosteric modification to the actin binding surface. Correlation analysis demonstrates that the SASA of polar residues on loop 2 is inversely related to the cleft angle measurement in ADP.Pi simulations, with the strongest relationship appearing in ADP.Pi simulation #2. The cleft is the most open in ADP.Pi simulation #2, and has the lowest polar residue SASA of Loop 2 (Supporting Information Fig. S2). There is very little correlation between cleft angle and Loop 2 SASA, which reflects the stability of these parameters in the dADP.Pi simulations. dADP.Pi stabilizes a closed cleft conformation, which in turn, stabilizes the exposure of polar residues on Loop 2. The resulting cluster of stable conformations with high polar residue exposure on Loop 2 and closed cleft can be seen in Supporting Information Figure S2. Further evidence of this relationship can be observed by examining the average Loop 2 polar SASA at different time intervals. Whereas the average Loop 2 polar SASA remains relatively constant over the entire 50 ns of all dADP.Pi simulations [mean ± SD SASA equal to 304 ± 10 Å2 from 0 to 10 ns, 309 ± 21 Å2 from 10 to 20 ns, 316 ± 31 Å2 from 20 to 30 ns, 319 ± 28 Å2 from 30 to 40 ns, 311 ± 10 Å2 from 40 to 50 ns], the average Loop 2 polar SASA starts to decrease shortly after the cleft opens in the ADP.Pi simulations [mean ± SD SASA equal to 320.9 ± 11.9 Å2 from 0 to 10 ns, 309.8 ± 21.9 Å2 from 10 to 20 ns, 283.2 ± 15.0 Å2 from 20 to 30 ns, 270.0 ± 36.0 Å2 from 30 to 40 ns, 259.1 ± 29.1 Å2 from 40 to 50 ns]. Others have demonstrated that increasing the charge of the actin binding surface on myosin can increase acto‐myosin interaction and ATPase,31, 33 and we hypothesize that the increase in charged surface area increases the affinity of myosin for actin in the presence of dADP.Pi, thereby increasing the probability of cross‐bridge attachment.
In vitro motility data support the contention that dADP.Pi increases the charge on myosin surface and affinity for actin
In vitro motility (IVM) assays are commonly used to study the actin–myosin interaction under different conditions (e.g. temperature, pH, and ionic strength). The ionic strength of motility solutions typically ranges from 40 to 100 mM because lower ionic strengths (compared to physiological) are required for actin to stay on myosin‐coated surfaces. Solutions with ionic strength over 100 mM tend to disrupt the electrostatic interactions between actin and myosin, resulting in dissociation of actin and loss of movement.31, 34 We hypothesized, based on the increased exposure of polar groups observed in the pre‐powerstroke state in silico, that more actin filaments should stay on myosin‐coated surfaces and become motile as the ionic strength is varied when 0.1 mM dATP is the substrate (vs. 0.1 mM ATP) because there would be increased electrostatic interactions between myosin‐dADP.Pi and actin than between myosin‐ADP.Pi and actin. This concentration is near the K m for actin motility in the assay for both ATP and dATP,4 and was selected to ensure that enough filaments would land and be motile on the surface for rigorous comparisons. The substrates are hydrolyzed by myosin to dADP and Pi (vs. ADP and Pi) prior to addition of actin, and enabled us to experimentally examine the propensity of pre‐powerstroke myosin bound to dADP.Pi to bind to actin.
We quantified the fraction of filaments moving (rather than characterizing their sliding speed or other motility property) because we were interested in the amount of acto‐myosin interaction at high ionic strength (Fig. 8). Some of the nonmotile filaments are likely actin filaments held to the surface by damaged myosin, but we expect the fraction of nonmoving filaments associated with damaged myosin to be the same at every ionic strength. Thus, the decreasing percentage of moving filaments (observed in both ATP and dATP systems) is likely attributable to charge masking. We propose, therefore, that the differences in percent moving between ATP and dATP reflect dATP‐induced alterations that increase the weak electrostatic interactions between actin and myosin. The increase in bound, moving filaments at high ionic strength suggests that dATP (dADP and Pi when hydrolyzed) enhances long‐range electrostatic interactions between actin and myosin and supports the idea that enhanced binding (electrostatic) between myosin and actin could occur in myofibrils at physiological ionic strength (∼200 mM). This effect is particularly noticeable at 150 mM where more than three times as many actin filaments were moving with dATP than with ATP (41% vs. 13%). The dADP.Pi‐induced increase in charge exposure on the actin binding surface may underlie the observed increases in myosin binding and force production in the presence of dATP.3, 7, 13 Moreover, the surface analysis of the dADP.Pi simulations suggests a structural correlate that may explain this observation, i.e. dADP.Pi may allosterically alter the actin binding surface of myosin, resulting in increased exposure of polar side chains that increases myosin binding to actin. Together, these data demonstrate a potential mechanism for dATP‐induced increases in cross‐bridge attachment and ultimately in increased force production. We acknowledge that the simulations and IVM experiments were run with different types of myosin, but propose that these data suggest that there is a common mechanism of action that occurs on a structural level with dADP.Pi binding. Future studies may be important to better understand species differences on a structural level, however we propose that the regions of myosin affected by dADP.Pi will be similar across different myosin species while the extent of change may differ, as we have seen enhanced contraction in mice, rats, rabbits, pigs, dogs, and human striated muscle.35, 36
Figure 8.
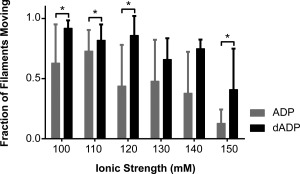
The fraction of moving filaments in vitro motility assay with ATP and dATP at increasing ionic strengths. The fraction of moving filaments at ionic strengths 100 to 150 mM are shown for conditions with ATP and with dATP. There were between 20 and 1600 filaments measured for each group (number of filaments decreased with increasing ionic strength) on 4–15 slides. Bars represent mean + SD of the fraction of moving filaments. *P < 0.05.
Materials and Methods
The sequence and residue numbering for Dictyostelium discoideum myosin II are used throughout this manuscript.
Model selection
The crystal structure for pre‐powerstroke myosin (PDB ID:1VOM), which was resolved at 1.90 Å was used as a starting structure for all simulations.15 VO4 was replaced in all structures by PO4 (Pi). PO4, ADP, and dADP were parameterized in the Levitt et al. force field37 using previously established bond length, bond angles, and torsion angles. Partial charges (Supporting Information Fig. S3) were calculated by running a geometry optimization of each structure in GAMESS.38 Loops missing from the 1VOM structure were replaced using homology modeling in MODELLER.39, 40, 41, 42 Dictyostelium discoideum myosin was chosen for these studies because it is the only myosin II that has been crystalized in multiple conformations at high resolution. We sought a myosin that was available in multiple conformations so that we can further study the effects of dATP and its hydrolysis products on other parts of the cross‐bridge cycle. Furthermore we chose high‐resolution structures because those are most suitable for the all‐atom MD simulations performed.
MD simulations
Both systems were prepared according to our standard protocols.43 All‐atom, explicit solvent MD simulations were performed at 37°C and neutral pH using in lucem molecular mechanics (ilmm).44 The NVE microcanonical ensemble (constant number of particles, volume, and energy) was used with periodic boundary conditions, and the Levitt et al. force field.37 Before running the simulations, the starting structures (Myosin + (d)ADP + Pi + Mg2+) were minimized for 1000 steps, and flexible three‐center (F3C) water molecules45 were added to a rectangular box The solvent density of the box was adjusted to 0.99336 g/mL, the experimentally determined density for that temperature.46 A time step of 2 femtoseconds (fs) was used, and after each picosecond (ps), a structure was saved for analysis. An 8 Å force‐shifted cutoff was employed for nonbonded interactions.43, 47 Three simulations of Myosin + ADP + Pi + Mg2+ and three simulations of Myosin + dADP + Pi + Mg2+ were performed for 50 ns each.
MD analysis
All analyses of MD trajectories were performed using ilmm unless otherwise indicated. Contacts between nonsequential atoms were classified as in contact if two carbon atoms were separated by less than 5.4 Å, or if two noncarbon atoms were separated by less than 4.6 Å. They were further classified as hydrophobic if they were between two carbon atoms less than 5.4 Å apart and each carbon was bound to at least one hydrogen atom. Contacts were considered hydrogen bonded if there was a donor (D‐H) and acceptor (A) atom that were within 2.6 Å of each other and where the D‐H‐A angle was between 45° and 135°. Lastly, contacts were defined as ‘other’ if there were two heavy atoms within 4.6 Å of each other and did not fit in either of the other two classifications. Distances were measured between specific atom pairs. Solvent accessible surface area (SASA) was calculated using the Lee and Richards algorithm.48 Protein images were generated using the UCSF Chimera package from the Computer Graphics Laboratory, University of California, San Francisco (supported by NIH P41 RR‐01081)49 except for Figure 5(A), for which VMD was used.50, 51
In vitro motility
In vitro motility assays were performed as previously described.52, 53 Briefly, skeletal muscle myosin used in this assay was prepared from back muscle of male New Zealand White rabbits according to Margossian and Lowey.54 It was stored at −20°C in 50% glycerol for no longer than 4 months. Heavy meromyosin (HMM) was prepared as described by Kron et al. and used within one week of preparation. F‐actin was prepared from rabbit back and leg muscle ether powder, labeled with Rhodamine‐phalloidin (RhPh; Molecular Probes, Eugene, OR), and stored on ice for no longer than six weeks.55, 56, 57, 58
Flow cells were constructed as described previously.59, 60 Two glass cover slips were used and separated by 2 mm foam adhesive strips. Total chamber volume was typically 50–70 μL. The lower slide surface was coated with 0.1% nitrocellulose in amylacetate (Sigma‐Aldritch, St. Louis, MO). The experimental procedure followed was similar to Gordon et al.60 Briefly, HMM (0.17– 0.4 mg/mL) was added to the flow cell for 3 min, and nonspecific protein binding to the surface was blocked with bovine serum albumin (BSA). Nonlabeled sheared F‐actin was then added to the chamber for 1 min followed by wash, after which RhPh F‐actin was then added for 1 min followed by infusion of the motility buffer. The motility buffer consisted of (in mM/L): 25 imidazole, 1 EGTA, 0.85 free Mg2+, 0.1 MgATP/MgdATP, and ionic strength was varied from 100 to 150 mM with KCl. Antioxidants (18 μg/mL catalase, 0.1 mg/mL glucose oxidase, 3 mg/mL d‐glucose, and 40 mm DTT) were added to minimize photobleaching.55 All motility assays were performed at 23.5 ± 0.5°C. At least six areas of each assay chamber were recorded for 10 sec each at 10 Hz with IVM Image Acquisition. Recordings were analyzed digitally, using custom software developed in house.
Conclusions
Previous experimental work assessing the functional effects of dATP on myosin and muscle contraction demonstrated that dATP significantly increases the rate of cross‐bridge cycling, the magnitude and rate of force development and the rate of relaxation.3, 7, 13 More specifically, these studies indicated that the primary effect of dATP is an increased rate of cross‐bridge attachment, resulting in an increased number of force‐generating cross‐bridges.3, 7, 13 These earlier studies suggest that dATP (and its hydrolysis products, dADP and Pi) increase the propensity of myosin binding to actin in the pre‐powerstroke state. However, little work has been done to understand the structural changes in myosin that elicit these dramatic functional effects.
Here, we employed MD simulations to address this gap in an attempt to elucidate the structural effect of dADP.Pi binding to pre‐powerstroke myosin. We chose to use myosin in the pre‐powerstroke conformation because it provides insight into the specific portion of the cross‐bridge cycle where we expect dATP to have the most dramatic effects. Upon the removal of a single oxygen atom, significant conformational alterations occurred in the binding pocket, which in turn were transmitted across the protein, stabilizing a closed cleft conformation and increasing the SASA of polar groups in Loop 2 on the actin binding surface. These analyses highlight the significance of a single atom and provide insight into how myosin translates conformational changes between the nucleotide binding site and the actin‐binding surface throughout the cross‐bridge cycle (Fig. 9). Furthermore, these simulations provide novel insight into how allosteric modifications occur in myosin at the atomic level and the structural mechanisms that underlie alterations to the cross‐bridge cycle.
Figure 9.
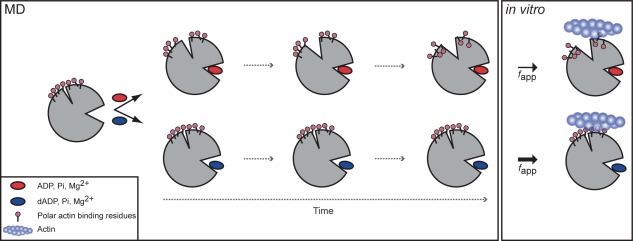
Schematic of dADP.Pi‐induced changes to myosin structure. Simplified overview of allosteric pathway of alterations over time to myosin structure with ADP.Pi (top) vs. dADP.Pi (bottom). ADP.Pi was stable in the binding pocket and maintained contacts with the switch regions, which stabilized a “closed” conformation of the binding pocket. This resulted in an opening of the actin binding cleft over time, which in turn reduced the exposure of polar residues on the actin‐binding surface. dADP.Pi, on the other hand, destabilized the binding pocket. Contacts were broken between the switch regions and the nucleotide, Pi and Mg2+, which stabilized an “open” conformation of the nucleotide‐binding pocket resulting in increased SASA of the nucleotide. This binding pocket conformation stabilized a closed cleft conformation, which in turn favored increased (relative to ADP.Pi simulations) exposure of charged residues on the actin binding surface of myosin bound to dADP.Pi. The model predicted that the increased charge on the actin binding surface would result in increased electrostatic interactions between myosin and actin, which was supported by the IVM data (Fig. 9) and previous in vitro studies,7 and suggests a mechanism by which dATP (and its hydrolysis products, dADP and Pi) increase actomyosin interaction and force production.
Our previous work4, 7 suggests that dATP binding to myosin increases the probability of cross‐bridge attachment. Results from our in vitro motility assays (Fig. 8) and simulations suggest this may occur by improving weak, electrostatic interactions between myosin and actin. Thus, based on both the experimental and MD studies, we propose that dADP.Pi‐induced alterations to the conformation and dynamics of myosin stabilize a myosin conformation that enables more energetically favored actomyosin interactions, providing a molecular framework for the large body of biochemical data available for this system. Thus, the data presented here leverage the atomic resolution of MD simulations and provide insight into the conformational changes that result from dADP.Pi binding to myosin. These simulations could potentially be used in concert with larger‐scale models of the acto‐myosin network to enable better understanding of how molecular‐level changes affect macro‐level performance and also inform the potential design of small molecules or other myosin substrates that could be used for therapeutic application in heart failure.
Supporting information
Supporting Information
Acknowledgments
We thank Dr. Michelle E. McCully, Dr. Clare L. Towse, and Dr. Chin Jung Cheng for their help with molecular dynamics simulation and analysis.
Contributor Information
Michael Regnier, Email: mregnier@uw.edu.
Valerie Daggett, Email: daggett@uw.edu.
References
- 1. Pate E, Nakamaye KL, Franks‐Skiba K, Yount RG, Cooke R (1991) Mechanics of glycerinated muscle fibers using nonnucleoside triphosphate substrates. Biophys J 59:598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. White HD, Belknap B, Jiang W (1993) Kinetics of binding and hydrolysis of a series of nucleoside triphosphates by actomyosin‐S1. Relationship between solution rate constants and properties of muscle fibers. J Biol Chem 268:10039–10045. [PubMed] [Google Scholar]
- 3. Regnier M, Martin H, Barsotti RJ, Rivera AJ, Martyn DA, Clemmens E (2004) Cross‐bridge versus thin filament contributions to the level and rate of force development in cardiac muscle. Biophys J 87:1815–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Regnier M, Lee DM, Homsher E (1998) ATP analogs and muscle contraction: mechanics and kinetics of nucleoside triphosphate binding and hydrolysis. Biophys J 74:3044–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Korte FS, Dai J, Buckley K, Feest ER, Adamek N, Geeves MA, Murry CE, Regnier M (2011) Upregulation of cardiomyocyte ribonucleotide reductase increases intracellular 2 deoxy‐ATP, contractility, and relaxation. J Mol Cell Cardiol 51:894–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Regnier M, Martyn DA, Chase PB (1998) Calcium regulation of tension redevelopment kinetics with 2‐deoxy‐ATP or low [ATP] in rabbit skeletal muscle. Biophys J 74:2005–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Regnier M, Homsher E (1998) The effect of ATP analogs on posthydrolytic and force development steps in skinned skeletal muscle fibers. Biophys J 74:3059–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kolwicz SC, Odom GL, Nowakowski SG, Moussavi‐Harami F, Chen X, Reinecke H, Hauschka SD, Murry CE, Mahairas GG, Regnier M (2015) AAV6‐mediated cardiac specific over‐expression of ribonucleotide reductase enhances myocardial contractility. Mol Therapy 24:240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kadota S, Carey J, Reinecke H, Leggett J, Teichman S, Laflamme MA, Murry CE, Regnier M, Mahairas GG (2015) Ribonucleotide reductase‐mediated increase in dATP improves cardiac performance via myosin activation in a large animal model of heart failure. Eur J Heart Fail 17:772–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nowakowski SG, Kolwicz SC, Korte FS, Luo Z, Robinson‐Hamm JN, Page JL, Brozovich F, Weiss RS, Tian R, Murry CE, Regnier M (2013) Transgenic overexpression of ribonucleotide reductase improves cardiac performance. Proc Natl Acad Sci USA 110:6187–6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moussavi‐Harami F, Razumova MV, Racca AW, Cheng Y, Stempien‐Otero, Regnier M (2015) 2‐Deoxy adenosine triphosphate improves contraction in human end‐stage heart failure. J Mol Cell Cardiol 79:256–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cheng Y, Hogarth KA, O'Sullivan ML, Regnier M, Pyle WG (2016) 2‐Deoxy‐adenosine triphosphate restores the contractile function of cardiac myofibril from adult dogs with naturally occurring dilated cardiomyopathy. Am J Physiol Heart Circ Physiol 310:H80–H91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Preller M, Manstein DJ (2013) Myosin structure, allostery, and mechano‐chemistry. Structure 21:1911–1922. [DOI] [PubMed] [Google Scholar]
- 14. Geeves MA, Holmes KC (2005) The molecular mechanism of muscle contraction. Adv Prot Chem 71:161–193. [DOI] [PubMed] [Google Scholar]
- 15. Smith CA, Rayment I (1996) X‐ray structure of the magnesium(II).ADP.vanadate complex of the Dictyostelium discoideum myosin motor domain to 1.9 A resolution. Biochemistry 35:5404–5417. [DOI] [PubMed] [Google Scholar]
- 16. Kintses B, Gyimesi M, Pearson DS, Geeves MA, Zeng W, Bagshaw CR, Málnási‐Csizmadia A (2007) Reversible movement of switch 1 loop of myosin determines actin interaction. Embo J 26:265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Naber N, Purcell TJ, Pate E, Cooke R (2007) Dynamics of the nucleotide pocket of myosin measured by spin‐labeled nucleotides. Biophys J 92:172–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cecchini M, Alexeev Y, Karplus M (2010) Pi release from myosin: a simulation analysis of possible pathways. Structure 18:458–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Geeves MA, Holmes KC (1999) Structural mechanism of muscle contraction. Annu Rev Biochem 68:687–728. [DOI] [PubMed] [Google Scholar]
- 20. Preller M, Holmes KC (2013) The myosin start‐of‐power stroke state and how actin binding drives the power stroke. Cytoskeleton 70:651–660. [DOI] [PubMed] [Google Scholar]
- 21. Furch M, Fujita‐Becker S, Geeves MA, Holmes KC, Manstein DJ (1999) Role of the salt‐bridge between switch‐1 and switch‐2 of Dictyostelium myosin. J Mol Biol 290:797–809. [DOI] [PubMed] [Google Scholar]
- 22. Reubold TF, Eschenburg S, Becker A, Kull FJ, Manstein DJ (2003) A structural model for actin‐induced nucleotide release in myosin. Nat Struct Biol 10:826–830. [DOI] [PubMed] [Google Scholar]
- 23. Cecchini M, Houdusse A, Karplus M (2008) Allosteric communication in myosin V: from small conformational changes to large directed movements. PLoS Comput Biol 4:e1000129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Llinas P, Isabet T, Song L, Ropars V, Zong B, Bensity H, Siriqu S, Morris C, Kikuit C, Safer D, Sweeney HL, Houdusse A (2015) How actin initiates the motor activity of myosin. Dev Cell 33:401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Muretta JM, Rohde JA, Johnsrud DO, Cornea S, Thomas DD (2015) Direct real‐time detection of the structural and biochemical events in the myosin power stroke. Proc Natl Acad Sci USA 112:14272–14277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Takács B, O'Neall‐Hennessey E, Hetényi C, Kardos J, Szent‐Györgyi AG, Kovács M (2011) Myosin cleft closure determines the energetics of the actomyosin interaction. Faseb J 25:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang Y, Gourinath S, Kovács M, Nyitray L, Reutzel R, Himmel DM, O'Neall‐Hennessey E, Reshetnikova L, Szent‐Györgyi AG, Brown JH, Cohen C (2007) Rigor‐like structures from muscle myosins reveal key mechanical elements in the transduction pathways of this allosteric motor. Structure 15:553–564. [DOI] [PubMed] [Google Scholar]
- 28. Fisher AJ, Smith CA, Thoden JB, Smith R, Sutoh K, Holden HM, Rayment I (1995) X‐ray structures of the myosin motor domain of Dictyostelium discoideum complexed with MgADP.BeFx and MgADP.AlF4. Biochemistry 34:8960–8972. [DOI] [PubMed] [Google Scholar]
- 29. Lorenz M, Holmes KC (2010) The actin–myosin interface. Proc Natl Acad Sci USA 107:12529–12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Behrmann E, Müller M, Penczek PA, Mannherz HG, Manstein DJ, Raunser S (2012) Structure of the rigor actin–tropomyosin–myosin complex. Cell 150:327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Furch M, Geeves MA, Manstein DJ (1998) Modulation of actin affinity and actomyosin adenosine triphosphatase by charge changes in the myosin motor domain. Biochemistry 37:6317–6326. [DOI] [PubMed] [Google Scholar]
- 32. Joel PB, Trybus KM, Sweeney HL (2001) Two conserved lysines at the 50/20‐kDa junction of myosin are necessary for triggering actin activation. J Biol Chem 276: 2998–3003. [DOI] [PubMed] [Google Scholar]
- 33. Joel PB, Sweeney HL, Trybus KM (2003) Addition of lysines to the 50/20 kDa junction of myosin strengthens weak binding to actin without affecting the maximum ATPase activity. Biochemistry 42:9160–9166. [DOI] [PubMed] [Google Scholar]
- 34. Homsher E, Wang F, Sellers JR (1992) Factors affecting movement of F‐actin. [JOURNAL INFO]. [DOI] [PubMed]
- 35. Regnier M, Rivera AJ, Chen Y, Chase PB (2000) 2‐deoxy‐ATP enhances contractility of rat cardiac muscle. Circ Res 86:1211–1217. [DOI] [PubMed] [Google Scholar]
- 36. Lundy SD, Murphy SA, Dupras SK, Dai J, Murry CE, Laflamme MA, Regnier M (2014) Cell‐based delivery of dATP via gap junctions enhances cardiac contractility. J Mol Cell Cardiol [VOL:PAGE #S]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Levitt M, Hirshberg M, Sharon R, Daggett V (1995) Potential energy function and parameters for simulations of the molecular dynamics of proteins and nucleic acids in solution. Comput Phys Commun 91:215–231. [Google Scholar]
- 38. Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S, Windus TL, Dupuis M, Montgomery JA (1993) General atomic and molecular electronic structure system. J Comput Chem 14:1347–1363. [Google Scholar]
- 39. Eswar N, Webb B, Marti‐Renom MA, Madhusudhan MS, Eramian D, Shen M‐Y, Pieper U, Sali A (2007) Comparative protein structure modeling using MODELLER. Curr Protoc Prot Sci Chapter 2:Unit 2.9. [DOI] [PubMed]
- 40. Martí‐Renom MA, Stuart AC, Fiser A, Sánchez R, Melo F, Sali A (2000) Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct 29:291–325. [DOI] [PubMed] [Google Scholar]
- 41. Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234:779–815. [DOI] [PubMed] [Google Scholar]
- 42. Fiser A, Do RK, Sali A (2000) Modeling of loops in protein structures. Protein Sci 9:1753–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Beck DA, Daggett V (2004) Methods for molecular dynamics simulations of protein folding/unfolding in solution. Methods 34:112–120. [DOI] [PubMed] [Google Scholar]
- 44. Beck DAC, McCully ME, Alonso DOV, Daggett V (2000. −2017). in lucem Molecular Mechanics (ilmm). Computer Program. University of Washington, Seattle.
- 45. Levitt M, Hirshberg M, Sharon R, Laidig KE, Daggett V (1997) Calibration and testing of a water model for simulation of the molecular dynamics of proteins and nucleic acids in solution. J Phys Chem B 101:5051–5061. [Google Scholar]
- 46. Kell GS (1967) Precise representation of volume properties of water at one atmosphere. J Chem Eng Data 12:66–69. [Google Scholar]
- 47. Beck DAC, Armen RS, Daggett V (2005) Cutoff size need not strongly influence molecular dynamics results for solvated polypeptides. Biochemistry 44:609–616. [DOI] [PubMed] [Google Scholar]
- 48. Lee B, Richards FM (1971) The interpretation of protein structures: estimation of static accessibility. J Mol Biol 55:379. IN4. [DOI] [PubMed] [Google Scholar]
- 49. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 50. Humphrey W, Dalke A, Schulten K (1996) VMD – Visual Molecular Dynamics. J Mol Graph 14:33–38. [DOI] [PubMed] [Google Scholar]
- 51. Eargle J, Wright D, Luthey‐Schulten Z (2006) Multiple alignment of protein structures and sequences for VMD. Bioinformatics 22:504–506. [DOI] [PubMed] [Google Scholar]
- 52. Racca AW, Beck AE, Rao VS, Flint GV, Lundy SD, Born DE, Bamshad MJ, Regnier M (2013) Contractility and kinetics of human fetal and human adult skeletal muscle. J Physiol 591:3049–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Razumova MV, Shaffer JF, Tu A‐Y, Flint GV, Regnier M, Harris SP (2006) Effects of the N‐terminal domains of myosin binding protein‐C in an in vitro motility assay: evidence for long‐lived cross‐bridges. J Biol Chem 281:35846–35854. [DOI] [PubMed] [Google Scholar]
- 54. Margossian SS, Lowey S (1982) Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzymol 85:55–71. [DOI] [PubMed] [Google Scholar]
- 55. Kron SJ, Toyoshima YY, Uyeda TQ, Spudich JA (1991) Assays for actin sliding movement over myosin‐coated surfaces. Methods Enzymol 196:399–416. [DOI] [PubMed] [Google Scholar]
- 56. Pardee JD, Spudich JA (1982) Purification of muscle actin. Methods Enzymol 85:164–181. [DOI] [PubMed] [Google Scholar]
- 57. Potter JD (1982) Preparation of troponin and its subunits. Methods Enzymol 85:241–263. [DOI] [PubMed] [Google Scholar]
- 58. Kron SJ, Uyeda TQ, Warrick HM, Spudich JA (1991) An approach to reconstituting motility of single myosin molecules. J Cell Sci Suppl 14:129–133. [DOI] [PubMed] [Google Scholar]
- 59. Clemmens EW, Entezari M, Martyn DA, Regnier M (2005) Different effects of cardiac versus skeletal muscle regulatory proteins on in vitro measures of actin filament speed and force. J Physiol 566:737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gordon AM, LaMadrid MA, Chen Y, Luo Z, Chase PB (1997) Calcium regulation of skeletal muscle thin filament motility in vitro. Biophys J 72:1295–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information