

Abstract
Based on our findings that PHD2 is a negative regulator of chondrocyte differentiation and that hypoxia signaling is implicated in the pathogenesis of osteoarthritis, we investigated the consequence of disruption of the Phd2 gene in chondrocytes on the articular cartilage phenotype in mice. Immunohistochemistry detected high expression of PHD2 in the superficial zone (SZ), while PHD3 and HIF-1α (target of PHD2) are mainly expressed in the middle-deep zone (MDZ). Conditional deletion of the Phd2 gene (cKO) in chondrocytes accelerated the transition of progenitors to hypertrophic (differentiating) chondrocytes as revealed by reduced SZ thickness, and increased MDZ thickness, as well as increased chondrocyte hypertrophy. Immunohistochemistry further revealed decreased levels of progenitor markers but increased levels of hypertrophy markers in the articular cartilage of the cKO mice. Treatment of primary articular chondrocytes, in vitro, with IOX2, a specific inhibitor of PHD2, promoted articular chondrocyte differentiation. Knockdown of Hif-1α expression in primary articular chondrocytes using lentiviral vectors containing Hif-1α shRNA resulted in reduced expression levels of Vegf, Glut1, Pgk1, and Col10 compared to control shRNA. We conclude that Phd2 is a key regulator of articular cartilage development that acts by inhibiting the differentiation of articular cartilage progenitors via modulating HIF-1α signaling.
Articular cartilage is a hyaline cartilage that provides a smooth and frictionless surface for bone movements within a joint. The thin layer of articular cartilage covers the bone surface and maintains its lubricating function for an individual’s lifetime. Damage to the articular cartilage is a significant clinical issue causing an enormous economic burden worldwide. The most common articular cartilage damage is osteoarthritis characterized by progressive degeneration of articular cartilage that leads to chronic pain and functional restrictions in the affected joints. Pathophysiological features of osteoarthritis include degeneration or loss of articular cartilage, sclerosis, an increase of subchondral bone density, and osteophyte formation1,2,3. The precise mechanisms that regulate the integrity of articular cartilage and the mechanisms of osteoarthritis development are still unclear and nonsurgical therapeutic interventions to articular cartilage diseases are limited to date3,4.
The articular cartilage is composed of several chondrocyte layers with unique cellular arrangements including a superficial zone (SZ), a middle zone, a deep zone, and a calcified cartilage zone. The SZ contains flat shaped articular cartilage progenitor cells robustly expressing proteoglycans such as aggrecan and lubricin5,6, while chondrocytes in the deep zone are in a columnar organization with some of them becoming hypertrophic and marked by expression of Col102. A number of growth factors including Wnt9A, TGFα, PTHrP, IGF-I and BMPs have been implicated in maintaining articular cartilage integrity by regulating the transit of articular cartilage progenitors towards differentiating chondrocytes6,7,8. It is known that increased chondrocyte hypertrophy, which leads to increased endochondral ossification, is frequently identified in early stage osteoarthritis samples9,10. Accordingly, studies have shown an inverse correlation between the onset of osteoarthritis and an increase in subchondral bone density11,12. Hence, understanding the molecular mechanisms controlling the transition of articular cartilage progenitors towards hypertrophic chondrocytes is important for the prevention and treatment of articular cartilage diseases.
Prolyl Hydroxylase Domain-containing Proteins (PHDs) are negative regulators of the hypoxia-inducible factors (HIFs) which moderate chondrocyte differentiation13,14,15. PHDs belong to the 2-oxoglutarate/iron-dependent dioxygenase superfamily. There are three members of Phds in mammals: Phd1 Phd2, and Phd3. All PHDs contain the highly conserved hydroxylase domain in the catalytic carboxy-terminal16. PHD2 is the major regulator of HIF-1α activity. In the presence of oxygen, PHD2 hydroxylates two proline residues (Pro-402 and Pro-564) in the C-terminal of HIF-1α which leads to the ubiquitin-mediated proteasomal degradation of HIF-1α17,18. HIF-1α is required for the maintenance and differentiation of chondrocytes in the hypoxic growth plate19. Conditional deletion of HIF-1α in mesenchymal cells resulted in abnormal cartilage and joint development20,21. On the other hand, we found that deletion of Phd2 in chondrocytes caused accelerated chondrocyte differentiation and increased endochondral bone formation in the metaphyses and epiphyses of long bones13,14. These data clearly demonstrate that Phd2 negatively regulates chondrocyte differentiation and endochondral bone formation through the PHD/HIF regulatory pathway.
Interestingly, in addition to the growth plate chondrocytes, Phd2 and Phd3 are also highly expressed the articular chondrocytes with distinct expression patterns. Since Phd2 is a negative regulator of chondrocyte differentiation, we hypothesize that Phd2 also inhibits the differentiation of articular cartilage progenitors, and deletion of Phd2 in chondrocytes promotes progenitors to differentiate into hypertrophic chondrocytes and thereby, reduce articular cartilage thickness. Since articular cartilage richly contains Type II collagen (Col2), we used Col2α1-Cre line to conditionally delete Phd2 gene in articular chondrocytes.
Results
PHDs and HIFs are expressed in distinct patterns in the femoral articular cartilage
To study the role of PHDs in articular cartilage development, we first evaluated the expression patterns of PHD2 and PHD3 and their targets (HIFs) using immunohistochemistry in the distal femoral articular cartilage in 2 week old mice, when articular cartilage formation is known to occur. PHD2 protein is highly expressed in the SZ of articular cartilage, but remains low in the middle-deep zone (MDZ) (Fig. 1A, blue is positive staining). By contrast, PHD3 is almost absent in the SZ, but highly expressed in the MDZ, where some of the chondrocytes are undergoing hypertrophy (Fig. 1B). HIF-1α protein, a known target of PHD2, appears mainly in the MDZ of articular cartilage, whereas PHD2 expression is low (Fig. 1C). On the other hand, HIF-2α is highly expressed in the SZ, but not in the MDZ of the articular cartilage, very similar to the expression pattern of PHD2 (Fig. 1D). HIF-3α is more ubiquitous than other HIFs and is expressed throughout different zones at relatively low levels (Fig. 1E). The high expression of Phd2 and Hif-2α in the progenitor containing SZ suggests a role for Phd2 and Hif-2α in maintaining the progenitor status of articular chondrocytes. On the other hand, the predominant expression of Phd3 and Hif-1α in MDZ implicates their role in articular chondrocyte differentiation.
Figure 1. Expression patterns of PHDs and HIFs in the articular cartilage.

Immunohistochemistry was performed on femur sections from 2 week old mice. Left panels show expression of each protein in whole femur heads and right panels are high magnifications of the articular cartilage. (A) PHD2 is highly expressed in the SZ, but remains low in the MDZ. (B) PHD3 is highly expressed in the MDZ, but remains low in the SZ. (C) HIF-1α protein is highly expressed in the MDZ. An arrow shows a hypertrophic chondrocyte. (D) HIF-2α protein is highly expressed in the SZ. (E) HIF-3α is ubiquitously expressed in the articular cartilage. Blue is positive staining. AC, articular cartilage; GP, growth plate; SZ, superficial zone; MDZ, middle-deep zone. Bar = 50 μm. Magnification: left panel, 40X; right panel, 200X.
Conditional deletion of Phd2 in the Col2-expressing cells ablated PHD2 expression in articular cartilage
To test the hypothesis that Phd2 plays a role in maintaining the progenitor status of the cells in the SZ by preventing their differentiation towards a hypertrophic stage, we used Col2α1-Cre transgenic mice to disrupt the Phd2 gene in articular chondrocyte progenitors that express high levels of Col2α1 (Fig. 2A). In skeletal tissue, Col2α1-Cre activity was specifically detected in chondrocytes and the specificity of the Col2α1-Cre line was documented in several studies22,23,24,25. We have also used this line to knockout Phd2 in chondrocytes to study Phd2 function in endochondral bone formation at the primary and secondary ossification centers13,14. The efficiency of the Col2α1-Cre line used for disruption of the Phd2 gene in articular chondrocytes was validated by immunohistochemistry. In Fig. 2B, PHD2 protein was detected in tibial articular cartilage of 4 week old control mice, but not in the conditional knockout (cKO) mice (Fig. 2B, blue is positive staining). By contrast, PHD2 protein expressed in bone marrow was intact in the cKO mice compared to the control mice (Fig. 2B).
Figure 2. Generating the conditional Phd2 knockout mice in articular chondrocytes.

(A) Conditional deletion of the Phd2 gene in Col2α1-expressing chondrocytes. (B) Immunohistochemistry analysis of PHD2 expression in tibiae of 4 week old control and cKO mice. PHD2 was expressed in the control but not in the cKO tibial articular cartilage (arrows). Expression of PHD2 in bone marrow was intact in the cKO mice (arrow heads). Blue is positive staining. BM, bone marrow.
Deletion of Phd2 in articular cartilage reduced the thickness of the superficial zone and increased chondrocyte hypertrophy in articular cartilage
To evaluate the effect of Phd2 deletion on articular cartilage development, the articular cartilage phenotype was evaluated by histology in the long bones of 4 week old mice. We measured the thickness of the SZ and MDZ in the tibial articular cartilage. The thickness of SZ was reduced by 25% (P < 0.05) in the cKO tibia compared to control tibia (Fig. 3A,B). By contrast, the thickness of MDZ was increased by 31% (P < 0.05) in the cKO tibia compared to controls (Fig. 3A,C). The number of hypertrophic chondrocytes was increased in the articular cartilage of the cKO mice compared to controls (Fig. 3A), indicating increased chondrocyte differentiation in the articular cartilage of the cKO mice. These data suggests that loss of Phd2 promoted the transition of articular cartilage progenitors in the SZ into differentiating chondrocytes.
Figure 3. Histological changes in the articular cartilage of the Phd2 cKO mice.

(A) Safranin-O staining of 4 week old control and cKO tibiae. Arrows show hypertrophic chondrocytes. (B) SZ thickness in control and cKO tibiae at 4 weeks of age. (C) MDZ thickness in control and cKO tibiae from 4 week old mice. (D) SZ thickness in 12 week old control and cKO tibiae. (E) MDZ thickness in 12 week old control and cKO tibiae. (F) Thickness of joint space in 12 week old control and cKO tibiae. (G) OARSI score in 12 week old control and cKO tibiae. *P < 0.05, **P < 0.01, n = 8/group in (B,C), n = 6/group in (D–G). Data were presented as the mean ± SEM. Bar = 50 μm.
To examine if the histological changes of the articular cartilage in the cKO mice were maintained in adult mice, we also measured the thickness of the SZ and the MDZ in 12 week old mice. The reduction in SZ thickness in the cKO tibia was 23% (P < 0.05) compared to control tibia in 12 week old mice (Fig. 3D). The thickness of the MDZ was slightly increased in the cKO mice compared to control mice, but this change did not reach statistical significance (p = 0.19) (Fig. 3E). We further evaluated an early stage osteoarthritis phenotype in these mice. We found that the joint space was increased by 44% (P < 0.05), and the Osteoarthritis Research Society International (OARSI) score was increased by 270% (P < 0.01) in the cKO mice compared to control mice (Fig. 3F,G), suggesting that a lack of Phd2 may increase the risk of osteoarthritis development.
Deletion of Phd2 in articular cartilage reduced expression of progenitor markers, but elevated expression of hypertrophy markers
Articular cartilage progenitors in the SZ express high levels of lubricin and aggrecan5,6. Consistent with the prediction that loss of Phd2 promoted chondrocyte differentiation, we found expression levels of both lubricin and aggrecan were markedly reduced in the cKO articular cartilage (Fig. 4A,B, blue is positive staining) while levels of PHD3 and HIF-1α, that are known to be highly expressed in hypertrophic chondrocytes, were elevated in the cKO articular cartilage compared to controls (Fig. 4C,D). We also examined the expression of COL10, a well-established marker of hypertrophic chondrocytes, and found that it was markedly increased in the cKO articular chondrocytes (Fig. 4E), thus suggesting elevated chondrocyte differentiation in the articular cartilage of the Phd2 cKO mice.
Figure 4. Expression of markers of articular cartilage progenitors and chondrocyte differentiation in control and cKO mice.

Immunohistochemistry analysis of markers in tibial articular cartilage of 12 week old control and cKO mice. (A,B) Expression of SZ progenitor markers, lubricin and aggrecan, in articular cartilage. (C) PHD3 expression in control and cKO articular cartilage. (D) HIF-1α expression in control and cKO articular cartilage. (E) Expression of the chondrocyte hypertrophy marker COL10 in control and cKO articular cartilage. Blue is positive staining. Arrows show positive stains of each protein in the articular cartilage. Bar = 50 μm.
Inhibition of PHD2 by IOX2 promoted cell differentiation and HIF-1α signaling in primary articular chondrocytes
In order to determine if the increased differentiation of articular chondrocytes in the Phd2 cKO mice is a direct consequence of loss of PHD2 function, we treated primary articular chondrocytes with a specific inhibitor of PHD2, IOX2, for 72 hours and then measured expression levels of markers of proliferation, articular cartilage progenitors, and chondrocyte differentiation. We found that expression of proliferation markers, p57 and cyclin D1, were both reduced by 28% (P < 0.05) (Fig. 5A). Accordingly, expression levels of markers of articular cartilage progenitors, tenascin c and clusterin, were reduced by 28% and 32%, respectively (P < 0.05) (Fig. 5B). Expression of Col2 was unchanged (Fig. 5C). By contrast, expression level of Col10 was increased by 126% (P < 0.01) (Fig. 5C). Loss of PHD2 activity is known to increase protein levels of HIF-1α via inhibition of prolyl hydroxylation of HIF-1α and its subsequent proteosomal degradation17,18. Activation of HIF-1α signaling is known to promote angiogenesis and glycolysis26,27,28. Accordingly, expression levels of Vegf, an angiogenic marker and a known target of HIF-1α, were increased by 8 fold (P < 0.01) (Fig. 5D). Epo is also a target of HIF-1α. Its expression was also increased, but the increase did not reach statistical significance. Expression levels of Glut1 and Pgk1, markers of glycolysis, were increased by 154% and 170%, respectively (P < 0.05) in IOX2 treated cells (Fig. 5D).
Figure 5. IOX2-mediated inhibition of PHD2 promotes differentiation of articular chondrocytes.

Primary articular chondrocytes were cultured with 20 μM PHD2 inhibitor IOX2 or vehicle (DMSO) for 72 hours and expression of markers were measured by Real time RT-PCR. (A) mRNA expression of cell proliferation marker p57Kip2 and cyclin D1. (B) mRNA expression of SZ progenitor marker tenascin C and clusterin. (C) mRNA expression of chondrocyte marker Col2 and chondrocyte hypertrophy marker Col10. (D) mRNA expression of HIF-1α targets, vascularization marker Vegf, Epo, and glycolysis marker Glut1 and Pgk1. *P < 0.05, **P < 0.01, n = 4/group. Data were normalized to controls and presented as the mean ± SEM.
Knockdown of Hif-1α reduced differentiation and glycolysis of primary articular chondrocytes
In order to determine the role of HIF-1α signaling in mediating Phd2 effects on chondrocyte differentiation, we evaluated the consequence of knockdown of Hif-1α in primary articular chondrocytes. We used lentiviral vectors containing shRNA specific to Hif-1α to knockdown HIF-1α. Hif-1α shRNA treatment knocked down Hif-1α mRNA expression by 92% (P < 0.01) compared to control shRNA (Fig. 6A). Expression of the HIF-1α target, Vegf, was also reduced by 75% (P < 0.01) (Fig. 6A). Furthermore, HIF-1α targets, the glycolytic enzymes Glut1 and Pgk1, were reduced by 41% and 55%, respectively (P < 0.05) (Fig. 6B). The chondrocyte hypertrophy marker Col10 was also reduced by 50% (P < 0.05), while Col2 expression was not changed (Fig. 6C). The findings that knockdown of Hif-1α inhibited angiogenesis, glycolysis, and chondrocyte differentiation, effects that are opposite to those of IOX2 mediated inhibition of PHD2 activity, suggests that the negative effect of Phd2 on chondrocyte differentiation in articular cartilage is perhaps mediated via inhibition of the HIF-1α signaling pathway.
Figure 6. shRNA knockdown of HIF-1α reduced expression of markers of glycolysis and chondrocyte differentiation.

Primary articular chondrocytes were treated with lentiviral vectors containing shRNA specific to Hif-1α or control shRNA. (A) Hif-1α mRNA expression was knocked down by 92%. Vegf mRNA expression was significantly reduced in Hif-1α knockdown cells. (B) mRNA Expression of glycolysis marker Glut1 and Pgk1 in the Hif-1α knockdown cells. (C) mRNA expression of Col2 and Col10 in the Hif-1α knockdown cells. *P < 0.05, **P < 0.01, n = 3/group. Data were normalized to control shRNA treated cells and presented as the mean ± SEM.
Discussion
In this study, we have found distinct spatial distribution of PHD2 and PHD3 in the articular cartilage, with PHD2 being mainly localized to the SZ progenitor cells while PHD3 to the differentiating chondrocytes in the MDZ. The low level of PHD2 in the MDZ was accompanied by a high level of HIF-1α in the MDZ, consistent with the well-known function of PHD2 to target HIF-1α to proteosomal-mediated degradation17,18. Conditional deletion of Phd2 in chondrocytes decreased the thickness of progenitors in the SZ and increased chondrocyte hypertrophy in the MDZ of the tibial articular cartilage, marked by down regulation of lubricin and aggrecan, and up regulation of HIF-1α and COL10. These findings suggest that high levels of Phd2 are required to maintain the progenitor status of cells in the SZ. Loss of functional PHD2 in the progenitor cells of the SZ promotes articular chondrocyte differentiation. This notion is further supported by a primary articular chondrocyte study showing that inhibition of PHD2 suppressed expression of tenascin C and clusterin, but elevated expression of Col10 and HIF-1α. The Phd2 effect on articular chondrocyte differentiation appears to be mainly mediated by HIF-1α signaling as revealed from our data from experiments involving knockdown of Hif-1α and PHD2 inhibitor.
Our study demonstrated a crucial role for Phd2 in articular chondrocyte regulation. Similar to its role in endochondral bone formation, Phd2 inhibits HIF-1α signaling and hence inhibits chondrocyte differentiation13,14. It is well established that HIF-1α is a positive regulator of chondrocyte differentiation and ossification, as conditional deletion of the von Hippel-Lindau gene (Vhl), a ubiquitin ligase that promotes proteolysis of HIFs, or overexpression of Vegf, a HIF-1α target, resulted in excessive endochondral bone formation29,30. HIF-1α signaling also appeared to negatively affect progenitor status of the articular chondrocytes, as Phd2 deletion in chondrocytes resulted in reduced articular cartilage SZ thickness. The negative regulation of HIF-1α signaling in progenitors was also supported by the Hif-1α knockdown experiment. Hif-1α knockdown reduced expression of markers of proliferation, as well as markers of progenitors. Based on these data, one would expect a negative effect of HIF-1α on the maintainence of articular cartilage, and higher levels of HIF-1α could contribute to the pathogenesis of osteoarthritis.
In addition to its established role in regulating chondrocyte differentiation, other studies have shown that Hif-1α is also required for chondrocyte growth arrest and survival. Lack of Hif-1α resulted in chondrocyte death19. Balb/C mice injected with HIF-1α inhibitor, 2-methoxyestradiol, resulted in progressive destruction of the articular cartilage, and HIF-1α inhibition in human osteoarthritic cartilage accelerates catabolic stress-induced apoptosis in vitro, thus suggesting a protective role for Hif-1α in osteoarthritis development31,32. However, this hypothesis contradicted research showing that stabilization of HIF-1α by dimethyloxaloylglycine did not prevent osteoarthritis development in the knee joints of STR/ORT mice31. Nevertheless, Hif-1α appeared to be clinically important in the development of osteoarthritis. Several studies reported elevation of HIF-1α signaling in human osteoarthritic samples32,33,34. The precise role of HIF-1α in articular cartilage development and the clinical significance of aberrant PHD2/HIF-1α signaling in osteoarthritis development needs to be investigated further.
Though PHD2 is the major regulator of HIF-1α, HIF-1α also interacts with PHD3. HIF-1α up regulates PHD3 through a feedback mechanism14. On the other hand, PHD3 hydroxylates HIF-1α, although PHD3 is the major regulator of HIF-2α35. In terms of clinical significance, HIF-2α is a catabolic factor for articular cartilage, and lack of HIF-2α is protective for osteoarthritis development36,37. We have observed elevated expression of PHD3 in the Phd2 cKO articular cartilage. This is consistent with our previous report that disruption of Phd2 in growth plate chondrocytes elevated Phd3 expression both in vivo and in vitro14. The role of Phd3 in articular cartilage development and mainanence need to be further investigated.
Based on our findings, we proposed a model for the regulation of Phd2 on articular cartilage (Fig. 7). PHD2 inhibits HIF-1α, which further inhibits PHD3. Lack of PHD2 promotes HIF-1α signaling and elevates PHD3 expression. HIF-1α translocates from the cytoplasm to the nucleus and binds to the hypoxia response element (HRE) of targeted genes including genes required for chondrocyte hypertrophy. Thus Phd2 is a key negative regular for articular chondrocyte differentiation (Fig. 7).
Figure 7. Model of Phd2 regulation of articular cartilage chondrocytes.
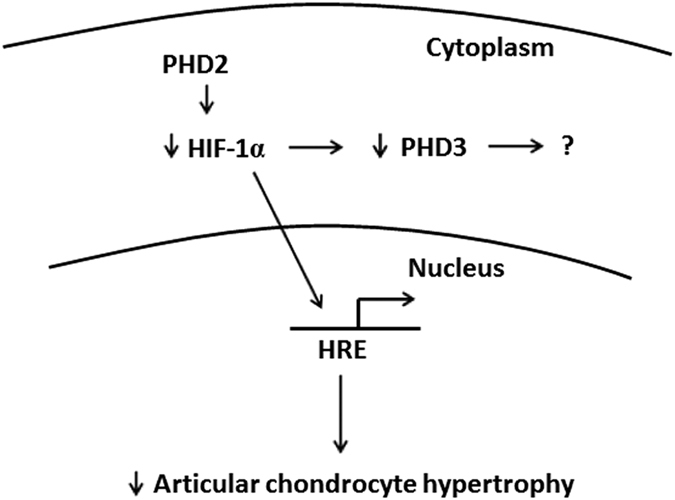
PHD2 inhibits HIF-1α which in turn inhibits PHD3 expression. In the absence of PHD2, HIF-1α translocates from the cytoplasm to the nucleus, binds to the hypoxia response element (HRE), and activates genes required for chondrocyte hypertrophy. In the presence of PHD2, HIF-1α undergoes prolyl hydroxylation and subsequent proteosomal degradation, and therefore, chondrocyte hypertrophy is reduced.
Methods
Animals
To generate Phd2 conditional knockout mice specifically in chondrocytes, Phd2 floxed mice (Phd2flox/flox) were first crossed with Col2α1-Cre mice38,39 to generate Cre positive, Phd2 loxp-heterozygous mice (Phd2flox/+; Col2α1-Cre). Phd2flox/+; Col2α1-Cre mice were then backcrossed with Phd2flox/flox mice, to yield Cre positive, loxp-homozygous (Phd2flox/flox; Col2α1-Cre) conditional knockout mice and Cre negative control littermates (Phd2flox/flox, Phd2flox/+). The genetic background of Phd2flox/flox and Col2α1-Cre mice is C57BL/6. Animals were housed according to the approved laboratory conditions in the VMU at VA Loma Linda Healthcare System (Loma Linda, CA).
Ethics Statement
Animals were housed according to the approved laboratory conditions in the animal facility unit at VA Loma Linda Healthcare System (Loma Linda, CA). All experimental procedures were evaluated and carried out in accordance with the protocols approved by the Institutional Animal Care and Use Committee of the VA Loma Linda Healthcare System. Isoflurane was used for anesthesia, and CO2 exposure was used for euthanasia followed by cervical dislocation. All procedures performed followed the ethical guidelines for animal studies.
Histomorphometry and immunohistochemistry
Four and 12 week old control and cKO mice were sacrificed and the femurs were fixed in 10% formalin for 4 days, decalcified, and processed for paraffin sectioning as previously described40. Three comparable sections of the knee joint from each animal were stained with Safranin O and counter stained with Fast Green to visualize the articular cartilage. Measurements of the thickness of the SZ and the MDZ were carried out using the OsteoMeasure software (Osteometrics, Inc. Decatur, GA). OARSI scoring was according to previously described41. Immunohistochemistry was performed using the VECTASTAIN ABC-AP kit (AK-5000, Vector Laboratories, Burlingame, CA) as previously described40. Antibodies and dilutions of each antibody were listed in Supplemental Table 1. Vector Blue was used as the AP substrate (SK-5300, Vector Laboratories, Burlingame, CA).
Primary chondrocyte culture and IOX2 treatment
Primary chondrocytes were isolated from articular cartilage of a 3 day old pup knee joint and cultured as previously described42. Cells were grown in DMEM/F12 medium containing 10% fetal bovine serum, penicillin (100 U/mL), and streptomycin (100 μg/mL) to 70% confluence followed by 24 hours of serum free (0.1% BSA DMEM/F12) treatment. The cells were then treated with the PHD2 specific inhibitor, IOX2 (Xcess Biosciences, San Diego, CA), at 20 μM or vehicle control (DMSO) for 72 hours.
shRNA knockdown
MISSION® shRNA Lentiviral Transduction Particles against Hif-1α (CCGGTGGATAGCGATATGGTCAATGCTCGAGCATTGACCATATCGCTATCCATTTTTG) and control non-target shRNA were purchased from Sigma-Aldrich (St. Louis, MO). Primary chondrocytes were grown to 25–30% confluence and transduced with Hif-1α shRNA or scramble control lentivirus at a multiplicity of infection (MOI) of 10 by adding a pre-made viral particle stock solution (1.4 × 107) into 6-cm culture plates in the presence of 8 μg/ml of polybrene for 2 days, followed by puromycin selection (10 μg/ml) for 3–5 days as previously described43. The puromycin selected cells werepropagated and used for gene expression experiments.
Quantitative RT-PCR
RNA was extracted from cells with Trizol reagent according to the manufacturer’s instructions (Invitrogen, Grand Island, NY). An aliquot of RNA (400 ng) from each sample was reverse-transcribed into cDNA and mRNA levels were followed by quantitative real time PCR as previously described44. Relative gene expression values were calculated using the ∆∆CT method with Ppia mRNA serving as an internal control44. Primer sequences are as following: p57Kip2, forward, 5′-GGAGCAGGACGAGAATCAAG-3′, reverse, 5′-GAAGAAGTCGTTCGCATTGG-3′, cyclin D1, forward, 5′-AATGTACTCTGCTTTGCTGAA-3′, reverse, 5′-ATGAGACCACTAGAGGTCG-3′, tenascin C, forward, 5′-TGACTGCAGCAACCAAGGAC-3′, reverse, 5′-AGTGACCCGCATCTCATTGT-3′, clusterin, forward, 5′-AGAAGGTGAAGATGACCGCA-3′, reverse, 5′-CTTGTACTGCTCTGTCAGCC-3′. Other primer sequences were listed in previous publications13,14,45.
Statistics
The student’s T-test was used for statistical analysis. P < 0.05 was considered statistically significant. All data were presented as mean ± SEM (standard error of the mean).
Additional Information
How to cite this article: Cheng, S. et al. Conditional Deletion of the Phd2 Gene in Articular Chondrocytes Accelerates Differentiation and Reduces Articular Cartilage Thickness. Sci. Rep. 7, 45408; doi: 10.1038/srep45408 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
All work was performed at facilities provided by the US Department of Veterans Affairs. The authors would like to thank Nancy Lowen for technical assistance and Dr. Donna Strong for proof reading the manuscript. SM is a recipient of a Senior Research Career Scientist Award from the Department of Veterans Affairs. This study was supported by funding from the Veterans Administration BLR&D merit review grant (101-BX-001396) to SM and the National Institutes of Arthritis and Musculoskeletal Diseases R01 grant (AR048139) to SM.
Footnotes
The authors declare no competing financial interests.
Author Contributions S.M. designed the study, interpreted the data and wrote the manuscript. S.C. performed the described experiments, analyzed the data, and wrote the manuscript. S.P. and C.A. performed study experiments and analyzed the data.
References
- Staines K. A., Pollard A. S., McGonnell I. M., Farquharson C. & Pitsillides A. A. Cartilage to bone transitions in health and disease. J Endocrinol 219, R1–R12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musumeci G. et al. Histochemistry as a unique approach for investigating normal and osteoarthritic cartilage. Eur J Histochem 58, 2371 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia B. et al. Osteoarthritis pathogenesis: a review of molecular mechanisms. Calcified tissue international 95, 495–505 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldring S. R. & Goldring M. B. Changes in the osteochondral unit during osteoarthritis: structure, function and cartilage-bone crosstalk. Nat Rev Rheumatol 12, 632–644 (2016). [DOI] [PubMed] [Google Scholar]
- Grogan S. P. et al. Zone-specific gene expression patterns in articular cartilage. Arthritis Rheum 65, 418–428 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Las Heras F., Gahunia H. K. & Pritzker K. P. Articular cartilage development: a molecular perspective. Orthop Clin North Am 43, 155–171, v (2012). [DOI] [PubMed] [Google Scholar]
- Decker R. S., Koyama E. & Pacifici M. Genesis and morphogenesis of limb synovial joints and articular cartilage. Matrix Biol 39, 5–10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto M., Ohta Y., Larmour C. & Enomoto-Iwamoto M. Toward regeneration of articular cartilage. Birth Defects Res C Embryo Today 99, 192–202 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi H. Endochondral ossification signals in cartilage degradation during osteoarthritis progression in experimental mouse models. Mol Cells 25, 1–6 (2008). [PubMed] [Google Scholar]
- van der Kraan P. M. & van den Berg W. B. Chondrocyte hypertrophy and osteoarthritis: role in initiation and progression of cartilage degeneration? Osteoarthritis Cartilage 20, 223–232 (2012). [DOI] [PubMed] [Google Scholar]
- Bobinac D., Spanjol J., Zoricic S. & Maric I. Changes in articular cartilage and subchondral bone histomorphometry in osteoarthritic knee joints in humans. Bone 32, 284–290 (2003). [DOI] [PubMed] [Google Scholar]
- Stok K. S. et al. Revealing the interplay of bone and cartilage in osteoarthritis through multimodal imaging of murine joints. Bone 45, 414–422 (2009). [DOI] [PubMed] [Google Scholar]
- Cheng S., Aghajanian P., Pourteymoor S., Alarcon C. & Mohan S. Prolyl Hydroxylase Domain-Containing Protein 2 (Phd2) Regulates Chondrocyte Differentiation and Secondary Ossification in Mice. Sci Rep 6, 35748 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S., Xing W., Pourteymoor S., Schulte J. & Mohan S. Conditional Deletion of Prolyl Hydroxylase Domain-Containing Protein 2 (Phd2) Gene Reveals Its Essential Role in Chondrocyte Function and Endochondral Bone Formation. Endocrinology 157, 127–140 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong G. H. & Takeda K. Role and regulation of prolyl hydroxylase domain proteins. Cell Death Differ 15, 635–641 (2008). [DOI] [PubMed] [Google Scholar]
- Epstein A. C. et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43–54 (2001). [DOI] [PubMed] [Google Scholar]
- Berra E., Ginouves A. & Pouyssegur J. The hypoxia-inducible-factor hydroxylases bring fresh air into hypoxia signalling. EMBO Rep 7, 41–45 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lando D., Peet D. J., Whelan D. A., Gorman J. J. & Whitelaw M. L. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 295, 858–861 (2002). [DOI] [PubMed] [Google Scholar]
- Schipani E. et al. Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev 15, 2865–2876 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarilio R. et al. HIF1alpha regulation of Sox9 is necessary to maintain differentiation of hypoxic prechondrogenic cells during early skeletogenesis. Development 134, 3917–3928 (2007). [DOI] [PubMed] [Google Scholar]
- Provot S. et al. Hif-1alpha regulates differentiation of limb bud mesenchyme and joint development. J Cell Biol 177, 451–464 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki W. et al. Cdc42 is critical for cartilage development during endochondral ossification. Endocrinology 156, 314–322 (2015). [DOI] [PubMed] [Google Scholar]
- Wu M. et al. Chondrocyte-specific knockout of Cbfbeta reveals the indispensable function of Cbfbeta in chondrocyte maturation, growth plate development and trabecular bone formation in mice. Int J Biol Sci 10, 861–872 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G. et al. PTEN deficiency causes dyschondroplasia in mice by enhanced hypoxia-inducible factor 1alpha signaling and endoplasmic reticulum stress. Development 135, 3587–3597 (2008). [DOI] [PubMed] [Google Scholar]
- Zhang J. et al. Smad4 is required for the normal organization of the cartilage growth plate. Dev Biol 284, 311–322 (2005). [DOI] [PubMed] [Google Scholar]
- Riddle R. C., Khatri R., Schipani E. & Clemens T. L. Role of hypoxia-inducible factor-1alpha in angiogenic-osteogenic coupling. J Mol Med (Berl) 87, 583–590 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. et al. The hypoxia-inducible factor alpha pathway couples angiogenesis to osteogenesis during skeletal development. J Clin Invest 117, 1616–1626 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobasheri A., Richardson S., Mobasheri R., Shakibaei M. & Hoyland J. A. Hypoxia inducible factor-1 and facilitative glucose transporters GLUT1 and GLUT3: putative molecular components of the oxygen and glucose sensing apparatus in articular chondrocytes. Histol Histopathol 20, 1327–1338 (2005). [DOI] [PubMed] [Google Scholar]
- Maes C. et al. Increased skeletal VEGF enhances beta-catenin activity and results in excessively ossified bones. EMBO J 29, 424–441 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng T. et al. Inactivation of Vhl in osteochondral progenitor cells causes high bone mass phenotype and protects against age-related bone loss in adult mice. J Bone Miner Res 29, 820–829 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelse K. et al. Role of hypoxia-inducible factor 1 alpha in the integrity of articular cartilage in murine knee joints. Arthritis Res Ther 10, R111 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudoh K., Nakamura H., Masuko-Hongo K., Kato T. & Nishioka K. Catabolic stress induces expression of hypoxia-inducible factor (HIF)-1 alpha in articular chondrocytes: involvement of HIF-1 alpha in the pathogenesis of osteoarthritis. Arthritis Res Ther 7, R904–914 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfander D., Cramer T. & Swoboda B. Hypoxia and HIF-1alpha in osteoarthritis. Int Orthop 29, 6–9 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfander D. et al. Vascular endothelial growth factor in articular cartilage of healthy and osteoarthritic human knee joints. Ann Rheum Dis 60, 1070–1073 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith B., Johnson R. S. & Simon M. C. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer 12, 9–22 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T. et al. Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat Med 16, 678–686 (2010). [DOI] [PubMed] [Google Scholar]
- Yang S. et al. Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat Med 16, 687–693 (2010). [DOI] [PubMed] [Google Scholar]
- Minamishima Y. A. et al. Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood 111, 3236–3244 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovchinnikov D. A., Deng J. M., Ogunrinu G. & Behringer R. R. Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis 26, 145–146 (2000). [PubMed] [Google Scholar]
- Cheng S. et al. Transgenic overexpression of ephrin b1 in bone cells promotes bone formation and an anabolic response to mechanical loading in mice. PLoS One 8, e69051 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasson S. S., Blanchet T. J. & Morris E. A. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage 15, 1061–1069 (2007). [DOI] [PubMed] [Google Scholar]
- Cheng S., Xing W., Zhou X. & Mohan S. Haploinsufficiency of Osterix in Chondrocytes Impairs Skeletal Growth in Mice. Physiol Genomics 45, 917–23 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing W., Pourteymoor S. & Mohan S. Ascorbic acid regulates osterix expression in osteoblasts by activation of prolyl hydroxylase and ubiquitination-mediated proteosomal degradation pathway. Physiol Genomics 43, 749–757 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S. et al. Targeted disruption of ephrin B1 in cells of myeloid lineage increases osteoclast differentiation and bone resorption in mice. PLoS One 7, e32887 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S., Xing W., Pourteymoor S. & Mohan S. Conditional disruption of the prolyl hydroxylase domain-containing protein 2 (Phd2) gene defines its key role in skeletal development. J Bone Miner Res 29, 2276–2286 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.