

Abstract
Aldosterone mediates many of the physiological and pathophysiological/cardio-toxic effects of angiotensin II (AngII). Its synthesis and secretion from the zona glomerulosa cells of the adrenal cortex, elevated in chronic heart failure (HF), is induced by AngII type 1 receptors (AT1Rs). The AT1R is a G protein-coupled receptor, mainly coupling to Gq/11 proteins. However, it can also signal through β-arrestin-1 (βarr1) or -2 (βarr2), both of which mediate G protein-independent signaling. Over the past decade, a second, Gq/11 protein-independent but βarr1-dependent signaling pathway emanating from the adrenocortical AT1R and leading to aldosterone production has become appreciated. Thus, it became apparent that AT1R antagonists that block both pathways equally well are warranted for fully effective aldosterone suppression in HF. This spurred the comparison of all of the currently marketed angiotensin receptor blockers (ARBs, AT1R antagonists or sartans) at blocking activation of the two signaling modes (G protein-, and βarr1-dependent) at the AngII-activated AT1R and hence, at suppression of aldosterone in vitro and in vivo. Although all agents are very potent inhibitors of G protein activation at the AT1R, candesartan and valsartan were uncovered to be the most potent ARBs at blocking βarr activation by AngII and at suppressing aldosterone in vitro and in vivo in post-myocardial infarction HF animals. In contrast, irbesartan and losartan are virtually G protein-“biased” blockers at the human AT1R, with very low efficacy for βarr inhibition and aldosterone suppression. Therefore, candesartan and valsartan (and other, structurally similar compounds) may be the most preferred ARB agents for HF pharmacotherapy, as well as for treatment of other conditions characterized by elevated aldosterone.
Keywords: Adrenal cortex, Adrenocortical zona glomerulosa cell, Aldosterone, Angiotensin receptor blocker, Angiotensin II type 1 receptor, β-arrestin-1, Heart failure, Suppression efficacy
Core tip: The angiotensin II type 1 receptor (AT1R) endogenously expressed in adrenocortical cells was known for decades to induce aldosterone production via a well-defined Gq protein-mediated signaling pathway. Over the past decade, a number of studies have elucidated another, β-arrestin-1 (βarr1)-dependent signaling cascade, which proceeds in parallel to, and independently of the Gq-mediated one, and also results in aldosterone synthesis and secretion from the adrenal cortex. Importantly, although all of the Food and Drug Administration-approved angiotensin receptor blocker (ARB) drugs (AT1R antagonists) are very effective at blocking the Gq-mediated pathway, as expected, since they were designed to do so (i.e., to block the G protein signaling of the AT1R), they seem to display varying efficacies at blocking this new, βarr1-dependent pathway, which translates into significant variation at aldosterone suppression efficacies. In that context, candesartan and valsartan appear the most effective agents at blocking also the βarr1 pathway emanating from the adrenocortical AT1R, and thus, these two agents may be the best aldosterone suppressors within the ARB drug class.
INTRODUCTION
Aldosterone is a mineralocorticoid hormone with several cardio-toxic actions, whose plasma levels are extremely high in chronic heart failure (HF) negatively affecting progression of the disease[1]. Amongst its main actions on the failing myocardium is overall promotion of adverse remodeling via maladaptive hypertrophy, chamber dilatation, collagen deposition and fibrosis, increased inflammation and reactive oxygen species production, etc. The net result of all of these effects is acceleration of cardiac functional decline[2-4]. The main source of circulating aldosterone is the adrenocortical zona glomerulosa (AZG) cells, which synthesize and secrete it in response to high serum K+ levels (hyperkalemia), since its main action on the kidneys is K+ excretion (along with Na+ and water reabsorption)[5]. Another powerful physiological stimulus for aldosterone secretion from AZG cells is the octapeptide hormone angiotensin II (AngII), which activates its type 1 receptors (AT1Rs), endogenously expressed in AZG cells[5,6].
The AT1R is a 7-transmembrane-spanning or G protein-coupled receptor (GPCR); upon agonist activation, it couples primarily to the Gq/11 family of G proteins[6]. Nowadays however, it is known to signal also through other types of G proteins, like Gi/o and Gs, as well as through G protein-independent pathways mediated by the universal GPCR adapter proteins β-arrestin-1 (βarr1) and βarr2 (also known as arrestin-2 and -3, respectively)[7-9]. The βarrs bind agonist-activated and GPCR-kinase (GRK)-phosphorylated GPCRs to uncouple them from G proteins (receptor desensitization) and to target them to clathrin-coated vesicles for internalization (receptor endocytosis). At the same time, they initiate their own, “second wave” of signal transduction independently of G proteins[10-13].
ANGII-DEPENDENT ALDOSTERONE PRODUCTION: THE SUM OF TWO SIGNALING MODALITIES
The Gq/11 protein-dependent signaling pathway elicited by the AngII-activated AT1R that culminates in aldosterone synthesis and secretion in AZG cells has been well characterized (Figure 1)[14]. More specifically, diacylglycerol (DAG) and inositol trisphosphate (IP3), the two second messengers produced by this pathway, ultimately lead to: (1) aldosterone secretion, via elevated intracellular free Ca2+ concentration, which directly stimulates exocytosis and hormonal (in the context of AZG cells, aldosterone) secretion; and (2) aldosterone synthesis, via extracellular signal-regulated kinase (ERK) MAPK activation, which, in turn, stimulate aldosterone biosynthesis in AZG cells by transcriptionally upregulating the StAR (steroidogenic acute regulatory) protein[14]. This protein mediates the mitochondrial uptake of the precursor of all adrenal steroids cholesterol and is the rate-limiting enzyme of aldosterone biosynthesis in AZG cells[14].
Figure 1.
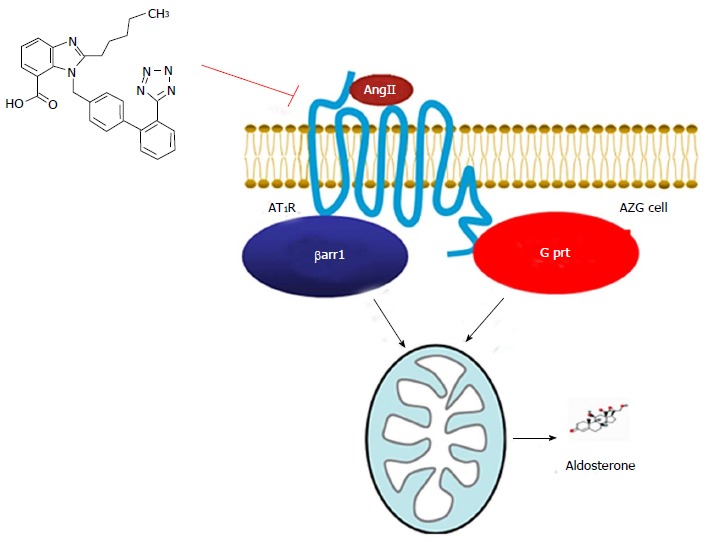
Angiotensin II type 1 receptor and aldosterone production. Schematic representation of the parallel G prt- and βarr1-mediated, AngII-bound AT1R signaling cascades that converge on mitochondrial aldosterone synthesis in adrenocortical zona glomerulosa (AZG) cells. The structure of the proposed AT1R antagonist (2-pentyl-1-({4-[2-(2H-1,2,3,4-tetrazol-5-yl)phenyl]phenyl}methyl)-1H-1,3-benzodiazole-7-carboxylic acid)[40], discussed in the text, capable of suppressing both pathways equally well, is also shown (upper left corner). See text for details. G prt: Gq protein; βarr1: β-arrestin1; AngII: Angiotensin II; AT1R: AngII type 1 receptor.
In the chronic HF setting, adrenal GRK2 is upregulated and, along with βarr1, hyperphosphorylates and severely desensitizes the sympatho-inhibitory α2-adrenergic receptors (ARs) of chromaffin cells in the adrenal medulla[15-21]. The result of this is chronic elevation of adrenal catecholamine secretion, which significantly contributes to the heightened sympathetic nervous system outflow and increased norepinephrine and epinephrine levels that further damage the failing heart[22-26]. Since aldosterone is also increased in HF and its production is stimulated by the AT1Rs of the adrenal cortex[1], which are also GRK2 and βarr1 substrates, it was theorized that the upregulated (in HF) adrenal GRK2 could lead to excessive interaction of βarr1 also with the AT1R in the adrenal cortex, thereby modulating aldosterone secretion in the chronic HF setting, as well. Indeed, this was found to be the case[27]. Via a combination of in vitro experiments in the human AZG cell line H295R and in vivo experiments in experimental rats developing HF following an acute, surgically induced myocardial infarction (MI), we were able to show that adrenal βarr1 actually promotes AngII-dependent aldosterone synthesis and secretion by also mediating AT1R signaling to ERK-dependent StAR upregulation independently of G proteins (Figure 1)[27,28]. This finding was somewhat surprising, given that βarr1 would normally be expected to reduce AngII-dependent aldosterone production thanks to desensitizing the AT1R (terminating its G protein-dependent signaling, see above). Nevertheless, it was discovered that, after abolishing the Gq-dependent signaling by the AT1R in AZG cells, AT1R-bound βarr1 initiated its own signaling to aldosterone synthesis by recruiting a DAG-kinase to the activated receptor[29], which converted the second messenger lipid DAG to phosphatidic acid (PA)[27]. PA can directly activate the small (monomeric) G protein Ras at the plasma membrane, which then initiates the cascade that results in ERK phosphorylation and activation[30]. Thus, AT1R-activated βarr1 elicits a “second (delayed) wave” of signaling leading to sustained ERK activation in AZG cells in its own right (i.e., independently of G proteins), which, as discussed above, promotes aldosterone production via StAR upregulation[27]. Importantly, since StAR regulates synthesis not only of aldosterone but of all adrenal steroids throughout the three anatomical zones of the adrenal cortex[14], adrenal βarr1 may also affect the synthesis of glucocorticoids and of androgens in the adrenal cortex.
Notably, adrenal βarr1 may not only stimulate the AT1R-dependent aldosterone synthesis via its “second wave” of signaling to ERK-dependent StAR upregulation but also facilitate the acute AT1R-dependent aldosterone secretion at the plasma membrane of AZG cells and in parallel to the G protein-mediated signaling by the receptor (Figure 1). Recent evidence in transfected heterologous systems suggests such a role in the “first wave” of GPCR signaling for the βarrs[31,32] and a very intriguing study, done specifically in the adrenal medulla, suggested an acute stimulation of catecholamine secretion and of Ca2+-dependent exocytosis by AT1R-activated βarr1 (but interestingly not by βarr2) in adrenal chromaffin cells, thanks to its direct interaction with the plasma membrane Ca2+ channel short transient receptor potential channel-3 (TRPC3)[33]. Thus, it is quite plausible that AT1R-bound βarr1 can directly stimulate TRPC3-dependent Ca2+ currents and hence, exocytosis, also in AZG cells, thereby acutely stimulating AngII-dependent aldosterone secretion within seconds of agonist binding (and in parallel to the Gq-mediated signaling by the AT1R). This interesting possibility of another signaling mechanism by which βarr1 can induce aldosterone production in AZG cells is definitely worthy of investigation in future studies.
Most importantly, adrenal βarr1-dependent aldosterone production has been documented to occur also in vivo, both under physiological (in normal, healthy animals) and pathophysiological (in the post-MI HF setting) conditions[27,28]. Specifically, adrenal-targeted βarr1 overexpression increased aldosterone serum levels in vivo in normal rats[27], and caused severe hyperaldosteronism also in post-MI rats on top of the circulating aldosterone elevation normally occurring due to the MI injury[28]. Importantly, in the latter animals, adrenal-specific βarr1 blockade in vivo with a βarr1 C-terminal fragment during post-MI HF progression helped stall the decline of cardiac function and even reversed several aspects/markers of adverse cardiac remodeling courtesy of normalization of circulating aldosterone levels[28]. What’s more, aldosterone levels remarkably show no increase in βarr1-knockout mice post-MI, which further highlights the importance of adrenal βarr1 in regulation of circulating aldosterone levels[34]. Together, these in vivo studies strongly suggest adrenal βarr1, in conjunction with GRK2, as an attractive therapeutic target for diseases associated with, and aggravated by hyperaldosteronism, such as post-MI HF[9,25]. Adding to its importance as a therapeutic target is also the fact that aldosterone can produce effects independently of its mineralocorticoid receptor (MR) (the so-called “non-genomic” actions of aldosterone)[4]. Obviously, these effects cannot be countered by MR antagonist drugs (e.g., eplerenone, finerenone, spironolactone) and thus, suppression of aldosterone production at its source, i.e., the adrenal cortex, via adrenal βarr1 blockade would be much more preferable from the therapeutic standpoint.
WHICH ARB DRUG WINS THE ALDOSTERONE SUPPRESSION “CONTEST”?
The realization that AngII-dependent aldosterone production from the adrenal cortex proceeds through two independent signaling modalities, i.e., Gq protein- and βarr1-dependent (Figure 1), signaled that complete blockade of both of these modalities is needed to attain full suppression of adrenal aldosterone production and effectively lower circulating aldosterone levels in HF and in other diseases. This, coupled with the fact that some AT1R antagonist drugs (angiotensin receptor blockers, ARBs, or sartans) appear ineffective at lowering aldosterone in HF, despite their full capacity to block AT1R-G protein coupling[35-38], prompted us to test the relative efficacy of the currently available ARBs at inhibiting the βarr1-dependent aldosterone production by the AT1R in an effort to identify the most effective agent(s). Indeed, the prototypic agent of this class, losartan, was found totally ineffective at preventing adrenal βarr1-dependent aldosterone production and combatting hyperaldosteronism post-MI due to very weak antagonism of βarr1 activation by the AT1R[28]. Interestingly however, the active metabolite of losartan EXP1374 was found quite effective at blocking AT1R-dependent aldosterone production and βarr1 activation[39,40].
Upon subsequent head-to-head testing of all the currently Food and Drug Administration (FDA)-approved ARB drugs, it was found that, although all ARBs (including losartan) are potent inhibitors of G protein activation by the AT1R, their potencies at preventing βarr1 activation by the human AT1R in vitro varied enormously[40]. Specifically, candesartan and valsartan appeared the most potent blockers of βarr1 activation and the most efficacious aldosterone suppressors in vitro and in vivo[39,40]. At the opposite end of the spectrum and in addition to losartan, was irbesartan, which was found to be a very weak βarr1 inhibitor and hence, a very ineffective aldosterone suppressor both in vitro and in vivo, despite its excellent G protein-blocking ability[39,40]. The rest of the class fell more or less in the middle of the βarr1 inhibition and AT1R-dependent aldosterone suppression scales, i.e., their potency values were lower than candesartan’s and valsartan’s but much higher than losartan’s and irbesartan’s[39,40]. Importantly, their effects on cardiac function of in post-MI HF animals in vivo were in complete concordance with their effects on circulating aldosterone levels; candesartan and valsartan induced significant improvements in cardiac function and remodeling post-MI, whereas irbesartan and losartan were not able to alter the course of progression of post-MI animals to full-blown HF[39].
IMPLICATIONS FOR HF PHARMACOTHERAPY
It is widely recognized nowadays that the members of the ARB drug class display significant variation in their pharmacological and clinical properties, which has significant repercussion for their use in HF pharmacotherapy[41]. In fact, certain agents have already been shown to afford larger improvements in morbidity and mortality of chronic HF than others[42-45]. Part of the reason for these differences among these agents that belong to the same pharmacological class and share the same mechanism of action (AT1R antagonism) may be differences in their efficacies at combating the hyperaldosteronism that accompanies and burdens chronic HF[1]. In other words, agents that suppress aldosterone effectively are bound to work better for HF therapy and, since adrenal βarr1 plays a pivotal role in regulation of this cardio-toxic hormone’s levels, the ARBs that are most effective at blocking the AT1R-βarr1 interaction in the adrenal cortex would be expected to be preferred agents. In that vein, our aforementioned recent findings that candesartan and valsartan are the most efficacious βarr1 inhibitors at the AT1R, coupled with their excellent efficacy at lowering aldosterone in vitro and in vivo, point to these two ARBs as being the most preferable agents of their class to use in HF treatment (and in other hyperaldosteronic conditions, e.g., salt-sensitive hypertension). In contrast, irbesartan and losartan were found very weak βarr1-dependent aldosterone inhibitors, a finding that may have some bearing on the lack of therapeutic benefit of these two agents demonstrated in HF with preserved ejection fraction (HF-PEF) and on their therapeutic inferiority to candesartan in terms of HF mortality reduction[44,45]. Of course, future trials providing data on the serum aldosterone levels of the ARB-treated HF patients are needed to confirm such a link between adrenal βarr1-dependent aldosterone suppression efficacy and clinical benefit for this important cardiovascular drug class.
On the other hand, failure of these agents to suppress aldosterone, otherwise referred to as “aldosterone breakthrough” or “aldosterone escape”, is a clinically well-documented phenomenon[46-49] and the efficacy of each agent at inhibiting βarr1-dependent aldosterone production may be inversely proportional to the probability of the ARB to exhibit it. In other words, the more potent βarr1-dependent aldosterone suppressor an ARB is, the lower the likelihood is that the treated patient will suffer from “aldosterone breakthrough”. Thus, candesartan and valsartan may be the safest ARB drugs to use in HF patients in terms of the risk of “aldosterone breakthrough”. However, large trials closely monitoring the circulating aldosterone levels of treated patients are again needed in order to confirm this hypothesis.
IMPLICATIONS FOR AT1R BLOCKER MEDICINAL CHEMISTRY
The studies on the relative potencies/efficacies of the currently FDA-approved ARBs at inhibiting AT1R-βarr1 interaction and βarr1-dependent aldosterone turnover provided some interesting medicinal chemistry and pharmacological insights, as well. Specifically, as far as the ARBs that are tetrazolo-biphenyl-methyl derivatives are concerned, which is a subgroup that includes losartan (and its metabolite EXP1374), irbesartan, candesartan, valsartan, and olmesartan, it was concluded that a substitution both bulky and negatively charged attached to the one side of the methylene group of the biphenyl-methyl backbone (the other end has the tetrazolo-biphenyl group attached) is needed to confer good inhibitory potency of βarr1 at the AT1R and consequently, effectively suppress aldosterone[40]. Indeed, both candesartan and valsartan, as well as EXP1374, have spacious, long aliphatic chain-containing and anionic (carboxylic acid) groups attached to that end of the biphenyl-methyl backbone[40]. In contrast, both losartan and irbesartan possess neutrally charged (unionizable) groups (albeit also bulky) at that biphenyl-methyl backbone end[40]. Finally, olmesartan, which also has an anionic (carboxylic acid) substitution but of intermediate bulkiness (i.e., less long aliphatic chain) compared to candesartan and valsartan on that side of its backbone, displays intermediate potency at inhibiting βarr1 activation and suppressing aldosterone[40]. Based on these observations, we have designed the compound 2-pentyl-1-({4-[2-(2H-1,2,3,4-tetrazol-5-yl)phenyl]phenyl}methyl)-1H-1,3-benzodiazole-7-carboxylic acid (Figure 1)[40], which carries a bulky, carboxyl acid group with a long aliphatic chain on the other end of the tetrazolo-biphenyl-methylene backbone, and we are currently testing both its potency at blocking βarr1 activation by the human AT1R and its efficacy at suppressing aldosterone secretion in vitro and in the post-MI HF setting in vivo. Our hope is that it will prove to be even more efficacious than candesartan and valsartan at suppressing aldosterone levels and thus, an even better drug for HF treatment than all currently available ARBs. Of course, the above mentioned ARB structure-activity relationship inferences have to be confirmed by crystal structure resolutions of the AT1R bound to βarrs. The first glimpse into the human AT1R crystal structure was recently provided and it was the first step towards that goal[50]. Unfortunately however, that crystal structure lacked the intracellular C-terminal tail of the receptor, which is exactly the AT1R region that interacts with βarrs [51].
CONCLUSION
A head-to-head comparison of the ARBs currently on the United States market identified candesartan and valsartan as the most potent βarr1 antagonists and the most efficacious aldosterone suppressors at the human AT1R. Conversely, irbesartan and losartan were found to be largely G protein-“biased” inhibitors, with minimal efficacy towards inhibition of AngII-dependent aldosterone production. Thus, from a therapeutic standpoint, candesartan and valsartan may be the most preferable agents of this drug class, as they provide the biggest benefit for cardiac function and patient survival in post-MI HF and have the lowest propensity to cause the “aldosterone escape” adverse effect (failure to suppress aldosterone). Future studies on this class of drugs and on the effects of βarrs at the adrenal AT1R will help solidify these inferences and will also provide additional important information regarding AngII/AT1R pharmacology for clinicians and medicinal chemists alike.
Footnotes
Conflict-of-interest statement: Both authors declare no conflict of interest related to this publication.
Manuscript source: Invited manuscript
Specialty type: Cardiac and cardiovascular systems
Country of origin: United States
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: August 12, 2016
First decision: October 20, 2016
Article in press: December 19, 2016
P- Reviewer: Lin GM, Said SAM, Ueda H S- Editor: Gong XM L- Editor: A E- Editor: Wu HL
References
- 1.Weber KT. Aldosterone in congestive heart failure. N Engl J Med. 2001;345:1689–1697. doi: 10.1056/NEJMra000050. [DOI] [PubMed] [Google Scholar]
- 2.Connell JM, Davies E. The new biology of aldosterone. J Endocrinol. 2005;186:1–20. doi: 10.1677/joe.1.06017. [DOI] [PubMed] [Google Scholar]
- 3.Marney AM, Brown NJ. Aldosterone and end-organ damage. Clin Sci (Lond) 2007;113:267–278. doi: 10.1042/CS20070123. [DOI] [PubMed] [Google Scholar]
- 4.Zhao W, Ahokas RA, Weber KT, Sun Y. ANG II-induced cardiac molecular and cellular events: role of aldosterone. Am J Physiol Heart Circ Physiol. 2006;291:H336–H343. doi: 10.1152/ajpheart.01307.2005. [DOI] [PubMed] [Google Scholar]
- 5.Ganguly A, Davis JS. Role of calcium and other mediators in aldosterone secretion from the adrenal glomerulosa cells. Pharmacol Rev. 1994;46:417–447. [PubMed] [Google Scholar]
- 6.de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000;52:415–472. [PubMed] [Google Scholar]
- 7.Lefkowitz RJ, Rajagopal K, Whalen EJ. New roles for beta-arrestins in cell signaling: not just for seven-transmembrane receptors. Mol Cell. 2006;24:643–652. doi: 10.1016/j.molcel.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 8.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 9.Lymperopoulos A, Bathgate A. Arrestins in the cardiovascular system. Prog Mol Biol Transl Sci. 2013;118:297–334. doi: 10.1016/B978-0-12-394440-5.00012-7. [DOI] [PubMed] [Google Scholar]
- 10.Lymperopoulos A, Negussie S. βArrestins in cardiac G protein-coupled receptor signaling and function: partners in crime or “good cop, bad cop”? Int J Mol Sci. 2013;14:24726–24741. doi: 10.3390/ijms141224726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lymperopoulos A, Bathgate A. Pharmacogenomics of the heptahelical receptor regulators G-protein-coupled receptor kinases and arrestins: the known and the unknown. Pharmacogenomics. 2012;13:323–341. doi: 10.2217/pgs.11.178. [DOI] [PubMed] [Google Scholar]
- 12.Capote LA, Mendez Perez R, Lymperopoulos A. GPCR signaling and cardiac function. Eur J Pharmacol. 2015;763:143–148. doi: 10.1016/j.ejphar.2015.05.019. [DOI] [PubMed] [Google Scholar]
- 13.Luttrell LM, Gesty-Palmer D. Beyond desensitization: physiological relevance of arrestin-dependent signaling. Pharmacol Rev. 2010;62:305–330. doi: 10.1124/pr.109.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rainey WE, Saner K, Schimmer BP. Adrenocortical cell lines. Mol Cell Endocrinol. 2004;228:23–38. doi: 10.1016/j.mce.2003.12.020. [DOI] [PubMed] [Google Scholar]
- 15.Lymperopoulos A, Brill A, McCrink KA. GPCRs of adrenal chromaffin cells & amp; catecholamines: The plot thickens. Int J Biochem Cell Biol. 2016;77:213–219. doi: 10.1016/j.biocel.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Jafferjee M, Reyes Valero T, Marrero C, McCrink KA, Brill A, Lymperopoulos A. GRK2 Up-Regulation Creates a Positive Feedback Loop for Catecholamine Production in Chromaffin Cells. Mol Endocrinol. 2016;30:372–381. doi: 10.1210/me.2015-1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lymperopoulos A, Rengo G, Koch WJ. Adrenal adrenoceptors in heart failure: fine-tuning cardiac stimulation. Trends Mol Med. 2007;13:503–511. doi: 10.1016/j.molmed.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 18.Lymperopoulos A, Rengo G, Funakoshi H, Eckhart AD, Koch WJ. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat Med. 2007;13:315–323. doi: 10.1038/nm1553. [DOI] [PubMed] [Google Scholar]
- 19.Rengo G, Lymperopoulos A, Zincarelli C, Femminella G, Liccardo D, Pagano G, de Lucia C, Cannavo A, Gargiulo P, Ferrara N, et al. Blockade of β-adrenoceptors restores the GRK2-mediated adrenal α(2) -adrenoceptor-catecholamine production axis in heart failure. Br J Pharmacol. 2012;166:2430–2440. doi: 10.1111/j.1476-5381.2012.01972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lymperopoulos A, Rengo G, Gao E, Ebert SN, Dorn GW, Koch WJ. Reduction of sympathetic activity via adrenal-targeted GRK2 gene deletion attenuates heart failure progression and improves cardiac function after myocardial infarction. J Biol Chem. 2010;285:16378–16386. doi: 10.1074/jbc.M109.077859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lymperopoulos A, Rengo G, Zincarelli C, Soltys S, Koch WJ. Modulation of adrenal catecholamine secretion by in vivo gene transfer and manipulation of G protein-coupled receptor kinase-2 activity. Mol Ther. 2008;16:302–307. doi: 10.1038/sj.mt.6300371. [DOI] [PubMed] [Google Scholar]
- 22.Rengo G, Lymperopoulos A, Leosco D, Koch WJ. GRK2 as a novel gene therapy target in heart failure. J Mol Cell Cardiol. 2011;50:785–792. doi: 10.1016/j.yjmcc.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rengo G, Lymperopoulos A, Koch WJ. Future g protein-coupled receptor targets for treatment of heart failure. Curr Treat Options Cardiovasc Med. 2009;11:328–338. doi: 10.1007/s11936-009-0033-5. [DOI] [PubMed] [Google Scholar]
- 24.Lymperopoulos A, Rengo G, Koch WJ. Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ Res. 2013;113:739–753. doi: 10.1161/CIRCRESAHA.113.300308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCrink KA, Brill A, Lymperopoulos A. Adrenal G protein-coupled receptor kinase-2 in regulation of sympathetic nervous system activity in heart failure. World J Cardiol. 2015;7:539–543. doi: 10.4330/wjc.v7.i9.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rengo G, Leosco D, Zincarelli C, Marchese M, Corbi G, Liccardo D, Filippelli A, Ferrara N, Lisanti MP, Koch WJ, et al. Adrenal GRK2 lowering is an underlying mechanism for the beneficial sympathetic effects of exercise training in heart failure. Am J Physiol Heart Circ Physiol. 2010;298:H2032–H2038. doi: 10.1152/ajpheart.00702.2009. [DOI] [PubMed] [Google Scholar]
- 27.Lymperopoulos A, Rengo G, Zincarelli C, Kim J, Soltys S, Koch WJ. An adrenal beta-arrestin 1-mediated signaling pathway underlies angiotensin II-induced aldosterone production in vitro and in vivo. Proc Natl Acad Sci USA. 2009;106:5825–5830. doi: 10.1073/pnas.0811706106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lymperopoulos A, Rengo G, Zincarelli C, Kim J, Koch WJ. Adrenal beta-arrestin 1 inhibition in vivo attenuates post-myocardial infarction progression to heart failure and adverse remodeling via reduction of circulating aldosterone levels. J Am Coll Cardiol. 2011;57:356–365. doi: 10.1016/j.jacc.2010.08.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nelson CD, Perry SJ, Regier DS, Prescott SM, Topham MK, Lefkowitz RJ. Targeting of diacylglycerol degradation to M1 muscarinic receptors by beta-arrestins. Science. 2007;315:663–666. doi: 10.1126/science.1134562. [DOI] [PubMed] [Google Scholar]
- 30.Rizzo MA, Shome K, Watkins SC, Romero G. The recruitment of Raf-1 to membranes is mediated by direct interaction with phosphatidic acid and is independent of association with Ras. J Biol Chem. 2000;275:23911–23918. doi: 10.1074/jbc.M001553200. [DOI] [PubMed] [Google Scholar]
- 31.Eichel K, Jullié D, von Zastrow M. β-Arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat Cell Biol. 2016;18:303–310. doi: 10.1038/ncb3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nuber S, Zabel U, Lorenz K, Nuber A, Milligan G, Tobin AB, Lohse MJ, Hoffmann C. β-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature. 2016;531:661–664. doi: 10.1038/nature17198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu CH, Gong Z, Liang ZL, Liu ZX, Yang F, Sun YJ, Ma ML, Wang YJ, Ji CR, Wang MJ, et al. Arrestin-biased GPCR agonism induces acute catecholamine secretion through TRPC3 coupling. Nat Commun. 2017;8:14335. doi: 10.1038/ncomms14335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bathgate-Siryk A, Dabul S, Pandya K, Walklett K, Rengo G, Cannavo A, De Lucia C, Liccardo D, Gao E, Leosco D, et al. Negative impact of β-arrestin-1 on post-myocardial infarction heart failure via cardiac and adrenal-dependent neurohormonal mechanisms. Hypertension. 2014;63:404–412. doi: 10.1161/HYPERTENSIONAHA.113.02043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nehme JA, Lacolley P, Labat C, Challande P, Robidel E, Perret C, Leenhardt A, Safar ME, Delcayre C, Milliez P. Spironolactone improves carotid artery fibrosis and distensibility in rat post-ischaemic heart failure. J Mol Cell Cardiol. 2005;39:511–519. doi: 10.1016/j.yjmcc.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 36.Benetos A, Lacolley P, Safar ME. Prevention of aortic fibrosis by spironolactone in spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol. 1997;17:1152–1156. doi: 10.1161/01.atv.17.6.1152. [DOI] [PubMed] [Google Scholar]
- 37.Borghi C, Boschi S, Ambrosioni E, Melandri G, Branzi A, Magnani B. Evidence of a partial escape of renin-angiotensin-aldosterone blockade in patients with acute myocardial infarction treated with ACE inhibitors. J Clin Pharmacol. 1993;33:40–45. doi: 10.1002/j.1552-4604.1993.tb03901.x. [DOI] [PubMed] [Google Scholar]
- 38.Struthers AD. Aldosterone escape during ACE inhibitor therapy in chronic heart failure. Eur Heart J. 1995;16 Suppl N:103–106. doi: 10.1093/eurheartj/16.suppl_n.103. [DOI] [PubMed] [Google Scholar]
- 39.Lymperopoulos A, Sturchler E, Bathgate-Siryk A, Dabul S, Garcia D, Walklett K, Rengo G, McDonald P, Koch WJ. Different potencies of angiotensin receptor blockers at suppressing adrenal β-Arrestin1-dependent post-myocardial infarction hyperaldosteronism. J Am Coll Cardiol. 2014;64:2805–2806. doi: 10.1016/j.jacc.2014.09.070. [DOI] [PubMed] [Google Scholar]
- 40.Dabul S, Bathgate-Siryk A, Valero TR, Jafferjee M, Sturchler E, McDonald P, Koch WJ, Lymperopoulos A. Suppression of adrenal βarrestin1-dependent aldosterone production by ARBs: head-to-head comparison. Sci Rep. 2015;5:8116. doi: 10.1038/srep08116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Michel MC, Foster C, Brunner HR, Liu L. A systematic comparison of the properties of clinically used angiotensin II type 1 receptor antagonists. Pharmacol Rev. 2013;65:809–848. doi: 10.1124/pr.112.007278. [DOI] [PubMed] [Google Scholar]
- 42.Eklind-Cervenka M, Benson L, Dahlström U, Edner M, Rosenqvist M, Lund LH. Association of candesartan vs losartan with all-cause mortality in patients with heart failure. JAMA. 2011;305:175–182. doi: 10.1001/jama.2010.1949. [DOI] [PubMed] [Google Scholar]
- 43.Svanström H, Pasternak B, Hviid A. Association of treatment with losartan vs candesartan and mortality among patients with heart failure. JAMA. 2012;307:1506–1512. doi: 10.1001/jama.2012.452. [DOI] [PubMed] [Google Scholar]
- 44.Shah RV, Desai AS, Givertz MM. The effect of renin-angiotensin system inhibitors on mortality and heart failure hospitalization in patients with heart failure and preserved ejection fraction: a systematic review and meta-analysis. J Card Fail. 2010;16:260–267. doi: 10.1016/j.cardfail.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 45.Massie BM, Carson PE, McMurray JJ, Komajda M, McKelvie R, Zile MR, Anderson S, Donovan M, Iverson E, Staiger C, et al. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med. 2008;359:2456–2467. doi: 10.1056/NEJMoa0805450. [DOI] [PubMed] [Google Scholar]
- 46.Sarzani R, Guerra F, Mancinelli L, Buglioni A, Franchi E, Dessì-Fulgheri P. Plasma aldosterone is increased in class 2 and 3 obese essential hypertensive patients despite drug treatment. Am J Hypertens. 2012;25:818–826. doi: 10.1038/ajh.2012.47. [DOI] [PubMed] [Google Scholar]
- 47.Horita Y, Taura K, Taguchi T, Furusu A, Kohno S. Aldosterone breakthrough during therapy with angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers in proteinuric patients with immunoglobulin A nephropathy. Nephrology (Carlton) 2006;11:462–466. doi: 10.1111/j.1440-1797.2006.00665.x. [DOI] [PubMed] [Google Scholar]
- 48.Bomback AS, Klemmer PJ. The incidence and implications of aldosterone breakthrough. Nat Clin Pract Nephrol. 2007;3:486–492. doi: 10.1038/ncpneph0575. [DOI] [PubMed] [Google Scholar]
- 49.Naruse M, Tanabe A, Sato A, Takagi S, Tsuchiya K, Imaki T, Takano K. Aldosterone breakthrough during angiotensin II receptor antagonist therapy in stroke-prone spontaneously hypertensive rats. Hypertension. 2002;40:28–33. doi: 10.1161/01.hyp.0000022606.52221.2f. [DOI] [PubMed] [Google Scholar]
- 50.Zhang H, Unal H, Gati C, Han GW, Liu W, Zatsepin NA, James D, Wang D, Nelson G, Weierstall U, et al. Structure of the Angiotensin receptor revealed by serial femtosecond crystallography. Cell. 2015;161:833–844. doi: 10.1016/j.cell.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balakumar P, Jagadeesh G. Structural determinants for binding, activation, and functional selectivity of the angiotensin AT1 receptor. J Mol Endocrinol. 2014;53:R71–R92. doi: 10.1530/JME-14-0125. [DOI] [PubMed] [Google Scholar]