

Abstract
Hereditary tyrosinemia type 1 (HT-1) is a metabolic disorder caused by a defect in tyrosine degradation. Without treatment, symptoms of hepatomegaly, renal tubular dysfunction, growth failure, neurologic crises resembling porphyrias, rickets and possible hepatocellular carcinoma can develop. The use of 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione and early diagnosis through newborn screening initiatives have resulted in a sharp decline in morbidity and mortality associated with this disease. We present a case report of a 7-year-old patient with HT-1 who was born prior to the addition of tyrosinemia to the newborn screening in her birth area. At her time of diagnosis, the patient had developed many of the symptoms associated with her disease, including chronic kidney disease, rickets, and myopathy that left her non-ambulatory. During her initial evaluation, she was also noted to have hepatocellular carcinoma. With cadaveric liver transplantation and nutritional support, her symptoms all either resolved or stabilized. Her case illustrates the severity of the disease if left untreated, the need for vigilance in populations who do not routinely receive newborn screens, and the markedly improved outcomes in patients following transplant.
Keywords: Tyrosinemia, Screening, Hepatocellular carcinoma, Liver transplantation
Core tip: Hereditary tyrosinemia type 1 is a metabolic defect resulting in several disease manifestations including life threatening hepatorenal disease, neurologic disease, and rickets. Although neonatal screening for this disorder has allowed early identification and medical treatment with nitisinone, the need for recognition of this disorder in older individuals remains since aggressive intervention, including medical treatment and possible liver transplantation, may be lifesaving and have profound effects on morbidity and mortality.
INTRODUCTION
Hereditary tyrosinemia type 1 (HT-1) is a metabolic disorder caused by a defect in tyrosine degradation due to a deficiency of fumarylacetoacetate hydrolase (FAH). In 1956, Baber provided one of the earliest descriptions of the disorder in her report of a 9 mo old child with cirrhosis, renal tubular dysfunction, and rickets[1]. As a result of advancements in amino acid analysis, a number of reports followed over the next decade in which infants with similar findings were noted to have elevations of plasma tyrosine and aminoaciduria[2-4]. In 1977, Lindblad was able to identify decreased activity of FAH as the enzyme defect responsible for the disorder[5].
The major disease manifestations of HT-1 are now well-defined and include hepatic dysfunction, renal tubular dysfunction, and peripheral nerve injury. The mechanism is most likely a result of increases in toxic metabolites, including fumarylacetoacetate, maleylacetoacetate, and succinoacetylacetate. Most affected children present in infancy with an acute form of the disease associated with failure to thrive, severe liver dysfunction, and death in infancy if untreated. A chronic form may also occur with hepatomegaly, renal tubular dysfunction, growth failure, neurologic crises resembling porphyrias, and rickets. Hepatocellular carcinoma appears to be a common finding in both forms of untreated disease and may be noted in up to 37% of children that survive beyond 2 years of age. The incidence of this tumor increases with age and is reported to be the cause of death in over 50% of untreated individuals[6].
We report our recent experience in a 7-year-old female who underwent liver transplantation for advanced hepatocellular carcinoma in association with a delayed diagnosis of HT-1.
CASE REPORT
A 7-year-old Hispanic female with an uneventful birth history presented to our referral institution for workup and management of balance and gait disturbance and an inability to walk that was first noted at 3 ½ years of age. She was noted to have increasing “clumsiness” in the months leading to her inability to walk. She was also noted to have chronic renal disease and liver disease with hepatomegaly and enlarged kidneys in the first 4 years of life. Her inability to walk was previously attributed to severe pain throughout her body.
At 6 years of age, she developed bilateral lower extremity fractures and an upper extremity fracture while attempting to stand. She was noted to have bowing of her lower extremities and elevated alkaline phosphatase and was subsequently diagnosed with rickets that was believed to be the result of chronic kidney disease. Shortly thereafter, she was referred to a local medical center where she underwent liver biopsy, kidney biopsy, and muscle biopsy. Her liver biopsy revealed cirrhosis with minimal chronic inflammation and her kidney biopsy revealed nonspecific glomerular and tubular changes with some parenchymal fibrosis. Her muscle biopsies revealed severe myopathic changes with myofiber atrophy. She was noted to have mitochondrial DNA quantification on her muscle biopsy which was reduced and less than 29% of controls. She was subsequently diagnosed with mitochondrial depletion syndrome.
When her weakness and strength worsened further, she was referred to our institution’s mitochondrial clinic where she underwent additional workup. Extensive metabolic evaluation revealed an elevated succinylacetone which led to a diagnosis of HT-1. Analysis of the FAH gene revealed a homozygous splice mutation known to be associated with HT-1. She ultimately underwent treatment with dietary modification and 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC), but she was immediately referred for cadaveric liver transplantation after further workup revealed imaging findings consistent with hepatocellular carcinoma. Following this initial pre-transplant treatment, her condition improved substantially with significant improvement in her amino acid profile, improvement in her mental status, and reduction in her alpha-fetoprotein from 2320 ng/mL to 1585 ng/mL. Despite this metabolic improvement, she required liver transplantation as a result of her hepatocellular carcinoma. Since this lesion was felt to be adjacent to major blood vessels and liver transplantation was not ideal without improved nutrition and rehabilitation, she underwent transarterial chemoembolization for a 1.5 cm × 1.6 cm lesion in her liver (Figure 1) after which time she had further reduction in her alpha-fetoprotein to 511 ng/mL. After continued improvement in nutrition and physical conditioning, she ultimately underwent liver transplantation 4 mo after her diagnosis was made. Explant of her liver revealed a yellow-orange and nodular liver, and histology revealed cirrhosis and multinodular well-differentiated hepatocellular carcinoma with bile duct proliferation felt to be consistent with tyrosinemia (Figure 2).
Figure 1.
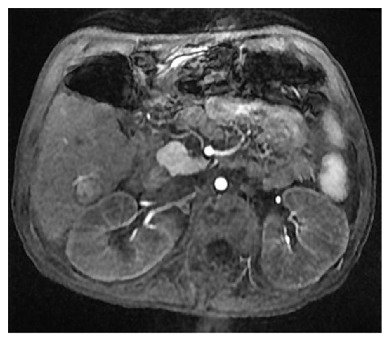
Magnetic resonance imaging of the liver with a large 1.5 cm × 1.6 cm lesion with arterial enhancement characterizing hepatocellular carcinoma and requiring chemoembolization.
Figure 2.

Explant of nodular and shrunken tyrosinemic liver obtained at the time of liver transplantation.
Her immediate postoperative course was unremarkable and, following transplant, she had further improvements in her cognitive function, nutrition, and physical conditioning. Within 6 mo, she was ambulating without assistance and attending regular school without difficulty. Her rickets had also improved and resolved with her bone mineral density Z-score in her hip improving from -4.1 prior to transplant to -1.1 fifteen months following transplant. Her renal function improved with her estimated GFR before and 6 mo after transplant noted to be 75 and 121 mL/min per 1.73 m2 respectively. She had no recurrence of tumor on follow-up imaging obtained 2 years following her transplantation, and her alpha-fetoprotein was normal during this time.
DISCUSSION
While HT-1 can result in a variety of multisystem and life-threatening complications, including hepatic and renal disease, recent approaches to management over the past few decades have resulted in a sharp decline in morbidity and mortality associated with this disease. Since Lindstedt first published his experience using NTBC in 5 patients with HT-1 in 1992, its incorporation into the standard treatment for HT-1 has resulted in improved effects on the long term outcome of individuals with this disease with improved metabolic control[7]. In addition, newborn screening for HT-1 is a common practice in most Western countries allowing early identification and treatment of affected individuals as early as the first month of life. In the large Quebec experience, infants beginning treatment with NTBC in the first month of life had no detectable liver lesions after more than 5 years of follow-up and no need for liver transplantation[8]. The combined effects of NTBC use and newborn screening have resulted in a significant reduction in the need for liver transplantation noted over the past decade[9].
Since our patient was born in 2006, one year prior to initiation of newborn screening in her region, her diagnosis was not identified early in life. Her presenting symptoms of developmental delay and weakness which were first noted at 3 years of age led to an underlying diagnosis of mitochondrial depletion syndrome. Interestingly, electron microscopy of tissue from individuals with HT-1 has revealed mitochondrial abnormalities with a relative loss of matrical bodies and decreased matrix density[10]. Like tyrosinemia, mitochondrial depletion syndromes may involve a variety of organs, including the liver, kidney, and peripheral nervous system. Her abnormal movements and unusual behavior could also be attributed to an underlying mitochondrial or primary neuromuscular disorder although these resolved after dietary and NTBC were started.
Marked elevations in alpha-fetoprotein were noted in our patient. Elevated alpha-fetoprotein is a typical finding in HT-1, even in the absence of hepatocellular carcinoma. Improvement in alpha-fetoprotein is also noted with improvement in metabolic control in HT-1 patients as we saw in our patient. Nevertheless, our patient required extensive workup to rule out hepatocellular carcinoma given the obvious risk of hepatocellular carcinoma in individuals with tyrosinemia, her continued markedly elevation alpha-fetoprotein, and her delayed age at diagnosis.
The presence of renal dysfunction in individuals with HT-1 requires consideration for combined liver-kidney transplantation in some individuals. Continued exposure to calcineurin inhibitors may cause significant deterioration in kidney function in some individuals receiving liver transplantation. In the Quebec experience, combined liver-kidney transplant may be warranted when the GFR < 40 mL/min per 1.73 m2[11]. Despite her late age at diagnosis, our patient had a GFR which was slightly below normal but still sufficient enough to make isolated liver transplant a reasonable option.
This case highlights a number of important points. The presence of liver disease in association with other uncharacteristic organ pathology warrants consideration for an underlying metabolic disorder. Despite great advancement in newborn screening, metabolic disorders such as HT-1 are still possible due to imperfection of neonatal screening and potential for missed populations. Recent political and economic upheaval has introduced a large number of migrants into Western countries who often may not have had screening for a variety of metabolic or genetic disorders. In addition, children receiving transplantation for metabolic liver disease have improved outcomes compared to children transplanted for other disorders, such as biliary atresia, with 1- and 5-year survival of 95% and 89%[12]. Finally, the presence of severe extrahepatic disease such as severe neuromuscular disease in our patient should not deter consideration for transplantation since liver transplantation can result in remarkable improvement and reversal in neurologic and cognitive dysfunction, particularly in children.
COMMENTS
Clinical diagnosis
The symptoms of hereditary tyrosinemia include hepatorenal dysfunction, neuromuscular symptoms, and rickets.
Differential diagnosis
The differential diagnosis of hereditary tyrosinemia includes disorders of carbohydrate metabolism, mitochondrial disorders, and Wilson’s disease.
Laboratory diagnosis
Laboratory diagnosis of tyrosinemia includes confirmation by noting the presence of succinylacetone in urine specimens or body fluids, identification of causative mutations, or enzyme analysis revealing decreased fumarylacetoacetate hydrolase.
Imaging diagnosis
Imaging in tyrosinemia may reveal complictations of this disorder, including cirrhosis, hepatomegaly, hepatocellular carcinoma, renal abnormalities, and rickets.
Pathological diagnosis
Pathological diagnosis of this disorder is obtained by laboratory methods described above.
Treatment
Treatment of tyrosinemia involved use of nitisinone and a protein restricted diet in conjunction with long term management by a specialist. Liver transplantation may be required for severe disease or complications of this disorder.
Experiences and lessons
A diagnosis of hereditary tyrosinemia should be considered in individuals with hepatorenal disease, rickets, and neuromuscular weakness since rapid initiation of aggressive treatment may be lifesaving.
Peer-review
Imseis E et al reported about a case of hepatocellular carcinoma in a patient with hereditary tyrosinemia, a kind of rare lesion, in the post-newborn screening era. This case report is very interesting.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: United States
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B, B, B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: IRB approval is not required for case reports involving one patient.
Informed consent statement: Informed consent was obtained from the patient and guardian for inclusion in this retrospective case report.
Conflict-of-interest statement: The authors whose names are listed on this manuscript have no affiliations with or involvement in any organization or entity with any financial or non-financial interest in the subject matter or materials discussed in this manuscript.
Peer-review started: October 12, 2016
First decision: November 11, 2016
Article in press: February 20, 2017
P- Reviewer: Han ZG, Kao JT, Long XD, Tsuchiya K, Tomizawa M S- Editor: Song XX L- Editor: A E- Editor: Li D
References
- 1.Baber MD. A case of congenital cirrhosis of the liver with renal tubular defects akin to those in the Fanconi syndrome. Arch Dis Child. 1956;31:335–339. doi: 10.1136/adc.31.159.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kitagawa T. Hepatorenal tyrosinemia. Proc Jpn Acad Ser B Phys Biol Sci. 2012;88:192–200. doi: 10.2183/pjab.88.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scriver CR, Silverberg M, Clow CL. Hereditary tyrosinemia and tyrosyluria: clinical report of four patients. Can Med Assoc J. 1967;97:1047–1050. [PMC free article] [PubMed] [Google Scholar]
- 4.Scriver CR, Larochelle J, Silverberg M. Hereditary tyrosinemia and tyrosyluria in a French Canadian geographic isolate. Am J Dis Child. 1967;113:41–46. doi: 10.1001/archpedi.1967.02090160091008. [DOI] [PubMed] [Google Scholar]
- 5.Lindblad B, Lindstedt S, Steen G. On the enzymic defects in hereditary tyrosinemia. Proc Natl Acad Sci USA. 1977;74:4641–4645. doi: 10.1073/pnas.74.10.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weinberg AG, Mize CE, Worthen HG. The occurrence of hepatoma in the chronic form of hereditary tyrosinemia. J Pediatr. 1976;88:434–438. doi: 10.1016/s0022-3476(76)80259-4. [DOI] [PubMed] [Google Scholar]
- 7.Lindstedt S, Holme E, Lock EA, Hjalmarson O, Strandvik B. Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet. 1992;340:813–817. doi: 10.1016/0140-6736(92)92685-9. [DOI] [PubMed] [Google Scholar]
- 8.Larochelle J, Alvarez F, Bussières JF, Chevalier I, Dallaire L, Dubois J, Faucher F, Fenyves D, Goodyer P, Grenier A, et al. Effect of nitisinone (NTBC) treatment on the clinical course of hepatorenal tyrosinemia in Québec. Mol Genet Metab. 2012;107:49–54. doi: 10.1016/j.ymgme.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 9.Arnon R, Annunziato R, Miloh T, Wasserstein M, Sogawa H, Wilson M, Suchy F, Kerkar N. Liver transplantation for hereditary tyrosinemia type I: analysis of the UNOS database. Pediatr Transplant. 2011;15:400–405. doi: 10.1111/j.1399-3046.2011.01497.x. [DOI] [PubMed] [Google Scholar]
- 10.Dehner LP, Snover DC, Sharp HL, Ascher N, Nakhleh R, Day DL. Hereditary tyrosinemia type I (chronic form): pathologic findings in the liver. Hum Pathol. 1989;20:149–158. doi: 10.1016/0046-8177(89)90179-2. [DOI] [PubMed] [Google Scholar]
- 11.Paradis K, Weber A, Seidman EG, Larochelle J, Garel L, Lenaerts C, Roy CC. Liver transplantation for hereditary tyrosinemia: the Quebec experience. Am J Hum Genet. 1990;47:338–342. [PMC free article] [PubMed] [Google Scholar]
- 12.Arnon R, Kerkar N, Davis MK, Anand R, Yin W, González-Peralta RP. Liver transplantation in children with metabolic diseases: the studies of pediatric liver transplantation experience. Pediatr Transplant. 2010;14:796–805. doi: 10.1111/j.1399-3046.2010.01339.x. [DOI] [PubMed] [Google Scholar]