

Abstract
Cholangiocarcinoma (CCA) is a highly aggressive epithelial malignancy still carrying a dismal prognosis, owing to early lymph node metastatic dissemination and striking resistance to conventional chemotherapy. Although mechanisms underpinning CCA progression are still a conundrum, it is now increasingly recognized that the desmoplastic microenvironment developing in conjunction with biliary carcinogenesis, recently renamed tumor reactive stroma (TRS), behaves as a paramount tumor-promoting driver. Indeed, once being recruited, activated and dangerously co-opted by neoplastic cells, the cellular components of the TRS (myofibroblasts, macrophages, endothelial cells and mesenchymal stem cells) continuously rekindle malignancy by secreting a huge variety of soluble factors (cyto/chemokines, growth factors, morphogens and proteinases). Furthermore, these factors are long-term stored within an abnormally remodeled extracellular matrix (ECM), which in turn can deleteriously mold cancer cell behavior. In this review, we will highlight evidence for the active role played by reactive stromal cells (as well as by the TRS-associated ECM) in CCA progression, including an overview of the most relevant TRS-derived signals possibly fueling CCA cell aggressiveness. Hopefully, a deeper knowledge of the paracrine communications reciprocally exchanged between cancer and stromal cells will steer the development of innovative, combinatorial therapies, which can finally hinder the progression of CCA, as well as of other cancer types with abundant TRS, such as pancreatic and breast carcinomas.
Keywords: Tumor microenvironment, Desmoplasia, Cancer-associated fibroblast, Inflammation, Tumor-associated macrophage, Lymphatic endothelial cell, Mesenchymal stem cell, Extracellular matrix
Core tip: Cholangiocarcinoma (CCA) is a typically worrisome malignancy, whose incidence has been steadily increasing. In CCA, as cancerous lesions are emerging, the surrounding stroma gradually undergoes a pathological remodeling, eventually becoming a paramount determinant of tumor growth and dissemination. Indeed, the different cell types populating the tumor microenvironment, also referred to as tumor reactive stroma, enable CCA cells to develop an aggressive phenotype, due to the secretion of a multitude of soluble factors. Therefore, functional insights into the harmful relationship between cancer and reactive stromal cells are of utmost importance, in order to unveil novel molecular targets amenable of therapeutic intervention.
INTRODUCTION
Cholangiocarcinoma (CCA) is a deadly malignancy originating from the epithelial cells lining the biliary tree, including the extrahepatic and intrahepatic portions either. Among primary liver cancers, it represents the second most common type after hepatocellular carcinoma, and its incidence and mortality rate have been steadily increasing for two decades. CCA still carries a very dismal prognosis (less than 5% of patients survives up to 5 years from diagnosis), due to a striking resistance to chemotherapy and a propensity for early intrahepatic or lymph node metastatization, making radical surgery suitable to less than one-third of patients. Furthermore, results from both surgical resection and liver transplant are limited by the high recurrence rates. To date, the pathophysiological mechanisms underlying CCA progression remain largely unknown, and consequently, the development of new effective treatments is a very awkward task[1-3].
Whilst the majority of CCAs are thought to be sporadic, several geographically heterogeneous risk factors have been identified, mostly related to an inflammatory bile duct injury. They include hepatobiliary fluke infestations (e.g., Opisthorchis viverrini, Clonorchis sinensis), hepatolithiasis, congenital abnormalities of the bile ducts (e.g., Caroli’s disease, choledochal cysts), primary sclerosing cholangitis (PSC), viral hepatitis B and C, and exposure to toxic compounds (e.g., thorium dioxide, naphthenic acids). Furthermore, CCA development has been associated with genetic and epigenetic alterations in well-known proto-oncogenes (e.g., KRAS GTPase, isocitrate dehydrogenases 1 and 2) and tumor suppressor genes (e.g., tumor protein p53, cyclin dependent kinase inhibitor 2A). More recent evidence indicates that cholangiocarcinogenesis is driven by chronic deregulation of various signaling pathways deeply involved in cholangiocyte biology, leading to uncontrolled proliferation, evasion of apoptosis, and loss of genome integrity. For instance, increased activity of cytokines and growth factors, such as interleukin (IL)-6, transforming growth factor (TGF)-β, tumor necrosis factor (TNF)-α, and platelet-derived growth factor (PDGF), is a common event in CCA, due to either enhanced production or increased cell responsiveness, and likely contributes to the malignant transformation of cholangiocytes[4,5].
Evidence is mounting that the aggressive behavior of CCA is greatly influenced by paracrine cues released from the inflammatory and mesenchymal cell types populating the tumor microenvironment[6]. Indeed, as cancerous lesions are emerging, the surrounding stroma gradually develops profound and complex changes, undergoing a switch from key player in tissue homeostasis to pathological niche supporting tumor growth and dissemination[7,8]. Therefore, an in-depth insight into the actual contribution of the stromal microenvironment to CCA progression is imperative, with the ultimate goal to pave the way for innovative combinatorial treatments targeting both stromal and cancer cells. Hopefully, this may lead to a more effective management of this devastating malignancy.
THE TUMOR REACTIVE STROMA: A SPECIALIZED COMPARTMENT ORCHESTRATING TUMOR PROGRESSION
Neoplastic bile ducts are tightly enveloped by a striking and diffuse desmoplastic, hypovascularized stroma that is usually referred to as tumor reactive stroma (TRS). This histopathological lesion is made up of a variety of stromal cells embedded in a dense collagenous extracellular matrix (ECM), encompassing myofibroblasts, inflammatory cells, endothelial cells and mesenchymal stem cells (MSCs)[6,9]. Once recruited, activated and co-opted by malignant cholangiocytes, the cellular components of the TRS can diffusely infiltrate the growing tumor and eventually support its progression by secreting a wide range of soluble factors. Indeed, these factors can directly trigger the emergence of malignant phenotypes and/or enhance the migration and aberrant activation of other stromal cells[10,11]. In addition to the plethora of cytokines, chemokines, growth factors and proteinases perpetually released by stromal cells, cell-extrinsic factors, such as hypoxia and abnormally remodeled ECM components, also provide the TRS with invasiveness-promoting properties[12]. Interestingly, it has been proposed that the pro-tumorigenic functions of the TRS could partially rely on the induction of epigenetic, and therefore heritable, changes in cancer cells[13,14]. In fact, gastric, ovarian and breast cancer cell lines co-cultured with fibroblasts isolated from the tumor milieu, were found to undergo gene-specific DNA hypermethylation events, generally coupled with increased migratory abilities[15-17].
Development of CCA is often associated with inflammation-related alterations, as observed in those cases arising in specific disease settings, such as PSC and congenital hepatic fibrosis. Moreover, recent observations indicate that in the last few years CCA is more often detected on a background of chronic inflammation associated to cirrhosis, regardless of its etiology[18]. As a general concept, the TRS may be regarded as an aberrant, over-healing reparative complex (“wound that does not heal” according to the old Dvorak’s paradigm), wherein various types of inflammatory and stromal cells are somehow hijacked by the malignant compartment, whose immunomodulatory functions and metabolic needs eventually prevail over the physiological homeostasis of the normal tissue[6,19]. This behavior also reflects the inherent plasticity of the naïve stroma, which enables it to comply quickly the evolution of the adjacent transformed epithelium, in contrast with the self-limited response occurring in the wound repair[8].
FUNCTIONAL INSIGHTS INTO THE INFLUENCE OF THE TRS ON CCA PROGRESSION
In CCA, increasing evidence has highlighted the prognostic relevance of the molecular alterations related to the generation of the TRS. Indeed, gene expression profiling of microdissected stroma from both tumoral and peritumoral areas of resected human CCA revealed a TRS-specific gene signature, encompassing 1073 genes involved in cell metabolism, cell cycle, cell signaling pathways and ECM biology. In particular, the overexpression of representative genes (namely KIAA0101, TGF-β2, laminin subunit γ2 and osteopontin) was found to significantly correlate, at different levels, with clinic-pathological features of CCA patients[20]. Andersen et al[21] undertook a similar approach in order to compare the epithelial and stromal transcriptomic profiles in CCA tissues. They identified a stromal gene signature associated with poor clinical outcome. Interestingly, the 1442 differentially expressed genes revealed a stromal dysregulation of both chemokine (CXCR4, CCR7, CCL2, CCL5, CCL19, CCL21) and IL (IL3RA, IL7R, IL10RA, IL18RAP, IL6, IL16, IL33) receptors and ligands. In the next paragraphs, we will discuss in detail how the main components of the TRS are supposed to promote CCA progression.
CANCER-ASSOCIATED FIBROBLASTS
The TRS is predominantly composed by a subpopulation of activated fibroblasts, called cancer-associated fibroblasts (CAFs). In stark contrast with the small number of lowly proliferating fibroblasts populating the naïve stroma, CAFs are present in an exaggerated high number, and exhibit a permanent state of activation, resulting in a broad release of both biochemical signals and ECM components, in particular fibronectin and collagen type I[6,10,12,22]. The main phenotypic markers of CAFs are alpha-smooth muscle actin (α-SMA), vimentin, S100A4 (also called fibroblast specific protein-1) and fibroblast activation protein alpha (FAP)[23]. CAFs are recognized as a heterogeneous population, likely reflecting the variety of cell precursors that they are supposed to originate from, including hepatic stellate cells (HSCs), portal fibroblasts and, to a lesser extent, bone marrow-derived MSCs[10]. The hypothesis that cancer cells themselves may represent an alternative source of CAFs by undergoing epithelial-to-mesenchymal transition (EMT) has gradually waned[14,24]. Nevertheless, neoplastic cells act in concert with inflammatory cells to secrete a vast array of growth factors, cytokines and chemokines ultimately responsible for the recruitment of fibroblasts to the TRS, as well as for their chronic activation state. In this regard, we recently showed that PDGF-DD is overexpressed by CCA cells under the effect of hypoxia, and acts as a key mediator of fibroblast recruitment nearby the tumoral mass. Indeed, PDGF-DD strongly induces fibroblast migration by binding its cognate receptor PDGFRβ (which is extensively expressed by CAFs), thereby activating the Rho GTPases, Rac1 (lamellipodia inducer) and Cdc42 (filopodia inducer), as well as the JNK pathway[24]. Furthermore, conditioned medium from CCA cells sustained the activation of both HSCs and liver myofibroblasts, which actually acquired a more elongated shape and up-regulated the expression of α-SMA, in vitro[25,26]. Among the multitude of soluble factors triggering the persistent activation of CAFs, TGF-β, fibroblast growth factor and, again, several PDGF family members, undoubtedly play a pivotal role[6,12].
Evidence for the pro-neoplastic effects exerted by CAFs
In CCA samples, the expression of α-SMA is barely detectable in fibroblasts populating the peritumoral areas, whereas most, if not all, fibroblasts embedded in the tumor stroma are α-SMA+[27]. Consistently, in a hamster model of CCA, the density of α-SMA+ fibroblasts within liver tissue was clearly shown to increase during cholangiocarcinogenesis[28]. There is a strong evidence that an increased density of CAFs within the TRS correlates with increased tumor growth and poor outcome in CCA patients. Indeed, high stromal expression of α-SMA was reported as an independent prognostic factor for overall and disease-free survival[27,29]. In line with these findings, both incubation of CCA cells with CAF conditioned medium and co-culture of CCA cells with CAFs resulted in increased cancer cell proliferation and migration, in vitro[27,30]. On the contrary, slighter pro-tumorigenic effects were elicited by liver fibroblasts isolated from the peritumoral areas, arguing for a deep biological gap between CAFs and their naïve counterpart (see below)[27]. Of further interest, Campbell et al[31] developed a three-dimensional organotypic culture model of CCA by embedding together within a collagen gel matrix clonal strains of CCA cells and CAFs, both derived from a syngeneic rat model of CCA generated by orthotopic inoculation of spontaneously transformed cholangiocytes. Clearly, these culture conditions more accurately reproduce the complex biological interactions occurring in vivo within the desmoplastic tumor. Interestingly, the authors observed that CCA cells co-cultured with CAFs exhibited markedly distinct growth features as compared to CCA cells cultured alone. In particular, the number of duct-like structures formed in the gel matrix by CCA cells dramatically increased in direct proportion to initial CAFs plating density. The in vitro ability of primary and established HSCs (that is, major CAF precursors) to boost CCA proliferation, survival and migration/invasion has been widely reported as well[25,29,32-36]. Moreover, it was shown that co-transplantation of CCA cells with either HSCs or liver myofibroblasts in immunodeficient mice resulted in accelerated tumor growth, compared with mice inoculated with cancer cells alone[25,26]. On the other hand, in a syngeneic rat model of CCA, selective CAF depletion in the tumor microenvironment, obtained by unleashing the specific CAF pro-apoptotic protein Bax by navitoclax, suppressed tumor growth and improved host survival[37]. Overall, these data indicate that myofibroblastic-like cells populating the tumor stroma are leading actors in fueling CCA progression.
Molecular players underlying the tumor-promoting effects of CAFs
Gene expression profiling of CAFs from human CCA samples revealed profound genetic changes as compared to normal liver fibroblasts. Most of the differentially expressed genes are involved in cell metabolism, likely reflecting the biologically active role of CAFs in supporting tumor growth. In addition, some of the up-regulated genes encode secreted proteins exerting pro-tumorigenic functions in multiple carcinomas (i.e., amphiregulin, epiregulin, Jagged 1, PDGF-AA, periostin, secretogranin 2 and ADAM metallopeptidase domain 12), thus emerging as potential candidates underlying the harmful cross-talk between CAFs and CCA cells[38]. Below, we will summarize the most prominent CAF-derived molecules fostering CCA aggressiveness. It is also worth noting that, beyond paracrine soluble factors, extracellular vesicles, especially exosomes, nano-sized molecular shuttles of about 40-100 nm of diameter, are also claimed to mediate the paracrine communications between cancer cells and neighboring stromal components. Indeed, exosomes can transfer functional proteins, lipids and nucleic acids from one cell to another, thereby modulating gene expression programs[11,19]. In this regard, it was recently showed that microRNA-loaded vesicles derived from myofibroblastic-like cells can selectively target CCA cells, thus influencing their neoplastic properties, both in vitro and in vivo[39]. However, a detailed characterization of their cargo is still missing, thus further studies are needed to better elucidate their role in tumor progression.
IL-1β: Chemokines can be secreted by many cell types, such as epithelial cells, fibroblasts and endothelial cells, either constitutively or upon inflammatory conditions. Besides their role in the immune system, chemokines are also implicated in tumor biology, owing to their ability to recruit specific subsets of leukocytes, stimulate angiogenesis, and directly promote cancer cell proliferation and invasiveness in an autocrine or paracrine fashion[40]. In particular, a mass spectrometry analysis of conditioned media from co-cultures of CCA cells and HSCs revealed a striking increase in C-X-C chemokine ligand (CXCL)5 production by cancer cells, as compared to mono-culture media. Consistently, CXCL5 expression by neoplastic bile ducts positively correlated with stromal expression of α-SMA, overall suggesting its active role in the interplay between tumor and stroma. In particular, IL-1β, a paramount inflammatory cytokine, has been pointed out as the most likely HSC-derived inducer of CXCL5 production. Interestingly, IL-1β secretion by HSCs can be further enhanced by CCA cells themselves through paracrine signals. Autocrine production of CXCL5 promotes CCA cell proliferation, migration and invasion, by activating phosphatidylinositol 3-kinase (PI3K)/Akt and extracellular signal-regulated kinases (ERK)1/2 pathways in a CXCR2-dependent manner, in vitro[41]. Moreover, CXCL5 provides cancer cells with the ability to massively recruit tumor-infiltrating neutrophils, which in turn enhance CCA growth and invasiveness, in vivo[42]. In line with these findings, high CXCL5 expression negatively affected the overall survival of CCA patients[41,42].
Stromal cell-derived factor 1: Stromal cell-derived factor (SDF)-1, also known as CXCL12, acts as ligand for the G protein-coupled receptors CXCR4 and CXCR7. SDF-1 binding to its receptors triggers a variety of downstream signaling pathways, governing cell proliferation, survival and chemotaxis. Besides its well-established role in embryogenesis and tissue homeostasis, the SDF-1/CXCR4 axis is also diffusely implicated in the pathogenesis of autoimmune and inflammatory diseases, as well as in cancer progression[43]. In CCA, SDF-1 is solely expressed by CAFs, and not by cancer cells, which overexpress its cognate receptor CXCR4. In contrast, fibroblasts in the peritumoral stroma weakly express SDF-1, suggesting that SDF-1 expression may markedly increase following their recruitment to the TRS, likely upon angiotensin II stimulation[33]. SDF-1 secretion by cultured HSCs was demonstrated to enhance CCA cell survival and invasiveness (along with EMT-like changes), via up-regulation of the anti-apoptotic protein Bcl-2, and activation of ERK1/2 and PI3K/Akt pathways, respectively[32,33]. In addition, SDF-1 could also promote the activation and proliferation of HSCs in an autocrine fashion, thus supporting further CAF enrichment. Consistent with these data, high stromal expression of SDF-1 predicted poor prognosis in CCA patients[33]. Noteworthy, CCA cells become hyper-responsive to SDF-1 due to the overexpression of CXCR4, likely induced by either TNF-α released from TAMs[44] or hepatocyte growth factor produced by CAFs[31]. This clearly outlines the wide web of communications sustaining the pro-tumorigenic function of the TRS, allowing multidirectional paracrine loops among its different cellular components, which support each other in speeding up tumor progression.
PDGF-BB: PDGF family includes five dimeric ligands (PDGF-AA, -BB, -AB, -CC, -DD), acting via two receptor tyrosine kinases (PDGFRα and PDGFRβ). The PDGF/PDGFR system is involved in various biological processes requiring mesenchymal cell activation, mostly related to tissue repair and wound healing. Moreover, overexpression of PDGF ligands and receptors has been documented in a huge variety of epithelial cancers, and usually predicts poor outcome[45]. Among growth factors commonly produced by cultured HSCs, PDGF-BB is one of the most abundantly expressed. HSCs secrete PDGF-BB at much higher levels compared with CCA cells, which, from their side, express its cognate receptor PDGFRβ. Co-culture experiments demonstrated that HSC-derived PDGF-BB promoted CCA cell resistance to TNF-related apoptosis-inducing ligand-mediated apoptosis, by activating the Hedgehog (Hh) signaling cascade[35,36], a morphogen pathway directing several cholangiocyte functions critical for liver repair[46,47]. Specifically, PDGF-BB binding to PDGFRβ increases intracellular levels of cyclic adenosine monophosphate, resulting in a protein kinase A-dependent translocation of the Hh signaling activator Smoothened (SMO) to the plasma membrane, which eventually leads to the activation of GLI transcription factors[35]. Importantly, both cyclopamine (SMO inhibitor) and imatinib mesylate (PDGFRβ inhibitor) were able to reduce tumor growth by promoting cancer cell apoptosis in an orthotopic syngeneic rat model of CCA[35,36]. Kim et al[34] further confirmed that paracrine signals from HSCs (which, actually, may include Sonic Hh as well) are of paramount importance for the activation of Hh signaling within CCA cells, whereas autocrine activation only plays a minor role. Furthermore, they also outlined the involvement of Hh signaling in CCA cell proliferation, migration and invasiveness.
Heparin-binding epidermal growth factor: In CCA, overexpression of epidermal growth factor receptor (EGFR) is one of the most common genetic aberrations, and, most relevantly, it was associated with poor survival and tumor recurrence after resection[48]. In CCA xenografts derived from subcutaneous co-injection of cancer cells and liver myofibroblasts, EGFR activation was shown to promote tumor growth and metastasis, and, above all, to be strictly dependent on the presence of activated fibroblasts. Indeed, cultured myofibroblasts secrete high amounts of heparin-binding epidermal growth factor (HB-EGF), a well-known EGFR ligand, thereby triggering the activation of EGFR signaling in CCA cells, in vitro. The HB-EGF/EGFR axis promotes CCA cell proliferation, migration and invasion, along with EMT-like changes, through activation of signal transducer and activator of transcription (STAT)-3 and ERK1/2 pathways. Of note, HB-EGF expression in fibroblasts can be further enhanced by TGF-β1 released from CCA cells, whose production is in turn triggered by EGFR activation, thus outlining the presence of a self-perpetuating paracrine loop[26].
The deleterious interplay between CAFs and endothelial cells: emerging evidence
Importantly, the paracrine signals released by CAFs not only directly exacerbate the malignancy of cancer cells, but also participate in the recruitment of other stromal components, including inflammatory cells and endothelial cells, thereby further supporting cancer growth and progression[6]. In particular, we recently unveiled that CAFs may cooperate with CCA cells in driving the development of a rich lymphatic vasculature within the tumor stroma. CCA is characterized by a striking expansion of the intratumoral and peritumoral lymphatic vessels, which represents a key determinant of the early metastasization to the regional lymph nodes, often precluding curative surgery[6]. Consistently, a high lymphatic microvessel density in CCA tissues correlated with significantly reduced overall and disease-free survival of patients[49]. Our recent findings demonstrated that, within the TRS, lymphatic endothelial cells (LECs) localize in close spatial relationship with either CCA cells or CAFs. Indeed, besides recruiting fibroblasts around the neoplastic ducts, PDGF-DD produced by CCA cells can also provide CAFs with the ability to secrete lymphangiogenic growth factors, namely vascular endothelial growth factor (VEGF)-A and VEGF-C, which eventually promote the recruitment of LECs, along with their tubular assembly in highly anastomosed structures[50]. Overall, these observations are consistent with the concept that CAFs are able to generate a pro-invasive microenvironment conducive to the lymphatic metastatic behavior of CCA.
It is important to note that in CCA, the large expansion of the lymphatic vasculature is not paralleled by an equal increase in blood vessels[6]. Nevertheless, angiogenesis has been also associated with a high risk of recurrence after surgery[51]. In this regard, it is likely that CAFs, especially those originated from HSCs, contribute to generate a pro-angiogenic microenvironment, as reported in other cancer types[52]. Indeed, HSCs likely behave as liver-specific pericytes, participating to vascular remodeling during both liver regeneration and tumor-associated angiogenesis. In this context, PDGF has been pinpointed as a relevant player. Specifically, PDGF ligands (especially PDGF-BB) released from the vascular endothelium are able to drive the recruitment of PDGFR-β-expressing HSCs, along with their subsequent adhesion to the vessel wall, similar to what occurring in embryogenesis. From their side, activated HSCs promote vascular tube formation by secreting VEGF ligands and angiopoietins under the effect of hypoxia, and mechanically stabilize the sprouting vessels by providing a tight envelope around the sinusoidal endothelial cell layer[53-56].
TUMOR-ASSOCIATED MACROPHAGES
Among the several immune cell types populating the TRS, macrophages are the most represented. Tumor-associated macrophages (TAMs) are mainly derived from circulating monocytes (CD14+/CD16+), rather than from resident macrophages (CD68+) or Kupffer cells in the liver. They are efficiently recruited to the tumor mass by a range of chemoattractants variably secreted by neoplastic and stromal cells, including C-C motif ligand (CCL) chemokines [e.g., monocyte chemoattractant protein (MCP)-1, also known as CCL2], colony stimulating factor (CSF)-1 and VEGF[6,57,58]. For instance, CAFs, especially FAP+ CAFs, are a major source of MCP-1[59]. In contrast to T cells, which can exert both tumor-promoting and tumor-suppressive functions, TAMs are almost exclusively implicated in boosting cancer aggressiveness, a function exemplified by their predominant localization at the tumor front. TAMs mostly display a M2 (or alternatively activated) phenotype, manipulated by paracrine signals originating from both malignant cells and specific subsets of T cells (including IL-10, CSF-1 and TGF-β), as well as by tumor hypoxia. Pro-tumorigenic effects of the M2 phenotype rely on a range of properties, including limited antigen-presenting functions, strong tissue remodeling and immune tolerance abilities, and production of pro-angiogenic and pro-lymphangiogenic growth factors; furthermore, TAMs directly provide cancer cells with pro-migratory inputs. TAMs are characterized by low expression of major histocompatibility complex class II molecules and IL-12, and high expression of IL-10, arginase-1 and multiple scavenging, mannose, and galactose receptors. Conversely, the so-called classically activated M1 macrophages, which are usually less represented within the TRS, possess strong antigen-presenting abilities, prime tissue destruction and anti-tumor immune responses, and possess tumoricidal activities[10,12,58,60-63].
Conditioned medium from CCA cells fostered the emergence of the M2 phenotype in cultured macrophages, which actually up-regulated the expression of the M2 specific marker CD163, as well as of the M2-related molecules IL-10 (immunosuppressive cytokine), TGF-β (pro-fibrotic cytokine), VEGF-A (pro-angiogenic growth factor) and matrix metalloproteinase (MMP)-2[64]. Growing interest has also been drawn on the interplay between TAMs and CAFs, as it was recently found that conditioned medium from HSCs affected the differentiation of macrophages, stimulating the production of pro-inflammatory (IL-6) and pro-fibrotic cytokines (TGF-β)[65]. Furthermore, within the CCA stroma, the density of M2 TAMs positively correlated with the number of regulatory T cells, suggesting that they contribute to macrophage polarization toward the pro-neoplastic phenotype[64]. Interestingly, cholangiocyte ability to finely orchestrate a macrophage-centric inflammatory response was also reported by our group in a mouse model of congenital hepatic fibrosis, a disease of the biliary epithelium at increased risk for CCA development. In this model, dysfunctional biliary epithelial cells (due to a genetic defect in the ciliary protein fibrocystin) secrete a range of chemokines (CXCL1, CXCL10, CXCL12) able to recruit and activate bone marrow-derived macrophages, which then progressively switch from an M1 to an M2 phenotype as the disease progresses[66]. However, it is worth considering that the macrophage phenotype is extremely plastic, showing a continuum of activation states, in which M1 and M2 types only represent the extreme points[62]. In line with this concept, many tumor-promoting cytokines that are actually M1 cytokines, such as IL-6, are even produced by TAMs[61]. Recently, Raggi et al[67] revealed that the CCA stem-like compartment is actively involved in both the recruitment of circulating monocytes and their differentiation into TAMs, owing to the release of IL-13, IL-34 and osteoactivin. Of note, cancer stem cell (CSC)-associated TAMs display unique phenotypic and functional features, namely mixed expression of M1 and M2 markers (e.g., M1-related chemokines CXCL9 and CXCL10, and M2-related chemokines CCL17 and CCL18), increased adhesive and invasive abilities, in vitro, and enhanced tumor-promoting functions, in vivo. This clearly highlights the existence of different TAM subsets within the tumor, depending on the multitude of microenvironmental cues originating from various cell niches.
Evidence for the pro-neoplastic effects exerted by TAMs
In CCA tissues, M2 macrophages are definitely much more abundant than in the peritumoral areas, and TAM are mostly located at the leading edge of the tumor[67]. Consistently, immunohistochemical analyses in Opisthorchis viverrini-associated CCA in a hamster model revealed a progressive, dramatic increase in M2 macrophages through carcinogenesis[28]. Studies from different groups showed that a high density of TAMs at the invasive front correlated with poor survival of CCA patients after resection[64,68,69]. However, it is important to underline that not all of these studies provided evidence that the observed TAM actually exhibited the pro-neoplastic, M2 phenotype. Whereas Subimerb et al[68] evaluated the expression of MAC387, a marker of recently infiltrated, bone marrow-derived, macrophages, Atanasov et al[69] evaluated the expression of resident, CD68+ macrophages. On the contrary, Hasita et al[64] sought to distinguish M2 TAMs from total resident macrophages based on their expression of CD163, in order to highlight their specific contribution to tumor progression. They found that, in CCA tissues, the number of CD163+ M2 cells was, as expected, lower than the number of CD68+ cells, and that high infiltration of M2 macrophages, but not of total macrophages, was significantly associated with poor disease-free survival of patients. Of further interest, the density of M2 macrophages within CCA stroma also correlated with the presence of extrahepatic metastases[28], the tumor pathological grade[67], and the microvascular density[64]. Although these findings are based on the evaluation of different phenotypic markers, overall, they suggest that TAMs strongly influence CCA progression, with a major role played by M2 TAMs, thus confirming what observed in other cancer types. In accordance with these immunohistochemical findings, conditioned medium from M2 macrophages boosted the migratory abilities of CCA cells by inducing EMT-like changes, in vitro[28].
As previously mentioned, recruitment of circulating monocytes, rather than proliferation of resident macrophages, is the mechanism responsible for TAM accumulation in the TRS[57]. In fact, in CCA patients, levels of circulating CD14+/CD16+ monocytes were increased, and correlated with high density of MAC387+ TAMs, and with poor survival rates. CD14+/CD16+ monocytes represent a minor subset of total monocytes, whose expansion is usually associated with acute or chronic inflammation. They are classically regarded as more mature cells than CD14+/CD16- monocytes, and thought to be the major precursors of tissue macrophages. Besides expressing a larger number of adhesion molecules, enabling them to strongly adhere to vascular endothelium, CD14+/CD16+ monocytes also up-regulate the EGFR ligand epiregulin, and the angiogenic chemokine CXCL3. Overall, these features are consistent with the adoption of a pro-tumorigenic phenotype, likely induced by tumor-derived molecules, which may also drive their recruitment into the TRS[70].
Molecular players underlying the tumor-promoting effects of TAMs
MMP-9: MMPs, in particular MMP-9, are the most important proteolytic enzymes in the context of tumor spread, and their overexpression tends to be predictive of worst outcome in human cancers. Besides underpinning cancer cell invasion through the selective deletion of ECM integrity, MMPs can also elicit the post-translational activation of growth factors and cytokines, thereby influencing key cellular processes[71]. Subimerb et al[68] found that TAMs (especially those located at the tumor-host interface) rather than cancer cells represent the main source of MMP-9 in CCA. Moreover, CCA patients with high numbers of MMP-9+ TAMs displayed significantly shorter survival times than those with low numbers, thus pointing out MMP-9 production as a key driver of CCA progression promoted by TAMs. Furthermore, a broad expression of other pivotal ECM remodeling-related genes, namely MMP-2, ADAM10, and ADAM17 was reported in CSC-associated TAMs[67].
TNF-α: TNF-α is a pleiotropic cytokine, acting as a central pro-inflammatory mediator in human carcinogenesis, wherein it was reported to play both anti-tumoral and pro-tumoral effects[72]. In CCA, as well as in the majority of carcinomas, TNF-α is widely expressed by macrophages located at the tumor edge, whereas it is only focally expressed by cancer cells[44]. Lipopolysaccharide (LPS)-activated macrophages were able to elicit EMT-like phenotypic changes in CCA cells (namely, down-regulation of the epithelial markers E-cadherin and cytokeratin 19, along with up-regulation of the mesenchymal markers S100A4 and MMP-9), probably mediated by a TNF-α-induced activation of Snail and ZEB2 transcription factors[73-75]. Consistently, upon TNF-α stimulation, CCA cells gained increased migratory functions in conjunction with the activation of ERK, Akt and nuclear factor (NF)-κB[74,76].
IL-6: Aberrant activation of the IL-6 classical downstream effector STAT3 is described in many epithelial cancers, and is currently regarded as a major oncogenic event[77]. For instance, in CCA patients, high expression of STAT3 by cancer cells was associated with poorly differentiated tumor phenotypes, as well as with low survival rates[78]. In particular, in CCA cells, increased cell survival by up-regulation of the anti-apoptotic protein myeloid cell leukemia-1 is the fundamental mechanism triggered by the IL-6/STAT3 axis[79]. In the hamster experimental model of Opisthorchis viverrini-induced CCA, STAT3 activation peaked at the pre-cancerous stage, in association with a high degree of inflammation. Consistently, conditioned medium from LPS-activated macrophages led to a robust STAT3 activation in CCA cells[78], likely mediated by IL-6, whose secretion is potently stimulated by LPS[73]. Although IL-6 can be secreted even by CCA cells themselves, paracrine signaling is probably essential to reach broad STAT3 activation[79], and TAMs may actually be central in this process.
YKL-40: YKL-40, also called chitinase 3-like 1, is a secreted glycoprotein, which is supposed to play key roles in different aspects of tumorigenesis, from cell proliferation and survival, to angiogenesis and ECM remodeling. Interestingly, YKL-40 serum levels are dramatically increased in patients with multiple chronic inflammatory diseases, such as liver cirrhosis, as well as in patients with several malignancies, including breast, lung and colorectal carcinomas[80]. In CCA patients, YKL-40 serum levels were actually much higher than those of healthy subjects, and also negatively correlated with overall survival. Importantly, within the tumoral area, CCA cells represent only minor contributors to YKL-40 production, which is primarily caused by infiltrating inflammatory cells, especially TAMs. Of further interest, exogenous YKL-40 stimulated CCA cell growth and migration, by triggering Akt and ERK1/2 activation[81].
Wnt3: Involvement of Wnt/β-catenin pathway in CCA pathogenesis and progression is a well-established concept for many years[82,83]. Wnt family ligands are secreted glycoproteins modulating fundamental transcriptional programs by stimulating the nuclear translocation of β-catenin. In the basal conditions, β-catenin is mainly located at the cell-cell junctions, whereas a minor pool is sequestered in the cytoplasm by a destruction complex, where phosphorylation at specific residues (Ser 33/37 and Thr 41) is a pre-requisite to allow its inactivation and proteasomal degradation. Binding of Wnt ligands to Frizzled receptors lets β-catenin to detach from the membrane, accumulate within the cytoplasm, and then translocate into the nucleus, where it interacts with several co-activators, among which T-cell factor and lymphoid enhancer-binding factor 1 are the main partner in gene regulation. β-catenin target genes encompass well-known proto-oncogenes relevant for CCA growth, such as c-Myc, cyclin D1 and ZEB1[84,85]. In CCA tissues, β-catenin is constitutively expressed at high levels either in the cytoplasm or in the nucleus of cancer cells, whereas its membranous expression is decreased, consistent with the activation of the Wnt signaling. Among Wnt ligands, whereas Wnt5a and Wnt7b are overexpressed by neoplastic bile ducts, TAMs represent a major source of Wnt3. Notably, conditioned medium from LPS-activated macrophages elicited β-catenin nuclear translocation in CCA cells, resulting in enhanced cell growth[86].
MSCs
MSCs are non-hematopoietic stem cells primarily resident in the bone marrow, where they are recruited from by chemotactic signals mainly originating from injured tissues and inflammatory sites. Indeed, MSCs are multipotent cells able to differentiate in a variety of cell types, thus being classically regarded as a valuable source of tissue replacement. However, under the influence of cancer-derived chemokines, MSCs can also home to primary tumor sites, wherein they eventually become an additional component of the tumor microenvironment. Tumor-resident MSCs have been also reported to perform several activities supporting cancer progression. For instance, they can interfere with anti-tumor immunity, promote angiogenesis, and directly enhance the aggressiveness of malignant cells through secreted factors[8,87,88].
In nude mice bearing subcutaneous human CCA xenografts, it was shown that, upon infusion into the venous circulation, MSCs were able to selectively reach both the primary tumor and the metastatic liver, thus confirming their pronounced tumor tropism. Furthermore, exposure of CCA cell to conditioned medium from MSCs resulted in increased proliferation, apoptosis resistance, and invasiveness, likely due to the activation of the Wnt/β-catenin signaling. Consistently, subcutaneous co-injection of CCA cells with MSCs in immunodeficient mice led to accelerated tumor growth, and higher incidence of liver metastases, compared with mice inoculated with cancer cells alone[87]. Interestingly, the ability of MSCs to promote CCA cell proliferation was further strengthened by preliminary exposure of MSCs to cancer cell-derived extracellular vesicles (with features consistent with exosomes). Indeed, these vesicles induced profound changes in the MSC secretome, including increased secretion of IL-6, CCL2/MCP-1, CXCL1/GRO-α, CX3CL1/Fractalkine and PDGF-AA. Besides directly favoring tumor cell growth, MSCs may also represent an additional (although minor) source of CAFs, as conditioned medium from CCA cells prompted a phenotypic switch from MSCs into myofibroblastic-like cells[88].
ECM
Besides providing a physical support to cells, the ECM (mainly consisting of collagens, glycoproteins and proteoglycans) also communicates straight with them, thereby modulating a variety of cellular functions, and acts as a paramount reservoir of cell-derived soluble factors[6]. Throughout carcinogenesis, the ECM gradually undergoes stiffening and profound compositional changes, resulting from the accumulation of secreted structural and non-structural proteins, in particular collagen type I and fibronectin, as well as of matrix modifying enzymes[19,22,89]. An abnormal ECM leads to a dysregulated behavior of both cancer and stromal cells, thereby affecting several processes related to tumor biology, including cancerous fibrogenesis, inflammation and angiogenesis[11,90]. Interestingly, ECM stiffness is emerging as a driving force behind cancer progression. As previously mentioned, tumor-associated ECM is typically stiffer than the normal matrix, due to a pathological remodeling mainly driven by neoplastic cells and CAFs. This stiff, collagen enriched ECM can signal to cells through specific mechanosensors, thus activating intracellular pathways regulating the acquisition of malignant phenotypic traits[90,91]. Among the intracellular sensors of ECM-driven mechanical stress, the transcriptional co-activator yes-associated protein (YAP) and its paralog, transcriptional co-activator with PDZ-binding motif (TAZ), are emerging as master directors of cancer cell reprogramming and enhanced invasiveness[92]. Indeed, high levels of cytoskeletal contractility, resulting from increased ECM rigidity, are generally coupled with the activation of YAP/TAZ, which can profoundly affect epithelial cell behavior, including the balance between proliferation and apoptosis[93,94]. Interestingly, in CCA, YAP overexpression was reported to enhance cancer cell proliferation, invasion (via EMT-like changes) and resistance to chemotherapeutic drugs, both in vitro and in tumor xenografts[95]. Therefore, it is tempting to speculate that, after being recruited by CCA cells through PDGF-DD[24], CAFs may gradually manipulate ECM stiffening within the TRS, thereby inducing YAP/TAZ activation in cancer cells, leading to the emergence of a particularly aggressive tumor phenotype.
In CCA, interactions between tumor cells and specific molecular components of the ECM may trigger additional pathways of tumor invasiveness. In fact, CCA cells cultured on a reconstituted basement membrane preparation (mainly composed of collagen type IV and laminin), showed enhanced invasive properties compared with cells grown on uncoated culture plates. This was dependent on the dysregulated expression of a wide range of proteins, especially L-plastin, which is an actin-bundling protein supporting cell motility and adhesion. L-plastin is dramatically up-regulated in many types of malignant cells and, in CCA tissue, it is primarily expressed at the tumor front, thereby indicating its involvement in tumor invasion[96]. The ability of the TRS-associated ECM to support cancer aggressiveness is also well exemplified by three fundamental non-structural ECM proteins, namely tenascin, periostin and osteopontin, reported as poor prognostic biomarkers for CCA patients. In CCA samples, tenascin is abnormally expressed in the intratumoral stroma, as well as at the tumor leading edge. Although CAFs undoubtedly represent the main source of tenascin, carcinoma cells can contribute to its biosynthesis. In CCA patients, aberrant deposition of tenascin at the invasive front positively correlated with tumor size and lymph node metastasis, and also predicted poor survival. It is worth noting that the expression pattern of tenascin roughly parallels that of EGFR, which tenascin can bind to, likely underpinning its tumor-promoting functions[97]. Similarly, high expression of periostin within the TRS, which is solely due to CAFs, was an independent prognostic factor for overall survival of CCA patients. Moreover, serum periostin levels were significantly higher in CCA patients compared with both healthy subjects and patients with other hepatic malignancies. Consistent with these findings, exogenous periostin induced CCA cell proliferation and invasion through its interaction with integrin receptors α5β1 and α6β4, leading to the activation of the PI3K/Akt pathway, in vitro[38,98,99]. High stromal expression of osteopontin is also an independent risk factor for reduced overall and disease-free survival in CCA patients, positively correlating with both tumor size and the presence of lymph node or macrovascular invasion[20].
THE TRS AS POTENTIAL THERAPEUTIC TARGET
Classically, anticancer therapies aim at targeting intrinsic traits of neoplastic cells, which, until recently, were actually seen as the only players deserving attention in the context of clinical management. However, in CCA, a lethal malignancy paradigmatic of the strong resistance to conventional chemotherapy, mounting evidence supports the role of tumor microenvironment in dictating tumor growth, progression and metastatic dissemination. Indeed, CCA cells establish intense, mutual, paracrine communications with neighboring stromal components, in particular CAFs and TAMs, which are a rich source of signals promoting malignancy (Figure 1). Therefore, combinatorial therapies that both directly tackle tumor growth and turn off the tumor-promoting functions of the TRS might represent an important step forward in anticancer treatment, especially in CCA. In addition to provide a number of druggable targets, TRS may help to identify (by gene expression profiling) molecular signatures serving as novel prognostic biomarkers, useful for predicting therapeutic response or monitoring tumor recurrence, as it could be the case with periostin[8,63,89]. It is worth noting that, unlike cancer cells, which undergo multiple genetic/epigenetic changes giving rise to a tremendously heterogeneous population, stromal cells represent a genetically stable, more uniform compartment, and thus stand out as viable and compelling therapeutic targets[12,37]. Basically, TRS-oriented therapeutic approaches should aim at: (1) hampering the recruitment of reactive stromal cells by counteracting tumor-derived chemokines; (2) promoting TRS depletion by eliciting apoptosis of its cellular components; (3) interfering with the intracellular pro-oncogenic pathways triggered by the TRS within the cancer cell; and (4) interfering with the paracrine communications between stromal and cancer cells, by neutralizing specific soluble factors or antagonizing their cognate receptors[6]. The study performed by Mertens et al[37] is an archetype of these potential new strategies. Using the BH3 mimetic navitoclax (a small molecule mimicking the pro-apoptotic protein Bad), the authors were able to selectively induce Bax-dependent apoptosis in CCA-derived CAFs, but not in normal fibroblasts or CCA cells, in vitro. By translating these findings in an in vivo, orthotopic syngeneic rat model of CCA, navitoclax markedly reduced tumor growth and metastasis, and significantly improved survival, an effect related to a quantitative depletion of CAFs from the stroma. Taking a different approach, the mammalian target of rapamycin inhibitor everolimus, in addition to directly reduce CCA cell proliferation and invasion[100], was reported to hamper the cross-talk between CAFs and CCA cells, by both impairing the activation of CAF-induced motogenic pathways in cancer cells, and inhibiting the secretion of tumor-promoting cyto/chemokines by CAFs[30]. Interestingly, everolimus is already an FDA-approved drug for the treatment of breast, neuroendocrine and renal cell carcinomas[101]. By turning to TAMs, it was shown that liposome-encapsulated clodronate, a selective macrophage-depleting agent, as well as GW2580 or AZD7507, small molecules preventing monocyte-to-macrophage differentiation, significantly reduced the growth of subcutaneous human CCA xenografts. Moreover, the tumor-suppressive effect of liposomal clodronate was also confirmed in a non-transgenic, thioacetamide-induced rat model of CCA, which faithfully reproduces the inflammatory and desmoplastic microenvironment associated with human CCA[102]. Noteworthy, besides priming TAMs for apoptosis or blocking monocyte recruitment, it might be possible to harness the inherent plasticity of macrophages in order to revert their polarization from the pro-neoplastic M2 phenotype to the anti-tumoral M1 phenotype[12,57]. However, the development of combinatorial therapies targeting both tumor and stromal cells must be rooted in a deep knowledge of the epithelial-mesenchymal interactions occurring within the CCA microenvironment, which is not possible without proper experimental models. In this regard, two-dimensional co-culture systems and, even more, three-dimensional organotypic culture models represent powerful tools for investigation, but, of course, they cannot fully reproduce the complexity of the TRS, which integrate a multitude of cell elements. On the other hand, rodent models of CCA more closely mimic the structural and functional heterogeneity of the TRS, even though the murine environment may not accurately reproduce the wide range of paracrine communications occurring in the human disease setting, all the more so in xenograft models, where the host is immunodeficient[8,11].
Figure 1.
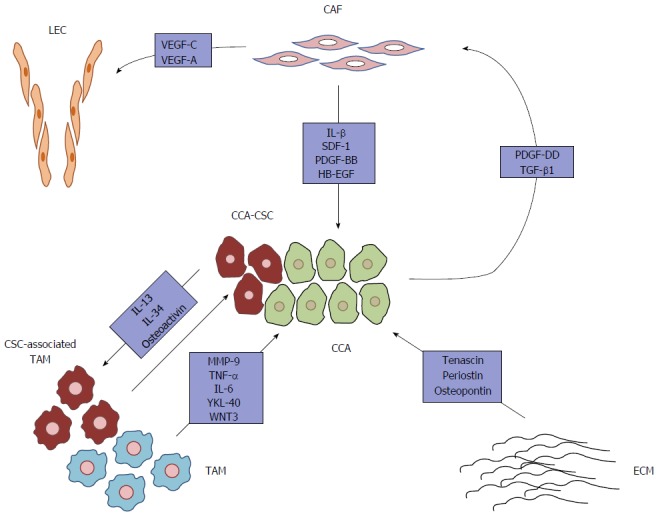
Soluble factors derived from reactive stromal cells, along with tumor-associated extracellular matrix, sustain cholangiocarcinoma cell malignancy. CCA cells shape the surrounding microenvironment to meet their highly demanding needs, thus providing CAFs and TAMs with the ability to secrete a broad range of cyto/chemokines, growth factors, morphogens and proteinases, which boost cancer cell proliferation, survival and invasiveness. In this model, CAFs are recruited by PDGF-DD released by CCA cells. In addition, TGF-β1, also derived by CCA cells, stimulates CAFs to produce the EGFR ligand, HB-EGF, which triggers the acquisition of malignant behaviors (i.e., EMT-like changes) by cancer cells. CAF-derived tumor-promoting molecules also include IL-1β, SDF-1 and PDGF-BB. Moreover, PDGF-DD induces CAFs to acquire pro-lymphangiogenic functions (exerted by VEGF-A and VEGF-C). On the other hand, TAMs, displaying a predominant M2 phenotype, also support tumor survival and invasiveness by secreting several soluble factors, including MMP-9, TNF-α, IL-6, YKL-40 and Wnt3. Of note, the CCA stem-like compartment molds a specific subset of TAMs (through secretion of IL-13, IL-34 and osteoactivin), displaying a mixed M1/M2 phenotype, to promote self-renewal and drug-resistance properties. In addition, non-structural proteins expressed by the abnormally remodeled ECM (tenascin, periostin, osteopontin) further enhance CCA aggressiveness. CAF: Cancer-associated fibroblast; CCA: Cholangiocarcinoma; CSC: Cancer stem cell; ECM: Extracellular matrix; LEC: Lymphatic endothelial cell; TAM: Tumor-associated macrophage; IL: Interleukin; TGF: Transforming growth factor; TNF: Tumor necrosis factor; PDGF: Platelet-derived growth factor; EGFR: Epidermal growth factor receptor; MMP-9: Matrix metalloproteinase 9.
CONCLUSION
Unravelling the complex mechanisms underlying the mutual interactions between the tumoral and stromal compartments is indeed a topic of great translational significance, worth being pursued further in the next future. Based on the data discussed above, specific targeting of the signals operating in the tumor microenvironment, coupled with conventional anticancer treatments, could actually open new promising and feasible therapeutic avenues in CCA, hopefully expandable to other aggressive desmoplastic epithelial malignancies, such as pancreas and breast carcinomas. It is tempting to speculate that these innovative, multitargeted therapies might more effectively eradicate tumor cells, owing to concurrent switching-off actions on intrinsic (cancer cell-dependent) as well as extrinsic (TRS-derived) tumor-promoting mechanisms, eventually leading to improved patient outcomes.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Italy
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: The authors declare no conflict of interests for this article.
Peer-review started: November 27, 2016
First decision: January 16, 2017
Article in press: February 20, 2017
P- Reviewer: Chuang WL, Sergi CM, Yildiz K S- Editor: Ji FF L- Editor: A E- Editor: Li D
References
- 1.Gatto M, Bragazzi MC, Semeraro R, Napoli C, Gentile R, Torrice A, Gaudio E, Alvaro D. Cholangiocarcinoma: update and future perspectives. Dig Liver Dis. 2010;42:253–260. doi: 10.1016/j.dld.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 2.Khan SA, Davidson BR, Goldin RD, Heaton N, Karani J, Pereira SP, Rosenberg WM, Tait P, Taylor-Robinson SD, Thillainayagam AV, et al. Guidelines for the diagnosis and treatment of cholangiocarcinoma: an update. Gut. 2012;61:1657–1669. doi: 10.1136/gutjnl-2011-301748. [DOI] [PubMed] [Google Scholar]
- 3.Zabron A, Edwards RJ, Khan SA. The challenge of cholangiocarcinoma: dissecting the molecular mechanisms of an insidious cancer. Dis Model Mech. 2013;6:281–292. doi: 10.1242/dmm.010561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al-Bahrani R, Abuetabh Y, Zeitouni N, Sergi C. Cholangiocarcinoma: risk factors, environmental influences and oncogenesis. Ann Clin Lab Sci. 2013;43:195–210. [PubMed] [Google Scholar]
- 5.Rizvi S, Gores GJ. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology. 2013;145:1215–1229. doi: 10.1053/j.gastro.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cadamuro M, Morton SD, Strazzabosco M, Fabris L. Unveiling the role of tumor reactive stroma in cholangiocarcinoma: An opportunity for new therapeutic strategies. Transl Gastrointest Cancer. 2013;2:130–144. doi: 10.3978/j.issn.2224-4778.2013.04.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coulouarn C, Clément B. Stellate cells and the development of liver cancer: therapeutic potential of targeting the stroma. J Hepatol. 2014;60:1306–1309. doi: 10.1016/j.jhep.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 8.Chen F, Zhuang X, Lin L, Yu P, Wang Y, Shi Y, Hu G, Sun Y. New horizons in tumor microenvironment biology: challenges and opportunities. BMC Med. 2015;13:45. doi: 10.1186/s12916-015-0278-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sirica AE, Gores GJ. Desmoplastic stroma and cholangiocarcinoma: clinical implications and therapeutic targeting. Hepatology. 2014;59:2397–2402. doi: 10.1002/hep.26762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leyva-Illades D, McMillin M, Quinn M, Demorrow S. Cholangiocarcinoma pathogenesis: Role of the tumor microenvironment. Transl Gastrointest Cancer. 2012;1:71–80. [PMC free article] [PubMed] [Google Scholar]
- 11.Hui L, Chen Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015;368:7–13. doi: 10.1016/j.canlet.2015.07.039. [DOI] [PubMed] [Google Scholar]
- 12.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang L, Frampton G, Liang LJ, Demorrow S. Aberrant DNA methylation profile in cholangiocarcinoma. World J Gastrointest Pathophysiol. 2010;1:23–29. doi: 10.4291/wjgp.v1.i2.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brivio S, Cadamuro M, Fabris L, Strazzabosco M. Epithelial-to-Mesenchymal Transition and Cancer Invasiveness: What Can We Learn from Cholangiocarcinoma? J Clin Med. 2015;4:2028–2041. doi: 10.3390/jcm4121958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurashige J, Mima K, Sawada G, Takahashi Y, Eguchi H, Sugimachi K, Mori M, Yanagihara K, Yashiro M, Hirakawa K, et al. Epigenetic modulation and repression of miR-200b by cancer-associated fibroblasts contribute to cancer invasion and peritoneal dissemination in gastric cancer. Carcinogenesis. 2015;36:133–141. doi: 10.1093/carcin/bgu232. [DOI] [PubMed] [Google Scholar]
- 16.Xu L, Deng Q, Pan Y, Peng M, Wang X, Song L, Xiao M, Wang Z. Cancer-associated fibroblasts enhance the migration ability of ovarian cancer cells by increasing EZH2 expression. Int J Mol Med. 2014;33:91–96. doi: 10.3892/ijmm.2013.1549. [DOI] [PubMed] [Google Scholar]
- 17.Lin HJ, Zuo T, Lin CH, Kuo CT, Liyanarachchi S, Sun S, Shen R, Deatherage DE, Potter D, Asamoto L, et al. Breast cancer-associated fibroblasts confer AKT1-mediated epigenetic silencing of Cystatin M in epithelial cells. Cancer Res. 2008;68:10257–10266. doi: 10.1158/0008-5472.CAN-08-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet. 2014;383:2168–2179. doi: 10.1016/S0140-6736(13)61903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhome R, Bullock MD, Al Saihati HA, Goh RW, Primrose JN, Sayan AE, Mirnezami AH. A top-down view of the tumor microenvironment: structure, cells and signaling. Front Cell Dev Biol. 2015;3:33. doi: 10.3389/fcell.2015.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sulpice L, Rayar M, Desille M, Turlin B, Fautrel A, Boucher E, Llamas-Gutierrez F, Meunier B, Boudjema K, Clément B, et al. Molecular profiling of stroma identifies osteopontin as an independent predictor of poor prognosis in intrahepatic cholangiocarcinoma. Hepatology. 2013;58:1992–2000. doi: 10.1002/hep.26577. [DOI] [PubMed] [Google Scholar]
- 21.Andersen JB, Spee B, Blechacz BR, Avital I, Komuta M, Barbour A, Conner EA, Gillen MC, Roskams T, Roberts LR, et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology. 2012;142:1021–1031.e15. doi: 10.1053/j.gastro.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 23.Augsten M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front Oncol. 2014;4:62. doi: 10.3389/fonc.2014.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cadamuro M, Nardo G, Indraccolo S, Dall’olmo L, Sambado L, Moserle L, Franceschet I, Colledan M, Massani M, Stecca T, et al. Platelet-derived growth factor-D and Rho GTPases regulate recruitment of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology. 2013;58:1042–1053. doi: 10.1002/hep.26384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okabe H, Beppu T, Hayashi H, Ishiko T, Masuda T, Otao R, Horlad H, Jono H, Ueda M, Phd SS, et al. Hepatic stellate cells accelerate the malignant behavior of cholangiocarcinoma cells. Ann Surg Oncol. 2011;18:1175–1184. doi: 10.1245/s10434-010-1391-7. [DOI] [PubMed] [Google Scholar]
- 26.Clapéron A, Mergey M, Aoudjehane L, Ho-Bouldoires TH, Wendum D, Prignon A, Merabtene F, Firrincieli D, Desbois-Mouthon C, Scatton O, et al. Hepatic myofibroblasts promote the progression of human cholangiocarcinoma through activation of epidermal growth factor receptor. Hepatology. 2013;58:2001–2011. doi: 10.1002/hep.26585. [DOI] [PubMed] [Google Scholar]
- 27.Chuaysri C, Thuwajit P, Paupairoj A, Chau-In S, Suthiphongchai T, Thuwajit C. Alpha-smooth muscle actin-positive fibroblasts promote biliary cell proliferation and correlate with poor survival in cholangiocarcinoma. Oncol Rep. 2009;21:957–969. doi: 10.3892/or_00000309. [DOI] [PubMed] [Google Scholar]
- 28.Thanee M, Loilome W, Techasen A, Namwat N, Boonmars T, Pairojkul C, Yongvanit P. Quantitative changes in tumor-associated M2 macrophages characterize cholangiocarcinoma and their association with metastasis. Asian Pac J Cancer Prev. 2015;16:3043–3050. doi: 10.7314/apjcp.2015.16.7.3043. [DOI] [PubMed] [Google Scholar]
- 29.Okabe H, Beppu T, Hayashi H, Horino K, Masuda T, Komori H, Ishikawa S, Watanabe M, Takamori H, Iyama K, et al. Hepatic stellate cells may relate to progression of intrahepatic cholangiocarcinoma. Ann Surg Oncol. 2009;16:2555–2564. doi: 10.1245/s10434-009-0568-4. [DOI] [PubMed] [Google Scholar]
- 30.Heits N, Heinze T, Bernsmeier A, Kerber J, Hauser C, Becker T, Kalthoff H, Egberts JH, Braun F. Influence of mTOR-inhibitors and mycophenolic acid on human cholangiocellular carcinoma and cancer associated fibroblasts. BMC Cancer. 2016;16:322. doi: 10.1186/s12885-016-2360-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Campbell DJ, Dumur CI, Lamour NF, Dewitt JL, Sirica AE. Novel organotypic culture model of cholangiocarcinoma progression. Hepatol Res. 2012;42:1119–1130. doi: 10.1111/j.1872-034X.2012.01026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gentilini A, Rombouts K, Galastri S, Caligiuri A, Mingarelli E, Mello T, Marra F, Mantero S, Roncalli M, Invernizzi P, et al. Role of the stromal-derived factor-1 (SDF-1)-CXCR4 axis in the interaction between hepatic stellate cells and cholangiocarcinoma. J Hepatol. 2012;57:813–820. doi: 10.1016/j.jhep.2012.06.012. [DOI] [PubMed] [Google Scholar]
- 33.Okamoto K, Tajima H, Nakanuma S, Sakai S, Makino I, Kinoshita J, Hayashi H, Nakamura K, Oyama K, Nakagawara H, et al. Angiotensin II enhances epithelial-to-mesenchymal transition through the interaction between activated hepatic stellate cells and the stromal cell-derived factor-1/CXCR4 axis in intrahepatic cholangiocarcinoma. Int J Oncol. 2012;41:573–582. doi: 10.3892/ijo.2012.1499. [DOI] [PubMed] [Google Scholar]
- 34.Kim Y, Kim MO, Shin JS, Park SH, Kim SB, Kim J, Park SC, Han CJ, Ryu JK, Yoon YB, et al. Hedgehog signaling between cancer cells and hepatic stellate cells in promoting cholangiocarcinoma. Ann Surg Oncol. 2014;21:2684–2698. doi: 10.1245/s10434-014-3531-y. [DOI] [PubMed] [Google Scholar]
- 35.Fingas CD, Bronk SF, Werneburg NW, Mott JL, Guicciardi ME, Cazanave SC, Mertens JC, Sirica AE, Gores GJ. Myofibroblast-derived PDGF-BB promotes Hedgehog survival signaling in cholangiocarcinoma cells. Hepatology. 2011;54:2076–2088. doi: 10.1002/hep.24588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fingas CD, Mertens JC, Razumilava N, Bronk SF, Sirica AE, Gores GJ. Targeting PDGFR-β in Cholangiocarcinoma. Liver Int. 2012;32:400–409. doi: 10.1111/j.1478-3231.2011.02687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mertens JC, Fingas CD, Christensen JD, Smoot RL, Bronk SF, Werneburg NW, Gustafson MP, Dietz AB, Roberts LR, Sirica AE, et al. Therapeutic effects of deleting cancer-associated fibroblasts in cholangiocarcinoma. Cancer Res. 2013;73:897–907. doi: 10.1158/0008-5472.CAN-12-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Utispan K, Thuwajit P, Abiko Y, Charngkaew K, Paupairoj A, Chau-in S, Thuwajit C. Gene expression profiling of cholangiocarcinoma-derived fibroblast reveals alterations related to tumor progression and indicates periostin as a poor prognostic marker. Mol Cancer. 2010;9:13. doi: 10.1186/1476-4598-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li L, Piontek K, Ishida M, Fausther M, Dranoff JA, Fu R, Mezey E, Gould SJ, Fordjour FK, Meltzer SJ, et al. Extracellular vesicles carry microRNA-195 to intrahepatic cholangiocarcinoma and improve survival in a rat model. Hepatology. 2017;65:501–514. doi: 10.1002/hep.28735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Verbeke H, Geboes K, Van Damme J, Struyf S. The role of CXC chemokines in the transition of chronic inflammation to esophageal and gastric cancer. Biochim Biophys Acta. 2012;1825:117–129. doi: 10.1016/j.bbcan.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 41.Okabe H, Beppu T, Ueda M, Hayashi H, Ishiko T, Masuda T, Otao R, Horlad H, Mima K, Miyake K, et al. Identification of CXCL5/ENA-78 as a factor involved in the interaction between cholangiocarcinoma cells and cancer-associated fibroblasts. Int J Cancer. 2012;131:2234–2241. doi: 10.1002/ijc.27496. [DOI] [PubMed] [Google Scholar]
- 42.Zhou SL, Dai Z, Zhou ZJ, Chen Q, Wang Z, Xiao YS, Hu ZQ, Huang XY, Yang GH, Shi YH, et al. CXCL5 contributes to tumor metastasis and recurrence of intrahepatic cholangiocarcinoma by recruiting infiltrative intratumoral neutrophils. Carcinogenesis. 2014;35:597–605. doi: 10.1093/carcin/bgt397. [DOI] [PubMed] [Google Scholar]
- 43.Chatterjee S, Behnam Azad B, Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res. 2014;124:31–82. doi: 10.1016/B978-0-12-411638-2.00002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohira S, Sasaki M, Harada K, Sato Y, Zen Y, Isse K, Kozaka K, Ishikawa A, Oda K, Nimura Y, et al. Possible regulation of migration of intrahepatic cholangiocarcinoma cells by interaction of CXCR4 expressed in carcinoma cells with tumor necrosis factor-alpha and stromal-derived factor-1 released in stroma. Am J Pathol. 2006;168:1155–1168. doi: 10.2353/ajpath.2006.050204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Appiah-Kubi K, Wang Y, Qian H, Wu M, Yao X, Wu Y, Chen Y. Platelet-derived growth factor receptor/platelet-derived growth factor (PDGFR/PDGF) system is a prognostic and treatment response biomarker with multifarious therapeutic targets in cancers. Tumour Biol. 2016;37:10053–10066. doi: 10.1007/s13277-016-5069-z. [DOI] [PubMed] [Google Scholar]
- 46.Omenetti A, Diehl AM. Hedgehog signaling in cholangiocytes. Curr Opin Gastroenterol. 2011;27:268–275. doi: 10.1097/MOG.0b013e32834550b4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strazzabosco M, Fabris L. Development of the bile ducts: essentials for the clinical hepatologist. J Hepatol. 2012;56:1159–1170. doi: 10.1016/j.jhep.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chong DQ, Zhu AX. The landscape of targeted therapies for cholangiocarcinoma: current status and emerging targets. Oncotarget. 2016;7:46750–46767. doi: 10.18632/oncotarget.8775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thelen A, Scholz A, Benckert C, Weichert W, Dietz E, Wiedenmann B, Neuhaus P, Jonas S. Tumor-associated lymphangiogenesis correlates with lymph node metastases and prognosis in hilar cholangiocarcinoma. Ann Surg Oncol. 2008;15:791–799. doi: 10.1245/s10434-007-9774-0. [DOI] [PubMed] [Google Scholar]
- 50.Cadamuro M, Vismara M, Brivio S, Bovo A, Strazzabosco M, Fabris L. Secretion of Vascular Endothelial Growth Factor-C by Cancer-Associated Fibroblasts is stimulated by Platelet-Derived Growth Factor-D and promotes lymphangiogenesis in cholangiocarcinoma. Proceedings of the Digestive Disease Week 2016; 2016 May 21-24; San Diego, USA. Gastroenterology. 2016;150:S1124. [Google Scholar]
- 51.Thelen A, Scholz A, Weichert W, Wiedenmann B, Neuhaus P, Gessner R, Benckert C, Jonas S. Tumor-associated angiogenesis and lymphangiogenesis correlate with progression of intrahepatic cholangiocarcinoma. Am J Gastroenterol. 2010;105:1123–1132. doi: 10.1038/ajg.2009.674. [DOI] [PubMed] [Google Scholar]
- 52.Tang D, Gao J, Wang S, Ye N, Chong Y, Huang Y, Wang J, Li B, Yin W, Wang D. Cancer-associated fibroblasts promote angiogenesis in gastric cancer through galectin-1 expression. Tumour Biol. 2016;37:1889–1899. doi: 10.1007/s13277-015-3942-9. [DOI] [PubMed] [Google Scholar]
- 53.Lee JS, Semela D, Iredale J, Shah VH. Sinusoidal remodeling and angiogenesis: a new function for the liver-specific pericyte? Hepatology. 2007;45:817–825. doi: 10.1002/hep.21564. [DOI] [PubMed] [Google Scholar]
- 54.Semela D, Das A, Langer D, Kang N, Leof E, Shah V. Platelet-derived growth factor signaling through ephrin-b2 regulates hepatic vascular structure and function. Gastroenterology. 2008;135:671–679. doi: 10.1053/j.gastro.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thabut D, Shah V. Intrahepatic angiogenesis and sinusoidal remodeling in chronic liver disease: new targets for the treatment of portal hypertension? J Hepatol. 2010;53:976–980. doi: 10.1016/j.jhep.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 56.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–1312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mantovani A, Germano G, Marchesi F, Locatelli M, Biswas SK. Cancer-promoting tumor-associated macrophages: new vistas and open questions. Eur J Immunol. 2011;41:2522–2525. doi: 10.1002/eji.201141894. [DOI] [PubMed] [Google Scholar]
- 58.Duong T, Koopman P, Francois M. Tumor lymphangiogenesis as a potential therapeutic target. J Oncol. 2012;2012:204946. doi: 10.1155/2012/204946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, Dang Y, Chu Y, Fan J, He R. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res. 2016;76:4124–4135. doi: 10.1158/0008-5472.CAN-15-2973. [DOI] [PubMed] [Google Scholar]
- 60.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 61.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sica A, Invernizzi P, Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology. 2014;59:2034–2042. doi: 10.1002/hep.26754. [DOI] [PubMed] [Google Scholar]
- 63.Raggi C, Invernizzi P, Andersen JB. Impact of microenvironment and stem-like plasticity in cholangiocarcinoma: molecular networks and biological concepts. J Hepatol. 2015;62:198–207. doi: 10.1016/j.jhep.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 64.Hasita H, Komohara Y, Okabe H, Masuda T, Ohnishi K, Lei XF, Beppu T, Baba H, Takeya M. Significance of alternatively activated macrophages in patients with intrahepatic cholangiocarcinoma. Cancer Sci. 2010;101:1913–1919. doi: 10.1111/j.1349-7006.2010.01614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chang J, Hisamatsu T, Shimamura K, Yoneno K, Adachi M, Naruse H, Igarashi T, Higuchi H, Matsuoka K, Kitazume MT, et al. Activated hepatic stellate cells mediate the differentiation of macrophages. Hepatol Res. 2013;43:658–669. doi: 10.1111/j.1872-034X.2012.01111.x. [DOI] [PubMed] [Google Scholar]
- 66.Locatelli L, Cadamuro M, Spirlì C, Fiorotto R, Lecchi S, Morell CM, Popov Y, Scirpo R, De Matteis M, Amenduni M, et al. Macrophage recruitment by fibrocystin-defective biliary epithelial cells promotes portal fibrosis in congenital hepatic fibrosis. Hepatology. 2016;63:965–982. doi: 10.1002/hep.28382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Raggi C, Correnti M, Sica A, Andersen JB, Cardinale V, Alvaro D, Chiorino G, Forti E, Glaser S, Alpini G, et al. Cholangiocarcinoma stem-like subset shapes tumor-initiating niche by educating associated macrophages. J Hepatol. 2017;66:102–115. doi: 10.1016/j.jhep.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Subimerb C, Pinlaor S, Khuntikeo N, Leelayuwat C, Morris A, McGrath MS, Wongkham S. Tissue invasive macrophage density is correlated with prognosis in cholangiocarcinoma. Mol Med Rep. 2010;3:597–605. doi: 10.3892/mmr_00000303. [DOI] [PubMed] [Google Scholar]
- 69.Atanasov G, Hau HM, Dietel C, Benzing C, Krenzien F, Brandl A, Wiltberger G, Matia I, Prager I, Schierle K, et al. Prognostic significance of macrophage invasion in hilar cholangiocarcinoma. BMC Cancer. 2015;15:790. doi: 10.1186/s12885-015-1795-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Subimerb C, Pinlaor S, Lulitanond V, Khuntikeo N, Okada S, McGrath MS, Wongkham S. Circulating CD14(+) CD16(+) monocyte levels predict tissue invasive character of cholangiocarcinoma. Clin Exp Immunol. 2010;161:471–479. doi: 10.1111/j.1365-2249.2010.04200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brown GT, Murray GI. Current mechanistic insights into the roles of matrix metalloproteinases in tumour invasion and metastasis. J Pathol. 2015;237:273–281. doi: 10.1002/path.4586. [DOI] [PubMed] [Google Scholar]
- 72.Nenu I, Tudor D, Filip AG, Baldea I. Current position of TNF-α in melanomagenesis. Tumour Biol. 2015;36:6589–6602. doi: 10.1007/s13277-015-3639-0. [DOI] [PubMed] [Google Scholar]
- 73.Techasen A, Loilome W, Namwat N, Dokduang H, Jongthawin J, Yongvanit P. Cytokines released from activated human macrophages induce epithelial mesenchymal transition markers of cholangiocarcinoma cells. Asian Pac J Cancer Prev. 2012;13 Suppl:115–118. [PubMed] [Google Scholar]
- 74.Techasen A, Namwat N, Loilome W, Bungkanjana P, Khuntikeo N, Puapairoj A, Jearanaikoon P, Saya H, Yongvanit P. Tumor necrosis factor-α (TNF-α) stimulates the epithelial-mesenchymal transition regulator Snail in cholangiocarcinoma. Med Oncol. 2012;29:3083–3091. doi: 10.1007/s12032-012-0305-x. [DOI] [PubMed] [Google Scholar]
- 75.Techasen A, Namwat N, Loilome W, Duangkumpha K, Puapairoj A, Saya H, Yongvanit P. Tumor necrosis factor-α modulates epithelial mesenchymal transition mediators ZEB2 and S100A4 to promote cholangiocarcinoma progression. J Hepatobiliary Pancreat Sci. 2014;21:703–711. doi: 10.1002/jhbp.125. [DOI] [PubMed] [Google Scholar]
- 76.Tanimura Y, Kokuryo T, Tsunoda N, Yamazaki Y, Oda K, Nimura Y, Naing Mon N, Huang P, Nakanuma Y, Chen MF, et al. Tumor necrosis factor alpha promotes invasiveness of cholangiocarcinoma cells via its receptor, TNFR2. Cancer Lett. 2005;219:205–213. doi: 10.1016/j.canlet.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 77.Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, Buettner R, Proia D, Kowolik CM, Xin H, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–497. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dokduang H, Techasen A, Namwat N, Khuntikeo N, Pairojkul C, Murakami Y, Loilome W, Yongvanit P. STATs profiling reveals predominantly-activated STAT3 in cholangiocarcinoma genesis and progression. J Hepatobiliary Pancreat Sci. 2014;21:767–776. doi: 10.1002/jhbp.131. [DOI] [PubMed] [Google Scholar]
- 79.Isomoto H, Kobayashi S, Werneburg NW, Bronk SF, Guicciardi ME, Frank DA, Gores GJ. Interleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology. 2005;42:1329–1338. doi: 10.1002/hep.20966. [DOI] [PubMed] [Google Scholar]
- 80.Eurich K, Segawa M, Toei-Shimizu S, Mizoguchi E. Potential role of chitinase 3-like-1 in inflammation-associated carcinogenic changes of epithelial cells. World J Gastroenterol. 2009;15:5249–5259. doi: 10.3748/wjg.15.5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Thongsom S, Chaocharoen W, Silsirivanit A, Wongkham S, Sripa B, Choe H, Suginta W, Talabnin C. YKL-40/chitinase-3-like protein 1 is associated with poor prognosis and promotes cell growth and migration of cholangiocarcinoma. Tumour Biol. 2016;37:9451–9463. doi: 10.1007/s13277-016-4838-z. [DOI] [PubMed] [Google Scholar]
- 82.Sugimachi K, Taguchi K, Aishima S, Tanaka S, Shimada M, Kajiyama K, Sugimachi K, Tsuneyoshi M. Altered expression of beta-catenin without genetic mutation in intrahepatic cholangiocarcinoma. Mod Pathol. 2001;14:900–905. doi: 10.1038/modpathol.3880409. [DOI] [PubMed] [Google Scholar]
- 83.Tokumoto N, Ikeda S, Ishizaki Y, Kurihara T, Ozaki S, Iseki M, Shimizu Y, Itamoto T, Arihiro K, Okajima M, et al. Immunohistochemical and mutational analyses of Wnt signaling components and target genes in intrahepatic cholangiocarcinomas. Int J Oncol. 2005;27:973–980. [PubMed] [Google Scholar]
- 84.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu LJ, Xie SX, Chen YT, Xue JL, Zhang CJ, Zhu F. Aberrant regulation of Wnt signaling in hepatocellular carcinoma. World J Gastroenterol. 2016;22:7486–7499. doi: 10.3748/wjg.v22.i33.7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Loilome W, Bungkanjana P, Techasen A, Namwat N, Yongvanit P, Puapairoj A, Khuntikeo N, Riggins GJ. Activated macrophages promote Wnt/β-catenin signaling in cholangiocarcinoma cells. Tumour Biol. 2014;35:5357–5367. doi: 10.1007/s13277-014-1698-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang W, Zhong W, Yuan J, Yan C, Hu S, Tong Y, Mao Y, Hu T, Zhang B, Song G. Involvement of Wnt/β-catenin signaling in the mesenchymal stem cells promote metastatic growth and chemoresistance of cholangiocarcinoma. Oncotarget. 2015;6:42276–42289. doi: 10.18632/oncotarget.5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Haga H, Yan IK, Takahashi K, Wood J, Zubair A, Patel T. Tumour cell-derived extracellular vesicles interact with mesenchymal stem cells to modulate the microenvironment and enhance cholangiocarcinoma growth. J Extracell Vesicles. 2015;4:24900. doi: 10.3402/jev.v4.24900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sirica AE, Almenara JA, Li C. Periostin in intrahepatic cholangiocarcinoma: pathobiological insights and clinical implications. Exp Mol Pathol. 2014;97:515–524. doi: 10.1016/j.yexmp.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dupont S. Role of YAP/TAZ in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp Cell Res. 2016;343:42–53. doi: 10.1016/j.yexcr.2015.10.034. [DOI] [PubMed] [Google Scholar]
- 91.DeClerck YA. Desmoplasia: a response or a niche? Cancer Discov. 2012;2:772–774. doi: 10.1158/2159-8290.CD-12-0348. [DOI] [PubMed] [Google Scholar]
- 92.Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell. 2016;29:783–803. doi: 10.1016/j.ccell.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 94.Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell. 2013;154:1047–1059. doi: 10.1016/j.cell.2013.07.042. [DOI] [PubMed] [Google Scholar]
- 95.Pei T, Li Y, Wang J, Wang H, Liang Y, Shi H, Sun B, Yin D, Sun J, Song R, et al. YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget. 2015;6:17206–17220. doi: 10.18632/oncotarget.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chaijan S, Roytrakul S, Mutirangura A, Leelawat K. Matrigel induces L-plastin expression and promotes L-plastin-dependent invasion in human cholangiocarcinoma cells. Oncol Lett. 2014;8:993–1000. doi: 10.3892/ol.2014.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Aishima S, Taguchi K, Terashi T, Matsuura S, Shimada M, Tsuneyoshi M. Tenascin expression at the invasive front is associated with poor prognosis in intrahepatic cholangiocarcinoma. Mod Pathol. 2003;16:1019–1027. doi: 10.1097/01.MP.0000086860.65672.73. [DOI] [PubMed] [Google Scholar]
- 98.Fujimoto K, Kawaguchi T, Nakashima O, Ono J, Ohta S, Kawaguchi A, Tonan T, Ohshima K, Yano H, Hayabuchi N, et al. Periostin, a matrix protein, has potential as a novel serodiagnostic marker for cholangiocarcinoma. Oncol Rep. 2011;25:1211–1216. doi: 10.3892/or.2011.1194. [DOI] [PubMed] [Google Scholar]
- 99.Utispan K, Sonongbua J, Thuwajit P, Chau-In S, Pairojkul C, Wongkham S, Thuwajit C. Periostin activates integrin α5β1 through a PI3K/AKTdependent pathway in invasion of cholangiocarcinoma. Int J Oncol. 2012;41:1110–1118. doi: 10.3892/ijo.2012.1530. [DOI] [PubMed] [Google Scholar]
- 100.Moolthiya P, Tohtong R, Keeratichamroen S, Leelawat K. Role of mTOR inhibitor in cholangiocarcinoma cell progression. Oncol Lett. 2014;7:854–860. doi: 10.3892/ol.2014.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xie J, Wang X, Proud CG. mTOR inhibitors in cancer therapy. F1000Res. 2016;5 doi: 10.12688/f1000research.9207.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Boulter L, Guest RV, Kendall TJ, Wilson DH, Wojtacha D, Robson AJ, Ridgway RA, Samuel K, Van Rooijen N, Barry ST, et al. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J Clin Invest. 2015;125:1269–1285. doi: 10.1172/JCI76452. [DOI] [PMC free article] [PubMed] [Google Scholar]