

Abstract
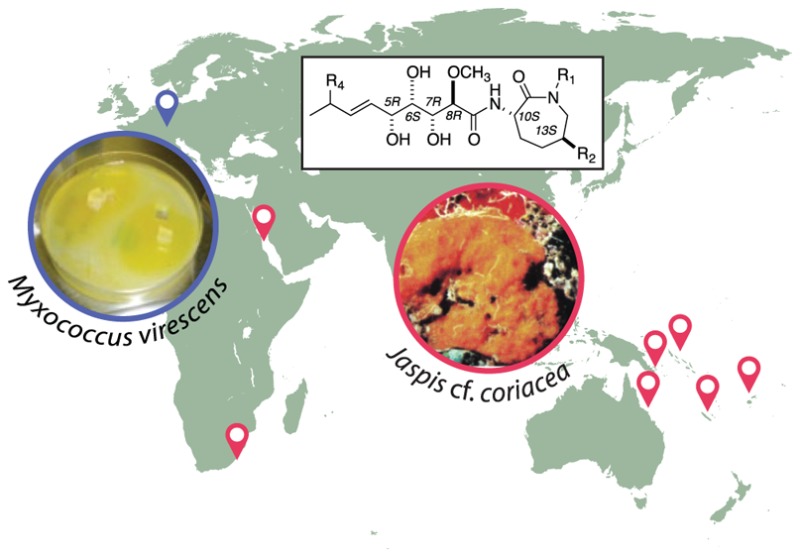
This review focuses entirely on the natural bengamides and selected synthetic analogues that have inspired decades of research. Bengamide A was first reported in 1986 from the sponge Jaspis cf. coriacea, and bengamide-containing sponges have been gathered from many biogeographic sites. In 2005, a terrestrial Gram-negative bacterium, Myxococcus virescens, was added as a source for bengamides. Biological activity data using varying bengamide-based scaffolds has enabled fine-tuning of structure–activity relationships. Molecular target finding contributed to the creation of a synthetic “lead” compound, LAF389, that was the subject of a phase I anticancer clinical trial. Despite clinical trial termination, the bengamide compound class is still attracting worldwide attention. Future breakthroughs based on the bengamide scaffold are possible and could build on their nanomolar in vitro and positive in vivo antiproliferative and antiangiogenic properties. Bengamide molecular targets include methionine aminopeptidases (MetAP1 and MetAP2) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). A mixed PKS/NRPS biosynthetic gene cluster appears to be responsible for creation of the bengamides. This review highlights that the bengamides have driven inspirational studies and that they will remain relevant for future research, even 30 years after the discovery of the first structures.
Introduction
The study of structurally unique small
molecules of the bengamide
class, first isolated from coral reef sponges, has been ongoing throughout
the world for more than three decades. The bengamide chronicle is
an example of research sustained by seeking new knowledge and outcomes
parallel to those realized from the chemical study of terrestrial
biota. The discovery of the bengamides illustrates that the shift
to examining the marine environment was a productive avenue for uncovering
biosynthetic products new to science and not discoverable from terrestrial
species. Such results have stimulated many laboratories to engage
in and develop the field of marine natural products. We view the finding
and continued study of the bengamides as a milestone in a subject
area rooted in part in the quest to build on past lessons learned
from studying communities of rainforest trees and microorganisms.
The research on the bengamides began through bioactivity-guided probing
of coral reef sponge metabolites and has been extended by examining
bengamide-producing terrestrial microorganisms. We predict and discuss
herein how such research will also continue into the future, driven
by the use of molecular genetic tools to examine the sponge and its
microbial associants for output of this compound class.
The story of the structure elucidation, biosynthetic makeup, and chemical biology studies of bengamide analogues through isolation, total synthesis, and biosynthetic engineering is the basis for this review. The bengamides are headed by bengamide A (C31H56N2O8) and bengamide B (C32H58N2O8). These compounds were originally isolated and described at the University of California, Santa Cruz (UCSC), in the early 1980s1a−1d during research on an abundant Indo-Pacific marine sponge, Jaspis cf. coriacea. Interestingly, the lead molecules of this class have a ratio of C/(N + O) = 3.1 in a framework containing six chiral centers. The bengamide molecule consists of a fused polyketide and amino acid moieties,1e and a current awareness similarity search in SciFinder (Chemical Abstracts Service, Columbus, OH, USA) reveals more than 221 structures, most of which are synthetic bengamide analogues.
Our focus is very different from a review published in 2014 by the Sarabia laboratory.2 We believe it is important to trace the pathway from discovery to therapeutic development of the bengamides. The bengamide total structures were first proposed and justified by a combination of NMR analyses, semisynthesis, and formation of chiral derivatives. Subsequently, the structural hypothesis was tested and verified through chiral total synthesis.3 In this review we will touch on how structure elucidation methods and molecular biology-targeted approaches can intersect to provide unexpected insights and prompt questions for future investigations. Another goal is to illustrate the complex and often tortuous path of investigating compounds isolated from sponges for which the true producers may be a consortium rather than a single macro- or microorganism.
Bengamide and Other Sponge Products as New Medicinal Chemistry Leads
Marine natural products research begun in the 1970s at UCSC was mostly discovery driven, and the intent was to focus on sponges. After much trial-and-error, the bengamides were discovered through a productive academic–industry collaboration (established in 1984) between the Crews laboratory at UCSC and that of Thomas R. Matthews, Ph.D., and his co-workers at the Institute of Antiviral and Antimicrobial Chemotherapy at Syntex Research, Inc., Palo Alto, California. The initial aim was to employ antiparasitic primary assays to screen Indo-Pacific sponge extracts. A number of antiparasitic compounds were subsequently purified and evaluated including the bengamides and bengazoles.4 The process involved aggressive compound purification to jumpstart the nascent program seeking to overlay marine natural products and pharmaceutical research. Today, the bengamides, for which the names were inspired by the collection location for this sponge [the Beqa Lagoon of Fiji (pronounced “benga”; still considered to be among the world’s premier barrier reefs)], continue to attract widespread interest as unique marine-derived molecular probes and therapeutic leads. In addition to anticancer activity in humans, the bengamides function as inhibitors of methionine aminopeptidase (MetAPs 1 and 2) and have also been shown to target the NF-κB pathway with the potential to function as potent and selective anti-inflammatory agents.
Sponges, such as those that produce the bengamides, represent wonderful study targets to initiate and extend multidisciplinary studies. Several facets of our work on the sponges producing bengamides are useful to briefly discuss because they illustrate general situations encountered by other research teams. Most of the initial stages of work on sponges, especially those producing bengamides, begin in Nature and sometimes in remote locations. Sponges rich in bengamide constituents are sessile, often prolific (in some but not all environments), have distinct recognizable morphologies (see discussion below), and are easily collected by scuba divers having a correct knowledge of their natural history. The physical characteristics of chemically rich sponges also suggest interesting roles for their constituent small-molecule products. Such sponges will grow toward one another but not on top of each other, and few organisms, with the exception of some nudibranchs and angelfish, graze on sponges despite their high visibility and inability to escape a potential threat! Another fundamental hypothesis is that sponges possess an innate defense mechanism that allows them to occupy and thrive in a fiercely competitive environment. A sampling of chemical scaffold diversity of sponge-derived molecules beyond the bengamide A and B structures includes unusual nucleoside and amino acid derivatives, polyketides, cyclic peptides, macrolides, cyclic polyethers, steroids and sterols, terpenoids, glycosides, and heterocyclic compounds. Recent reviews have been published reinforcing the power of the phylum Porifera as a source of biosynthetic products.
The path to contribute sponge-derived “medicines from the sea” is extremely challenging and continues to be a major stimulus in research focused on the bengamides from sponges, microorganisms, and synthesis. To date, noteworthy contributions have arisen based on using a few marine sponge products to treat diseases afflicting humans. Summarized in Figure 1 are four significant sponge-derived/inspired molecules in current use as medicines; however, the bengamides have not risen to be included in this category. The collection encompasses (a) two unusual nucleosides, ara-A (1) and ara-B (2), for which the storied history has been reviewed,5 (b) eribulin mesylate (3), aka Halaven,6 a synthetic analogue inspired by halichondrin B that blocks microtubules and was approved by the U.S. FDA in 2010,7 and (c) NVP-LBH5898 (4), a broad-spectrum HDAC inhibitor possessing a hydroxamate functionality that was inspired by the structure of the sponge-derived psammaplins, which have potent HDAC activity.9 On the horizon as a new medicine is PM184 (5). It is of mixed biogenetic origin and is in current phase II clinical trials.10
Figure 1.

Sponge-derived biosynthetic products approved for therapeutic use or currently in clinical trials.
The bengamides are among sponge-derived products evaluated but dropped from clinical trials. The current list of 12 such compounds is shown in Figure 2, and one of these, LAF389 (10), was inspired by the bengamide A scaffold. A more detailed discussion on the pre- and postclinical trial developments of LAF389 and its congeners is deferred until later in this review.11 As a prelude to this topic it is important to note that clinical trials are often discontinued due to either insufficient efficacy or off-target toxicity. In the case of LAF389, a bengamide analogue derived by total synthesis, the phase I trial was discontinued due to unanticipated cardiotoxicity. In looking toward the next steps, another structurally different bengamide derivative was synthesized in 2015 (Table 3, benzLAF389) and has demonstrated nanomolar cellular potency, high metabolic stability, and in vivo antitumor efficacy at nontoxic doses in a melanoma mouse model.
Figure 2.

Summary of sponge-derived or inspired chemical entities for which the clinical trials were discontinued.
Table 3. Inhibitory Data for Compounds on MetAP Enzyme Activity and Cell Proliferation Potencies Against Cancer Cell Lines.

| IC50 (μM) |
IC50 (nM) |
|||
|---|---|---|---|---|
| compound | MetAp1 | MetAp2 | MDA-MB-435 | HCT-116 |
| BGM-A53,54 | 0.754/253 | 0.454/1153 | 11e,35 | 0.4,53 161e |
| BGM-O53 | 353 | >5053 | 0.31e | 0.853 |
| LAF389 | 0.754 | 0.454 | 4035 | 4054 |
| LBM648 | >1054 | 0.454 | 14054 | |
| benzLAF389 | 3.838 | 4438 | ||
| FMG | 0.258 | 54 (MB-231)44 | ||
| IV-43 | 0.449/1.561 | –58/>30061 | 300 (HT1080)61 | |
| PZQ | 0.149 | >10049 | ||
There are many structural and functionality differences accompanied by a few heteroatom similarities in the group of sponge-derived/inspired compounds dropped from clinical trials and summarized in Figure 2. The chemical space occupied by all of these entries is quite diverse, and none overlap that of the bengamide-inspired LAF389 (10).12 All structures but one have multiple chiral centers. The achiral compound is NVP-LAQ-824 (17),13 and girolline (7)14 is the least complex. In broad terms this collection can be clustered into five major groupings based on heteroatom diversity: (a) compounds with multiple oxygens, manoalide (6)15a,15b and ILP-576092 (8);16 (b) compounds with multiple oxygens and one nitrogen, discodermolide (9)17 and KRN-7000 (15);18 (c) a compound with one oxygen and multiple nitrogens, girolline (7); (d) compounds with multiple oxygens and multiple nitrogens, LAF389 (10),13 arenastatin A (11),19 HTI-286 (13),20 E7974 (14),21 and NVP-LAQ-824 (17); and (e) a compound with multiple oxygens, multiple nitrogens, and halogens, LY355703 (12)22 and Zalypsis (16).23 The 17 compounds of Figures 1 and 2 clearly show the vital contribution of sponge natural products in the pathway to the discovery of unique bioactive chemical entities.
There are further dimensions to the topic of bioactive and/or novel sponge-derived products discovery. The reader is encouraged to examine sections of recent reviews highlighting the structures of Figure 2 and other compounds as models for the design of drug candidates.24a−24d The content in these publications and the cited literature illustrates victory for the idea of “drugs from the sea” first raised in meetings on this subject in the 1960s and highlighted in several articles published in 1999.25a,25b
Natural History of the Bengamide-Producing Sponge Jaspis cf. coriacea
In 1980, the UCSC chemistry team began conducting annual expeditions to Indo-Pacific coral reef environments. The initial goal was straightforward and involved developing a natural history understanding about abundant coral reef sponges and then establishing which taxa were consistently rich in natural products. Today, such an environmental sampling endeavor would be jumpstarted by consulting guidebooks written by experts. However, at that time, the resources to properly orient scientific diver collection teams did not exist and only appeared around 1995–1996.26a−26c Our initial expeditions from 1980 to 1983 to the Kingdom of Tonga27 afforded disappointing results, prompting a recalibration of the strategies used. After engaging in discussions with marine ecologists, our focus was shifted to Fiji. We believed that sponges from this ecological zone would be higher in diversity than those of the former area. This assumption proved to be correct and was verified during comparisons of the inventory of abundant sponge specimens observed and/or collected during a total of five six-week expeditions to both areas.
Underwater pictures of a representative Fijian sample (collected from 20 to 100 feet) that was prioritized early on is shown in Figure 3 (panel A); this material proved to be a source of the bengamides. Close inspection of the sponge surface revealed distinctive ultrastructural characteristics that were useful in obtaining additional specimens for chemical study. Highlights of the distinctive characteristics for this sponge, now classified as Jaspis cf. coriacea, are (a) a dull orange color; (b) encrusting (0.5–4.0 cm thickness) growth often under ledges; (c) rubbery easily torn texture; (d) flat, sometimes invisible, oscules (unlike the highly raised oscules of Jaspis splendens, also growing nearby and a source of jasplakinolide, and (e) an underside (Figure 3, panel A right) in many but not all populations penetrated by small crustaceans. A full taxonomic description of this material appeared in the experimental section of our 2001 publication.1e Our specimens are closest to that of Jaspis coriacea Carter 1886 classification: class Demospongiae, order Tetractinellida, family Ancorinidae. It is important to note that the specimens studied at UCSC (Figure 3) are not discussed or shown in any current field guides.
Figure 3.

Natural history of the bengamide-producing sponge Jaspis cf. coriacea (suborder Astrophorida; family Ancorinidae), a distinctive orange sponge on benthic substrates from three Indo-Pacific zones: (A) Fiji (the appearance of this encrusting sponge is shown to the left, and when removed from its substrate, the underside of the sponge in many but not all populations is penetrated by crustaceans; (B) Papua New Guinea; (C) Indonesia.
The beginning of the bengamide story can be traced to a brief expedition by the UCSC team to Fiji in 1983. The first environmental site we visited containing Jaspis cf. coriacea was the Beqa lagoon, which was home to numerous colorful freestanding and encrusting sponges of varying morphologies. In 1984, the UCSC team returned to this location in search of an abundant encrusting orange sponge observed the prior year. The material (shown in Figure 3) was found again and collected, but at that juncture it was not clear if any novel secondary metabolites would be isolated from this specimen. However, as further discussed below, during biological screening work it was clear that very bioactive compounds were present in this specimen. Rapidly, bengamides A and B were obtainable in high yield, but it took four years to complete the total compound structure elucidations.
Early lessons learned about the natural history of this particular sponge, discussed above, guided multiple follow-up expeditions to many Indo-Pacific sites where this sponge could be obtained, and these included several locations in Fiji (Beqa Lagoon, Somo Strait, Taveuni/Tasman Strait, and Ngau Island), the Solomon Islands, Papua New Guinea, and Indonesia. Underwater photos of these various specimens are also shown in Figure 3.
Secondary Metabolites of Jaspis cf. coriacea
Over several decades of surveying biosynthetic products from sponges with the morphology shown in Figure 3, the UCSC team always found multiple bengamide analogues.1a,1b,1e However, when two additional classes of nitrogen-containing metabolites were isolated during second-generation studies, it became clear that additional biosynthetic machinery was at work. These added frameworks, shown in Table 1, included the bengazoles and simple diketopiperazines.27−32 At first glance, it is obvious that the biosynthetic pathway producing the bengamides must involve both polyketide synthase (PKS) and nonribosomal peptide synthetase (NRPS) modules. This view has been fine-tuned in recent years and will be discussed more thoroughly later in the review. Clearly, the diketopiperazines are cyclic dipeptide adducts, while the bengazoles are composed of two isoxazoles coupled to a chiral triketide. Surprisingly, at this point, little is actually known about the biosynthetic machinery that assembles the bengazoles. The patterns shown in Table 1 intimate that the pathways producing both compounds seem to be at work in many different sponge species.
Table 1. Molecular Frameworks Isolated from Sponge Sources Headed by Jaspis cf. coriacea as a Function of the Coral Reef Collection Locale and the Taxonomic Names Assigned in Each Investigation (by Taxonomists) to the Collected Sponge.

| taxonomy | color | collection site | chemistry | year(s) |
|---|---|---|---|---|
| Jaspis cf. coriacea (Diaz/van Soest) | orange | Fiji, Indonesia, Papua New Guinea, Solomon Islands | BGM, BGZ, DKP | 1986–19971e |
| Jaspis digonoxeaa (van Soest) | unknown | South Africa | BGM, BGZ, DKP | 199429b |
| Jaspis sp.a (Harper) | unknown | Australia | BGZ | 199629c |
| Jaspis carteri (Hooper/Sanders) | orange | New Caledonia | BGM | 199730 |
| Pachastrissa sp.,a revised to Jaspis (Sanders) | orange | Red Sea | BGM, BGZ | 199931 |
| Jaspis sp.a (van Soest) | unknown | Australia | BGM, BGZ | 199929d |
| Dorypleres splendensa (NA) revised to Jaspis (this review) | orange | Fiji | BGM, BGZ | 200832 |
| Stelletta sp.a (NA) | unknown | Australia | BGM, BGZ, DKP | 201133 |
Original taxonomy not rigorously described.
The luster of the bengamide structure was recognized by others once the UCSC results were published. Eventually seven other laboratories collected and examined specimens and described findings (from 1994 to 2011) parallel to those published by UCSC. The bengamides and bengazoles can be considered “signature” compounds, and overall outcomes from the worldwide examination of sponges producing this pair of substances are shown in Table 1. An interesting pattern appears within these data because the “signature” compounds are produced either by (a) eight different sponge species or (b) closely related sponges for which the taxonomic assignments were not rigorously determined. Overall, six of the eight identifications listed for the genera of Table 1 (coded by a) do not seem credible, as vague taxonomic descriptions are contained in the experimental sections of these publications. It appears that situation (a) is valid for the J. coriacea and J. carteri entries because voucher specimens were assessed side by side,1e and they were concluded to be distinct species. In addition, inspection of the voucher specimen of Pachastrissa sp. supported its revision to the genus Jaspis, but no conclusion about its actual species could be made.1e It is highly likely that the Fijian orange sponge described as Dorypleres splendens (aka Jaspis splendens) was misidentified as this species and is a reliable source of jasplakinolides28 and not bengamides. Finally, and somewhat surprisingly, the 2014 review on the bengamides2 incorrectly showed an underwater picture of J. splendens (and not that of J. coriacea) as the source organism for the bengamides. This situation illustrates the difficulty in correlating sponge taxonomy to a secondary metabolite profile.
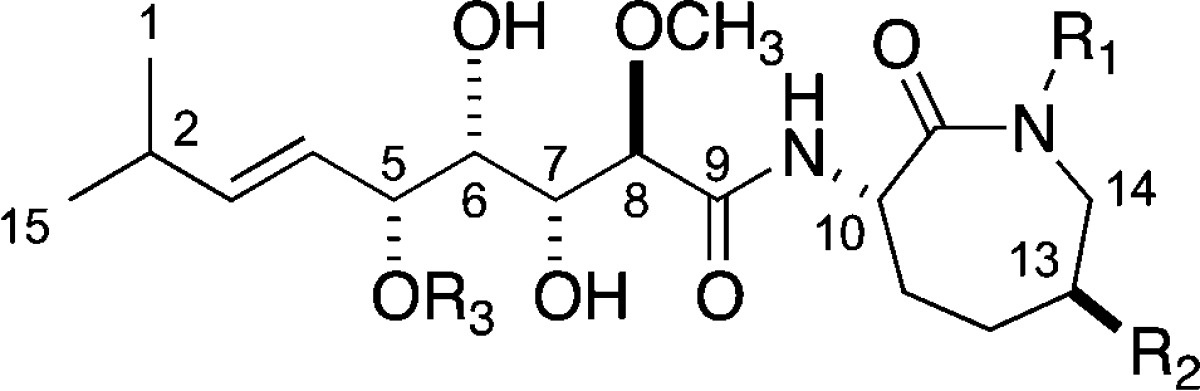
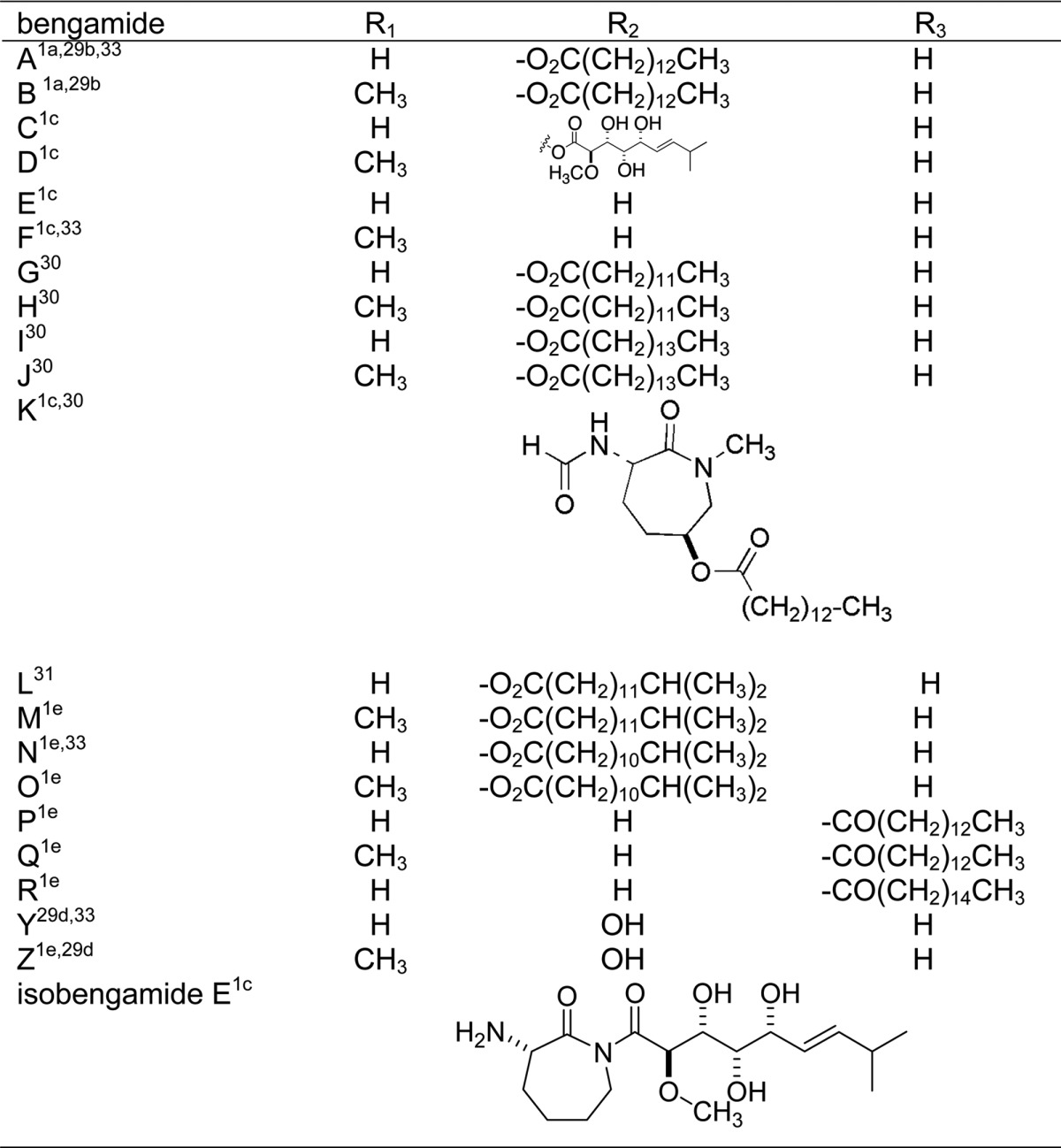
The bengamide core scaffold possesses significant structural variety, illustrating the versatile biosynthetic capabilities of these sponges (listed in Table 1).29−33 To date, 21 different bengamide analogues have been isolated from marine sponges and are listed in Table 2. Regular variation includes a free or methylated caprolactam amide nitrogen (as in bengamides A and B, C and D, for example), variation of the functional group, if present, on C-13 (R2 position), and the presence or lack of an ester at C-5 (R3 position). Bengamide K and isobengamide E fall outside of this regular classification: bengamide K lacks the entire polyketide portion of the molecule and instead possesses an N-formyl group, and isobengamide E is a structural isomer of bengamide E in which the polyketide region is attached to the side chain nitrogen of the caprolactam as opposed to the traditional backbone nitrogen. Interestingly, the first compounds isolated, bengamides A and B,1a persist as the two most biologically interesting natural compounds in the class. The configurations at each of the chiral centers for all the natural compounds follow those shown above in the structures of bengamides A/B.
Table 2. Sponge-Derived Bengamide Structures.
A Timeline Rich in Discoveries
There were two developments responsible for propelling research forward leading to the first publication of the bengamides. First was the chemistry and pharmacology program noted above that began in 1984 between Syntex and UCSC. Second was the UCSC campaign to collect and evaluate sponges from coral reefs of the mid-South Pacific (also initiated in 1984). The initial series of annual summer expeditions to Fijian coral reefs were undertaken to provide extracts for parallel biological and chemical evaluation. One of the most prized sponge samples gathered during 1984–1985 (collection codes 84-20 [0.5 kg] and 85-09 [1.4 kg]) was “an abundant, finger-like orange sponge, which is an undescribed member of the Jaspidae family”.1a Excitement grew for this project because the crude extract (collection code 84-20) exhibited complete in vitro cytotoxicity to larynx epithelial carcinoma cells at 1.0 μg/mL, along with activity against the bacterium Streptococcus pyrogenes and the nematode Nippostrongylus braziliensis. Poor yields obtained from the first extract necessitated examination of re-collected material, which provided an extract subsequently concentrated to an oil (7.4 g). Examination of this extract by 1H NMR spectroscopy (CDCl3, 300 MHz) showed many signals including prominent low-field diagnostic peaks: δ 5.78, 5.44, 4.60, 4.21; all are assignable to regions of the bengamide structure core (see Figure 3 of ref (1e)). The promising initial bioactivity and easily visualized signature 1H NMR peaks portended significant therapeutic potential and provided an NMR handle to identify members of the bengamide family that facilitated many decades of continued discovery. A timeline appears in Figure 4 of the outcomes based on an exploration trajectory including (a) examination of producing organisms, (b) compound discovery, (c) evaluation for potential as a molecular tool, (d) therapeutic development, and (e) formulating hypothesis for compound biogenesis.
Figure 4.

Timeline continuum for the bengamide discovery, exploration, and development as a therapeutic lead and cell biology tool. Three decades of sustained attention have been devoted to the broad-based study on its scaffold.
Enigmatically, it was not until 1992 that the UCSC team sent samples of bengamides A, B, and P (see Table 2) for evaluation in vitro in the National Cancer Institute’s (NCI) 60 cell line screen. The bengamides were found to have a unique activity profile when compared to other agents in the NCI’s database.1e This initial investigation into the cytotoxicity properties of the bengamides elevated the class to a high priority as a potential therapeutic lead and spurred further investigation once a National Cooperative Drug Discovery Groups (NCDDG) project was formalized with Sandoz in 1995. In the late 1990s, the oncology department of Novartis Pharmaceuticals Corporation, in collaboration with UCSC, confirmed that the bengamides possess significant antiproliferative activity against transformed and nontransformed cells.34a,34b This was the beginning of the most dedicated exploration of the bengamide class of compounds as potential commercial therapeutics. The unveiling of the therapeutic potential of the bengamides sparked an explosion of bengamide-related citations starting in the mid 1990s. Shown in Figure 5 is the time course for the publication of 94 papers from multiple laboratories that all explore facets of the chemistry and biology of the bengamides. There are three categories of papers shown: those that focus only on natural products (12), meeting abstracts (8), and chemical-biology research articles (74).
Figure 5.
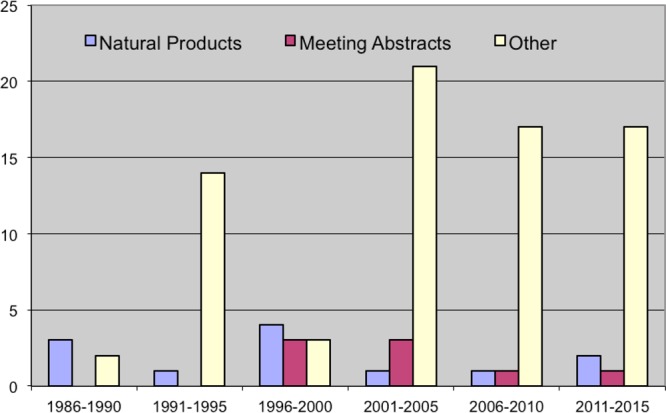
Literature on bengamides covering three decades in five-year increments (1986–2015): natural products publications, meeting abstracts, and other reports (syntheses, bioactivity, medicinal chemistry, patents).
Significant re-collections of J. coriacea sponge from five different locations in Fiji were acquired in 19971e to further therapeutic investigations at Novartis. The sponge collections provided approximately 88 g of a crude mixture of bengamides, afforded sufficient amounts of bengamides B, E, and Z to test against MDA-MB-435 carcinoma cells grown as xenografts in nude mice,35 and provided additional new analogues such as bengamides M–R. The most significant outcome of this preliminary study was the observation that bengamides with a lipophilic ester moiety on the caprolactam (C-13/R2 position), such as bengamides A, B, M, and O, are >100-fold more potent than the nonesterified counterparts and that methylation of the caprolactam nitrogen has little effect on in vitro potency.
Unfortunately, the very poor water solubility of bengamide B (0.002 ng/mL at pH 6.8) limited intravenous administration. This information, coupled with a finite natural supply and a daunting 14-step, low-yielding total synthesis, precluded bengamide B from further preclinical testing. Insights gleaned from the systematic analysis of natural structures and their bioactivities allowed for synthetic efforts to begin. This effort was spearheaded by Kinder at the U.S. Novartis Institute for Biomedical Research (NIBR) and led to the discovery of the lead structure, LAF389.3,35 At the top of the list of subsequent developments was the launch of the phase I clinical trial in 2000 on LAF389. This was made possible by an optimized convergent large-scale chiral synthesis carried out by the Process Research and Development team at Novartis.12 It involved seven linear steps and provided the LAF389 polymorph E (133.6 g, mp 148–9 °C) in 79% yield. The additional milestone developments shown from 2003 to the present will be discussed below. These will focus on the continuing development of insights on the bengamide molecular targets (such as the MetAPs), the isolation of bengamides from terrestrial Gram-negative bacteria, and understanding of the biosynthetic enzymes giving rise to this family.
Insights on the Biosynthetic Assembly of the Bengamdies
All chiral sponge-derived natural products, including the bengamides, arise from genetically encoded processes. By contrast, understanding of the true source organisms responsible for the production of sponge-derived compounds continues to be unresolved. Four distinct hypotheses have appeared in publications describing the pathway responsible for the assembly of the bengamides, and these are shown in Figure 6. An initial biosynthetic proposal appeared in 1989 and involved dissection of the iso-bengamide E structure1c into three constitutive units: S-Lys-diketide-Leu. The idea of a Leu moiety was favored over that involving a branched ketide to explain the isopropyl-containing terminus.
Figure 6.

Evolving hypotheses on the biosynthetic assembly of the bengamides.
Subsequently in 2001, but not shown in Figure 6, it was shown that the structures of bengamide A and E could be annotated with similar biosynthetic subunits.1e This was inspired by the experimental proof showing that barbamide A36 arises from the union of Leu-monoketide-Cys-Phe. In 2014, this 2001 analysis was recycled in the Garcia-Ruiz and Sarabia2 review as shown by the entry in Figure 6 and a passage from that paper, “...biosynthetic origin of the bengamides seems to be a result of a symbiotic interaction between this class of sponges and bacteria...”. Alternatively, in 2006 it seemed plausible to the Crews group that the insights of Challis et al.37 could be exported to create the view shown in Figure 6, consisting of the union of isobutyryl-CoA-diketide-Lys.
The early assumptions about the bengamide biosynthetic machinery proved to be rudimentary and in part an oversimplification. In 2015, the Muller–Bronstrup team published the definitive results that were described in Figure 4 of the paper.38 Molecular genetics experiments resulted in isolation of the bengamide gene cluster (a 731 kDa protein of about 25 kbp and nine genes: coded BenA to BenG) from cultures of the Gram-negative bacterium Myxococcus virescens. This organism produces four bengamides (E/E′, F/F′) and was isolated initially in 2005 from a terrestrial soil sample by scientists at Sanofi Adventis,39 and subsequently a sample of this strain was obtained and cultured at UCSC.40 The idea of a polyketide-nonribosomal peptide hybrid as being responsible for the production of the bengamides was validated, as was the assumption that a lysine unit was incorporated late in the pathway. Totally stunning was the finding that the entire gene cluster could be transferred into a more robust myxobacterial host, Myxococcus xanthus, to yield bengamide analogues in yields up to 5–10 mg/L. The success of three different groups in obtaining useable yields of bengamides from laboratory culturing of Gram-negative microorganisms is an important development.38−40 It also sets the stage for future investigations of the microbiome of Jaspis cf. coriacea.
At this juncture, it is tempting to conclude an organism related to the so-called “metabolically talented” bacterium endosymbiont Entotheonella, believed to be responsible for the production of many mixed biogenetic molecules isolated from Theonella sponges,41a could also be at work in producing the bengamides. The current state of understanding pertaining to the enzyme-mediated assembly of the bengamides underscores the potential future value of using taxa summarized in Table 1 as systems for future sponge-microbiome-based research.41b
Structure–Activity Relationship Cytotoxicity Activity Patterns for 5R,6S,7R,8R,10S-Bengamide E/Bengamide A and Analogues Have Provided a Valuable Landscape for Lessons Learned
The natural bengamides useful for assessing initial structure–activity relationship (SAR) cytotoxicity trends can be divided into four different categories (see Table 2) according to substitution patterns on the caprolactam. The groupings depend on the presence or absence of functional groups shown on the general structure at the top of Table 2 as follows: type I, lipophilic esters attached at R2, headed by bengamides A and B; type II, polyketide esters attached at R2, headed by bengamides C and D; type III, no substituent at R2, headed by bengamides E and F, and type IV, attached to the caprolactam ring nitrogen, exclusively as iso-bengamide E.1e While most of the preclinical development and subsequent phase I clinical trial work was focused on the type I scaffold, important initial insights were gained by examining in vitro IC50 potencies of structures from the type III category. A first key overview on functionality and configurations required for optimal in vitro activity within type III is shown in the boxed structure at the bottom of Figure 7.
Figure 7.

SAR cytotoxicity activity patterns for 5R,6S,7R,8R-bengamide E and selected analogues provide a landscape for many lessons learned.
An appreciation of structural features in the type III bengamide scaffold that impart activity, or inactivity, can be assessed from examining a selection of 10 type III structures shown in Figure 7.1e,3,38,42−48 Compounds with single-digit nanomolar or subnanomolar activity against MDA-MB-435 or HCT-116 tumor cell lines represent important inputs for the design of a potential clinical candidate. Alternatively, minor modifications of the polyketide OCH3 group, changing the absolute configurations at positions C-5 to C-8, or replacing the caprolactam with unoptimized subunits can be disastrous. In this regard, the following structures comprise the set of potent entities and include (a) a natural E3,4-bengamide (IC50 = 3.8 nM)/synthetic Z3,4-bengamide E not shown in Figure 7 (IC50 = 14 nM45); (b) the 5S,6R,7S,8S synthetic bengamide E (IC50 = 4 nM); (c) a benzene-fused synthetic analogue (IC50 = 9 nM); (d) cyclopentyl-substituted synthetic bengamide E (IC50 = 0.4 nM45); and (e) the synthetic N-substituted bengamide E (IC50 = 4 nM). There are several remarkable features associated with these data including the following: (i) the methoxy group at position 8 is essential; (ii) one entry represents the most potent in-vitro-active (IC50 = 0.4 nM) bengamide analogue ever examined; (iii) carbon atoms at positions 11–13 can be deleted (but only when the ring nitrogen has also been modified), as shown by comparing the trio of compounds at the bottom of Figure 7; and (iv) benzene ring annulation to C-13 or C-14 can be beneficial.
It is important to revisit a fundamental conclusion discussed above for the polyketide side chain and highlighted in the box of Figure 7. The in vitro nanomolar IC50 value activity can be greatly diminished by small changes in the configurations of the side chain attached at C-10. Similar potency is observed for 5R,6S,7R,8R,10S (natural bengamide E IC50 = 3.8 nM) and 5S,6R,7S,8S,10S (synthetic bengamide E diastereomer, IC50 = 4 nM), but no activity is observed for 5S,6R,7S,8S,10R (synthetic ent-bengamide E, HCT115 IC50 > 50 μM). The results in the report on ent-bengamide E48 are curious. First, the activity for bengamide E vs HCT-116 IC50 = 600 nM indicates potency but not on par with data against MDA-MB-435 (see Figure 7). Second, the activity parameters for ent-bengamide E vs those for the 5S,6R,7S,8S,10S bengamide E diastereomer are dramatically different and merit further investigations. The key requirement of the 5R,6S,7R,8R,10S configuration for potency in the type III structures served as a starting point in all SAR studies of the type I analogues. The ester-bearing type I compounds are supremely active and are headed by the 1–2 nM activity of bengamides A and B against MDA-MB-435 cells. As discussed in detail below, this was the scaffold chosen for development in the phase I clinical trial. Highlights of the additional structure–activity relationships from type I compounds are shown in the box of Figure 8.The most important new insights involve the SAR impact of chemical space changes in two regions: (a) O vs N substitution at C-13 and (b) deletion of the caprolactam ring while retaining the myristate.
Figure 8.

Additional SAR cytotoxicity activity patterns for 5R,6S,7R,8R,10S,13R bengamides A and B and selected analogues provide a landscape for many added lessons learned.
Trends for in vitro and in vivo responses of type I bengamides against MDA-MB-435 human tumors were carefully examined by Kinder et al.35 and were the basis for selecting LAF389 for the phase I clinical trial. Examining the original data for the 17 compounds shown in the Kinder publication (Figure 1 and Tables 2 and 3) is essential. Relative to bengamide B, there were two structural changes: (a) replacement of isopropyl with tert-butyl and (b) retention or inversion of the 15-myristoyloxy with 14 other ester arrays. These results led to the important conclusion “...over 2/3 of the analogues...inhibited in vivo tumor growth as well as or better than that of bengamide B...”. This prompted an extensive investigation of the antitumor properties of LAF389.
There is another dimension to the SAR understanding that was accumulated for type I analogues. The essential role of the ester attached at C-13 was discussed above, and, relative to the potency of tert-butyl bengamide (13R) (IC50 = 10 nm), there were nine compounds with similar potencies. Overall, modification of the myristoyl ester is generally well tolerated, as long as the ester and a sufficient number of aliphatic carbons are present, which presumably increases membrane permeability. Alternatively, replacement of the C-13 ester with an amide (bengamde A 13S amide) obliterates activity.47 Dramatic reduction of potency was observed with the synthetic compound of Figure 8 (IC50 = 190 nm), where the myristoyl ester is retained but the ring is opened.48−52
The journey to designate the lead compound, LAF389 (10, Figure 2), for clinical trial took many years of bench-scale syntheses, where its chiral total synthesis was optimized to be safe and convergent. The important next step involved development of a large-scale pilot-plant-type synthesis.12 This task proceeded via a key modification that used a modified Julia olefination50 rather than the formerly employed Takai procedure.51 Conversion of the olefinic isopropyl group in the natural sponge-derived bengamide compounds to a tert-butyl group, as in LAF389, was a strategic, common modification that greatly increased synthetic yields in the olefination reaction and had little effect on activity.12,35
LAF389 possesses a labile ester that is hydrolyzed intracellularly to the alcohol.11 Interestingly, the in vivo potency of the alcohol is lower than that of LAF389, presumably based on the increased membrane permeability of the lipophilic cyclohexyl ester. LAF389 was found to have comparable activity to bengamide B and bengamide Z in vitro and superior activity in vivo, causing significant tumor regression at 100 μmol/kg during preclinical investigation. Animal studies demonstrated that repeat bolus administration of LAF389 was effective and well tolerated, and thus, in 2000, the phase I clinical investigation of LAF389 began.11
Phase I study began in 2000 with assessment of the safety, tolerability, and pharmacokinetic profile of LAF389 and to determine the maximum tolerated dose.11 Patients were those with a histological or cytological diagnosis of advanced cancer for whom no standard therapy existed. LAF389 was provided by Novartis Pharma (Basel, Switzerland) as a 30 mg/mL solution in propylene glycol, which was diluted 30 times with saline to afford a 1 mg/mL solution and administered intravenously or via a central indwelling catheter as a slow bolus injection. A total of 33 patients were treated from May 2000 to April 2002, and 31 of these patients reported adverse effects. Additionally, eight patients suffered unexpected cardiovascular toxicity not predicted by preclinical testing in rats and dogs. The phase I clinical investigation of LAF389 was terminated for safety considerations together with the lack of clear evidence of clinical effectiveness.
Molecular Targets of the Bengamides
By 2001, it was clear that the natural bengamides, especially A/B (MDA-MB-435 IC50 = 1.0/2.4 nM) and a synthetic bengamide, LAF389 (MDA-MB-435 IC50 = 40 nM), were exquisitely potent against cancer cell lines.1e An important next study of these promising therapeutic leads involved molecular target identification guided by proteomics. The experimental design was successful and involved challenging H1299 human non-small-cell lung carcinoma cells with bengamide E and LAF389.53 Gel electrophoresis (2D) showed modulation of the family of 14-3-3 γ proteins in comparison to that of untreated cells. Specifically, the 14-3-3 γ family of ubiquitous cytosolic proteins lacked N-terminal processing due to the retention of the initiator methionine, suggesting that LAF389 reduced the activity of methionine aminopeptidases (MetAP). Experimental and in silico binding experiments involving human MetAP2 and bengamide analogues were also carried out as shown in Figure 9. Panel B shows a nice fit of LAF389 in a putative binding pocket. More definitive insights came from cocrystallization studies shown in Figure 9 (panel C), which involved the bengamide analogue LAF153 and provided visualization of the interaction between the three OH groups and the dicobalt moiety in the active site. Eventually, it was demonstrated that LAF389 nonselectively inhibited both isoforms of human methionine aminopeptidases, MetAP1 (IC50 = 0.7 μM) and MetAP2 (IC50 = 0.4 μM). A surprising outcome was that another synthetic bengamide, LBM648 (Figure 9), was MetAP2 selective [MetAP1 (>10 μM) and MetAP2 (IC50 = 0.38 μM)].55 Additional second-generation experiments54 were driven by siRNA depletion of MetAP2 without inhibition of endothelial cell growth. The conclusion advanced by Phillips et al. in 2004 that “MetAP2 function is not required for endothelial cell proliferation”54 implies that the idea the MetAP2 inhibition is not the full story and that further target finding experiments are needed.
Figure 9.

Summary of preclinical efficacy and putative therapeutic target of bengamide-inspired analogues. (A) Structures of synthetic bengamides (i) (LAF389) explored in advance preclinical studies against HCT-116 tumor cells and (ii) LAF153 and LBM648 explored in target finding with MetAP1/MetAP2. (B) In silico docking of LAF389 with MetAP2. (C) X-ray cocrystal structure of LAF153 (a product of serum esterase action on LAF389) bound to hMetAP2, resolution 1.6 Å (adapted from J. Biol. Chem.2003, 278, 52964), which shows interaction of OR groups with several residues of the protein. Panels B and C are adapted from a lecture given by R. Versace, Novartis Pharmaceuticals, New Jersey, USA. Also compare these results to additional MetAP enzyme inhibition data in Table 3.
Additional insights were subsequently gained three years later.53 Liu and co-workers believed that MetAP1 may also be an important cancer target and have suggested (unpublished) that small molecules selective for MetAP1 vs MetAP2 might represent a better therapeutic lead. This team used natural bengamides (A, B, G, M, N, O) to probe the downstream physiological functions of MetAPs.53 Interesting results were obtained during the IC50 profiling of this set against MetAP1 vs MetAP2 as follows: (a) similar IC50 μM data were observed for bengamide A vs B (M1 vs M2 bengamide A = 2, 11; bengamide B = 29, 18, and (b) selectivity was observed against MetAP1 (M1 vs M2 bengamide O = 3, >50). These results demonstrated MetAP1 selectivity. Overall, these results are similar to those (see above) obtained by Phillips et al.54 for LAF389 (MetAP unselective) but different than that observed for LBM648 (MetAP2 selective). It was also shown that nonselective inhibition of MetAPs by bengamide A alters the subcellular concentrations of the proto-oncogene c-Src, which is essential for tumor growth. Bengamide A significantly decreased the tyrosine kinase activity of c-Src and caused a delay in cell cycle progression through the G-2/M phase. As a final important conclusion, the Liu team stated, “c-Src...dysfunction is likely to account for the cell cycle effects of MetAP inhibitors including bengamide A.” Examined in a separate section below are key SAR effects of a variety of bengamides; the outcomes of this work are essential for the creation of effective next-generation clinical therapeutics based on the bengamide scaffold.
A collection of several recent papers provides some new understandings about the modulation of both human and bacterial MetAPs by the bengamides. The reader is directed to an interesting quote contained in a 2015 Angewandte Chemie press release shown in the bottom of Figure 10. The complex network of structures, arrows, and boxes below this quotation can be explained as follows. After the discovery that the bengamides are also produced by the terrestrial myxobacterium Myxococcus virescens ST200611 (DSM 15898),38−40 a campaign to supply bengamides by fermentation was launched, resulting in elucidation of the responsible biosynthetic gene cluster.38 Further analysis of the biosynthetic pathway showed that Leu154 of the myxobacterial MetAP confers bacterial self-resistance to bengamide inhibition via steric hindrance. The entire gene cluster was then successfully transferred to a robust strain of M. xanthus DK1622 for large-scale fermentation. Subsequent semisynthesis of microbially produced bengamides and total synthesis yielded the optimized bengamide derivative benzLAF389 (Table 3). This new clinical candidate exhibits nanomolar potency, high metabolic stability, and an improved pharmacokinetic profile. Moderate in vivo efficacy (T/C = 31% at the highest nontoxic dose) was demonstrated in a murine tumor bearing model (early stage B16 melanoma) with a limited therapeutic window between toxicity and antitumor efficacy.
Figure 10.

Key discoveries and developments in the understanding of inhibition of human and bacterial methionine aminopeptidases by bengamides.
Another potential application for bengamide derivatives is the treatment of tuberculosis, a disease caused in humans mainly by Mycobacterium tuberculosis. Unlike eukaryotic cells, most bacteria have only a single essential MetAP gene (type 1) that may serve as a target for the development of broad-spectrum antibiotics. In particular, M. tuberculosis has two MetAP genes (MtMetAP1c and MtMetAP1a), and both belong to type 1 MetAPs. Seven MetAP inhibitors were designed and synthesized based on natural bengamides and tested against purified tubercular MetAPs, and some also demonstrated initial antitubercular activity.55 Two of the newly designed inhibitors were complexed with MtMetAP1c to elucidate the binding mode and to optimize inhibitor design. In subsequent work, three new X-ray structures of MtMetAP1c in complex with two different inhibitors in the Mn(II) form and the Ni(II) form were presented.56 Four of the seven designed MetAP inhibitors were also complexed with a human MetAP1 (HsMetAP1) in the Mn(II) form, and the X-ray structures were solved at high resolution (1.47–1.75 Å).57 The binding mode of the bengamide derivatives in these four structures is significantly different from the previous HsMetAP1 structures in the protein databank. Overall, the structural information gleaned from studies of the interactions of the various inhibitors at the active sites in the five X-ray structures of tubercular MtMetAP1c and the four X-ray structures of human HsMetAP1 has provided additional important insights for the design of bengamide inhibitors with improved selectivity and potency in both antibacterial and anticancer applications.58
There is yet another development in the quest to fully define the bengamide molecular targets. A UC Santa Cruz–UC Berkeley collaboration identified the bengamides as an interesting new class of immune modulators through use of a nuclear factor-κB (NF-κB) luciferase assay.40 The bengamides showed comparable or superior activity to the known NF-κB inhibitor celastrol (Table 3 of ref (40)). NF-κB has also been implicated in the development and metastasis of some human cancers.59 The bengamides may thus prove to be a valuable new scaffold for the development of treatments of immune disorders and cancers that are shown to have elevated NF-κB activity.
Next-Generation Campaigns and Concepts to Reinvigorate Development of Bengamide-Based Therapeutics
The confluence of results from various disciplines is now pointing the way for the future use of bengamides as molecular tools or agents for clinical therapeutics. Some essential issues to be considered for future research are (1) maintaining nanomolar potency of new analogues; (2) reduction of human toxicity by enhancement of target selectivity (i.e., MetAP); (3) reduction of off-target effects (a topic not rigorously addressed to date); (4) pharmacophore simplification; (5) addressing metabolic stability; and (6) exploiting the potential of synthetic biology-based production of new analogues. While three decades of research by hundreds of scientists have provided the framework for many achievements, new technologies and insights are now needed to make the next groundbreaking advances.
Many advancements that have been made during the study on the bengamides have come from teams of collaborators including academic groups at multiple institutions or from interactions between academic and corporate scientists. The synergy between these groups was essential to make and maximize important scientific outcomes. As noted in the Introduction, the initial discovery of the bengamides was made possible through a UCSC Chemistry and Syntex Institute collaboration. Additional milestone results arose through the UCSC–Novartis collaboration, leading to the description of in vitro nanomolar active bengamides eventually shown to have in vivo efficacy. A three-way collaboration between UCSC, UC Berkeley, and the Josephine Ford Cancer Center revealed NF-κB as a molecular target of the bengamides. Recently, the collaboration between laboratories in Germany consisting of University of Saarlandes and Sanofi Research and Development enabled the characterization of the biosynthetic path leading to the bengamides from the culturing of Myxococcus virescens.
Venerable work exploring the production of bengamides in Nature, summarized in Figure 11, is always cited and discussed in papers describing outcomes catalyzed by these compounds. Powerful SAR insights have been derived through the study of 21 natural analogues from sponges (shown in Table 2) and were augmented by the four additional compounds isolated by the culture of Myxococcus virescens (DSM 15898), a Gram-negative bacterium.38−40 There are approximately 116 non-natural bengamides, reviewed by Garcia-Ruiz and Sarabia2 and prepared during campaigns to test innovations in total organic synthesis, verify hypotheses of absolute configuration assignments, and expand the understanding of SAR patterns. Added to this are greater than 50 bengamides that were prepared by the Novartis–Sandoz teams, but information about the structures and their properties remains buried in confidential corporate reports.
Figure 11.

Next-generation results for the development of bengamide-inspired analogues and insights for obtaining compounds from sponges and micro-organisms (see Figures 9 and 10 for structures).
After decades of exploration using the natural material and synthetic compound libraries, could the trajectory of obtaining useful findings be at an end? Possibly, but new opportunities offered by molecular genetics studies may provide some fresh stimuli, especially from insights on the bengamide E/F biosynthetic machinery.38,40 The recent characterization from Myxococcusvirescens of the bengamide gene cluster provides powerful insights about how this process proceeds and could be further manipulated.38 The first biosynthetic step involves (see Figure 11) loading of a 2-methylbutyrl-CoA starter unit, which is subsequently modified by a BenA–D assembly of the key glycolate unit and further transformed by BenE–H. This latter cassette installs the functionality and chirality at bengamide positions C-5 to C-8 required for nanomolar bioactivity (see Figures 7 and 8). Synthetic biology efforts envisioning the creation of modifications in this region using BenE–H that can be expressed in a different host (see Figure 11) were explored, but without success.38
It is tempting to speculate on additional opportunities that could provide the next generation of discoveries. Disentangling toxic side effects from the therapeutic advantages associated with the MetAp target of the bengamides continues to be a challenge. The MetAP isoforms, 1 and 2, are present in all eukaryotic cells and catalyze N-terminal methionine excision, an essential pathway of cotranslational protein maturation. There is intense interest and debate about how to exploit compounds that inhibit these enzymes for drug development. In 2013, Liu et al. highlighted the MetAP–bengamide situation with the following analysis: “...the clinical study of...LAF389 in patients with advanced cancer was terminated due to its cardiovascular toxicity, which might be caused by the simultaneous inhibition of MetAP1 and MetAP2”.60 A current hypothesis is that compounds selective for MetAP1 over MetAP2 will be successful as nontoxic therapeutics. The reproducible MetAP inhibition IC50 data in Table 3 for bengamide A and LAF389 provide the benchmark data illustrating that both MetAPs are inhibited equally by both compounds. In contrast, there are natural products and synthetic compounds that have been identified with selectivity against each MetAP isoform. The list of potential examples is growing, and some are summarized in Table 3 including (a) MetAP2 selective: LBM648, fumagillin,49 and its analogue beloranib (not shown) and (b) MetAP1 selective: bengamide O, IV-43 (aka ZNQ-6),49 and piperazinquinazoline.61 The fumagillin analogue beloranib, a MetAP2 angiogenesis inhibitor, was recently (July 2016) withdrawn from a phase III clinical trial (antiobesity) by Zafgen because of two patient deaths possibly due to the drug treatment. An obvious path forward is to now focus on MetAP1-selective agents with nanomolar cytotoxicity properties, and bengamide O is a potential example.
Increased metabolic stability and pharmacophore redesign will be essential for future bengamide therapeutic lead development. The Muller group38 has created a vision of the ideal future clinical candidate and has proposed a compound shown in Table 3, named here as benzLAF389. This compound exhibited nanomolar cellular potency, improved pharmacokinetic properties, good metabolic stability, and promising in vivo activity in a mouse model. However, the MetAP selectivity of this compound is currently unknown. Shifting the focus back to type III bengamide structures that do not possess the labile ester moiety, headed by bengamide E/E′, also merits consideration. Several examples of bengamide analogues that bind to MtMetAP1 and also possess reduced pharmacophore complexity are shown at the bottom of Figure 10. The obvious next steps for this chemical space is to assess their in vitro cytotoxicity and measure MetAP selectivity. It is important to note that a complete understanding of the various methionine aminopeptidase isoforms in humans and in Mycobacterium tuberculosis and their role(s) as anticancer or antitubercular targets is still under investigation. A current awareness search in the Chemical Abstracts Service Sci-Finder tool on the topic “MetAP” revealed 899 peer-reviewed publications and patents, and there were 30 citations from the year 2016 alone. We conclude that the relevance of this target and how to harness it for drug discovery is still evolving at a rapid pace.
Conclusions
In this review, we have highlighted a special circumstance involving the bengamide family of PKS–NRPS hybrid natural products first published in 1986. Overall, these moderately complex compounds have provided excellent opportunities for multidisciplinary natural products discovery and development projects. Work to build on the initial bengamide structure elucidation reports began quickly in many laboratories because the absolute configuration of the six chiral centers and the geometry of the double bond were accurately established at the outset. Subsequently, numerous total synthetic and medicinal chemistry efforts provided the basis for LAF389, the phase I anticancer clinical candidate.
The persistent interest and continued investigations of the bengamides and bengamide synthetics suggest that, in the next decade, a new analogue will be included among the successful sponge-inspired therapeutics shown in Figure 1. However, the trajectory for such an outcome can be lengthy. For example, it took 26 years to capitalize on the discovery of halichondrin B, isolated from the sponge Halichondria okadai and published in 1985. Its structure paved the way to the creation of eribulin mesylate (aka Halaven), which was approved by the U.S. FDA in 2010 as a clinical anticancer agent. The process to develop marine natural products with significant biological activity and structural complexity into useful entities, such as a molecular tools and therapeutics, takes patience, persistence, and luck. Setbacks always arise and must be overcome. In the case of eribulin mesylate, early supply problems and poor understanding about its true pharmacophore were dealt with through heroic synthetic optimization endeavors. We are hopeful for the same outcome in the coming years with the bengamides, but time could be running out.
The success by the UCSC group in gaining an understanding of the natural history of the bengamide-producing sponge provides an example of how such insights can overcome challenges involved in collecting or re-collecting specimens that possess important biosynthetic products. Grappling with the distinctive sponge phenotypes and features in the environment near Jaspis cf. coriacea has not been easy. Many of the publications on the bengamides contain evidence that the scientific divers involved in those projects were not fully versed on how to collect and properly assign the taxa of bengamide-containing sponges. In this review we have attempted to set the record straight. Our review also highlights the value of collaboration among teams located at various institutions, in this case academic research groups, the National Cancer Institute’s Developmental Therapeutics Program (NCI-DTP), Syntex, Sandoz, and Novartis Pharmaceuticals Corporation. Another factor stimulating bengamide research discoveries was the motivation and achievment that came through support provided by the NIH National Cooperative Drug Discovery Group’s (NCDDG) initiative.
Biochemical tools for sequencing, analyzing, and mining genomic data extended discoveries and provided valuable insights into the assembly of the bengamides. These methods allowed for the advancement of knowledge from the initial 1989 hypothetical analysis of the biosynthetic pathway to the impressive 2015 elucidation of the true biosynthetic gene cluster (BGC). The new in-depth understanding of this pathway will advance bengamide research as well as that generally focused on the origin of polyketide–nonribosomal peptide hybrid compounds. The successful transfer of the bengamide BGC to a robust production host, Myxococcus xanthus, addresses the “supply issue”, which is a common obstacle for many sponge-derived natural products shown in Figure 2.
The synthetic and medicinal chemistry contributions to the bengamide story are massive. Approximately 200 synthetic bengamide analogues have been created and evaluated against cancer cell lines, allowing for a detailed SAR scenario to be constructed. The SAR has been significantly buttressed by several (at least 10) cocrystal structures of bengamide analogues in complex with various human and bacterial methionine aminopeptidases.
There are several lessons that have been learned from the phase I clinical trial of LAF389. The unanticipated cardiotoxicity and other off-target effects encountered were very unfortunate, but recent developments in predictive model systems using various cell types to test candidate compounds will help to alleviate this problem in the future. Predicting and preventing the cardiotoxicity of cancer therapy is another rapidly evolving field in and of itself, and next generation concepts will help to reinvigorate the development of bengamide-based compounds.
The highly oxgenated and stereochemically rich polyketide region and a novel cyclic amino acid residue merge in the bengamide class to produce a beautiful molecular framework. Paired with meaningful results in MetAP and NF-κB activity screens, the bengamides remain attractive lead compounds for therapeutic discovery, molecular biology, and biosynthetic research endeavors. It is for this reason that today, more than three decades after the UCSC team of Quinoa, Adamczeski, and Crews initiated the first phase in the bengamide structure elucidation, this family persists in inspiring research ventures. The interesting natural history of the sponges, the isolation of a bengamide-producing bacterium, and the elucidation of the biosynthetic production mechanisms add depth and new applications to this powerful story of natural products discovery. The authors are confident that the record of meaningful findings is not complete. In particular, new samples of Jaspis cf. coriacea will undoubtedly be obtained and pursued for molecular genetics experiments. Thus, the bengamides will continue to endure as an inspiration to chemists and biologists alike for many more years.
Acknowledgments
The preparation of this review was supported by grants from the NSF CHE 1214065 (P.C.) and NIH R01 CA 47135 (P.C.). We acknowledge funding from an American Society of Pharmacognosy research starter grant (K.N.W.). The authors would like to thank all UCSC co-workers from 1983 to the present for their commitment to bengamide projects, and their names are listed in refs 1a–e, 4, 34, 35, and 40 of this review.
The authors declare no competing financial interest.
Dedication
Kimberly N. White and Karen Tenney dedicate this review to Professor Phillip Crews, of the University of California, Santa Cruz, for his pioneering work on bioactive natural products.
References
- a Quiñoà E.; Adamczeski M.; Crews P. J. Org. Chem. 1986, 51, 4494–4497. 10.1021/jo00373a036. [DOI] [Google Scholar]; b Adamczeski M.; Quiñoà E.; Crews P. J. Am. Chem. Soc. 1988, 110, 1598–1602. 10.1021/ja00213a037. [DOI] [Google Scholar]; c Adamczeski M.; Quiñoà E.; Crews P. J. Am. Chem. Soc. 1989, 111, 647–654. 10.1021/ja00184a037. [DOI] [Google Scholar]; d Adamczeski M.; Quiñoà E.; Crews P. J. Org. Chem. 1990, 55, 240–242. 10.1021/jo00288a039. [DOI] [Google Scholar]; e Thale Z.; Kinder F. R.; Bair K. W.; Bontempo J.; Czuchta A. M.; Versace R. W.; Phillips P. E.; Sanders M. L.; Wattanasin S.; Crews P. J. Org. Chem. 2001, 66, 1733–1741. 10.1021/jo001380+. [DOI] [PubMed] [Google Scholar]
- Garcia-Ruiz C.; Sarabia F. Mar. Drugs 2014, 12, 1580–1622. 10.3390/md12031580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinder F. R. Org. Prep. Proced. Int. 2002, 34, 559–583. 10.1080/00304940209355783. [DOI] [Google Scholar]
- Crews P.; Hunter L. M. In Marine Biotechnology, Vol. 1, Pharmaceutical and Bioactive Natural Products; Attaway D. H.; Zaborsky O. R., Eds.; Plenum Press: New York, 1993; Chapter 9, pp 343–389. [Google Scholar]
- Carrol J.; Crews P. In Natural Product Chemistry for Drug Discovery; Buss A. D.; Butler M. S., Eds.; Royal Society of Chemistry: Cambridge, U.K., 2010; Chapter 6, pp 174–214. [Google Scholar]
- www.halaven.com/.
- Huyck T. K.; Gradishar W.; Manuguid F.; Kirkpatrick P. Nat. Rev. Drug Discovery 2011, 10, 173–174. 10.1038/nrd3389. [DOI] [PubMed] [Google Scholar]
- http://www.us.farydak.com/.
- Pina I. C.; Gautschi J. T.; Wang G. Y.; Sanders M. L.; Schmitz F. J.; France D.; Cornell-Kennon S.; Sambucetti L. C.; Remiszewski S. W.; Perez L. B.; Bair K. W.; Crews P. J. Org. Chem. 2003, 68, 3866–3873. 10.1021/jo034248t. [DOI] [PubMed] [Google Scholar]
- Martinez-Diez M.; Guillen-Navarro M. J.; Pera B.; Bouchet B. P.; Martinez-Leal J. F.; Barasoain I.; Cuevas C.; Andreu J. M.; Garcia-Fernandez L. F.; Diaz J. F.; Aviles P.; Galmarini C. M. Biochem. Pharmacol. 2014, 88, 291–302. 10.1016/j.bcp.2014.01.026. [DOI] [PubMed] [Google Scholar]
- Dumez H.; Gall H.; Capdeville R.; Dutreix C.; van Oosterom A. T.; Giaccone G. Anti-Cancer Drugs 2007, 18, 219–225. 10.1097/CAD.0b013e328010ef5b. [DOI] [PubMed] [Google Scholar]
- Xu D. D.; Waykole L.; Calienni J. V.; Ciszewski L.; Lee G. T.; Lie W.; Szewczyk J.; Varga K.; Prasad K.; Repic O.; Blacklock T. J. Org. Process Res. Dev. 2003, 7, 856–865. 10.1021/op0341162. [DOI] [Google Scholar]
- Remiszewski S. W. Curr. Med. Chem. 2003, 10, 2393–2402. 10.2174/0929867033456675. [DOI] [PubMed] [Google Scholar]
- Tsukamoto S.; Yamashita K.; Tane K.; Kizu R.; Ohta T.; Matsunaga S.; Fusetani N.; Kawahara H.; Yokosawa H. Biol. Pharm. Bull. 2004, 27, 699–701. 10.1248/bpb.27.699. [DOI] [PubMed] [Google Scholar]
- a deSilva E. D.; Scheuer P. Tetrahedron Lett. 1980, 21, 1611–1614. 10.1016/S0040-4039(00)77766-5. [DOI] [Google Scholar]; b Soriente A.; DeRosa M. M.; Scettri A.; Sodano G.; Terencio M. C.; Paya M.; Alcaraz M. J. Curr. Med. Chem. 1999, 6, 415–431. [PubMed] [Google Scholar]
- Kasserra C. E.; Harris P.; Stenton G. R.; Abraham W.; Langlands J. M. Pulm. Pharmacol. Ther. 2004, 17, 309–318. 10.1016/j.pupt.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Gunasekera S. P.; Gunasekera M.; Longley R. E.; Schulte G. K. J. Org. Chem. 1990, 55, 4912–4915. 10.1021/jo00303a029. [DOI] [Google Scholar]
- Yu K. O. A.; Porcelli S. A. Immunol. Lett. 2005, 100, 42–55. 10.1016/j.imlet.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Kobayashi M.; Aoki S.; Ohyabu N.; Kurosu M.; Wang W.; Kitagawa I. Tetrahedron Lett. 1994, 35, 7969–7972. 10.1016/0040-4039(94)80024-3. [DOI] [Google Scholar]
- Loganzo F.; Discafani C. M.; Annable T.; Beyer C.; Musto S.; Hari M.; Tan X.; Hardy C.; Hernandez R.; Baxter M.; Singanallore T.; Khafizova G.; Poruchynsky M. S.; Fojo T.; Nieman J. A.; Ayral-Kaloustian S.; Zask A.; Andersen R. J.; Greenberger L. M. Cancer Res. 2003, 63, 1838–1845. [PubMed] [Google Scholar]
- Kuznetsov G.; TenDyke K.; Towle M. J.; Cheng H.; Liu J.; Marsh J. P.; Schiller S. E.; Spyvee M. R.; Yang H.; Seletsky B. M.; Shaffer C. J.; Marceau V.; Yao Y.; Suh E. M.; Campagna S.; Fang F. G.; Kowalczyk J. J.; Littlefield B. A. Mol. Cancer Ther. 2009, 8, 2852–2860. 10.1158/1535-7163.MCT-09-0301. [DOI] [PubMed] [Google Scholar]
- Sessa C.; Weigang-Kohler K.; Pagani O.; Greim G.; Mora O.; DePas T.; Burgess M.; Weimer I.; Johnson R. Eur. J. Cancer 2002, 38, 2388–2396. 10.1016/S0959-8049(02)00489-6. [DOI] [PubMed] [Google Scholar]
- Petek B. J.; Jones R. L. Molecules 2014, 19, 12328–12335. 10.3390/molecules190812328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Newman D. J.; Cragg G. M. Curr. Med. Chem. 2004, 11, 1693–1713. 10.2174/0929867043364982. [DOI] [PubMed] [Google Scholar]; b Molinski T. F.; Dalisay D. S.; Lievens S. L.; Saludes J. P. Nat. Rev. Drug Discovery 2009, 8, 69–85. 10.1038/nrd2487. [DOI] [PubMed] [Google Scholar]; c Sashidara K. V.; White K. N.; Crews P. J. Nat. Prod. 2009, 72, 588–603. 10.1021/np800817y. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Mayer A. M. S.; Glaser K. B.; Cuevas C.; Jacobs R. S.; Kem W.; Little R. D.; McIntosh J. M.; Newman D. J.; Potts B. C.; Shuster D. E. Trends Pharmacol. Sci. 2010, 31, 255–265. 10.1016/j.tips.2010.02.005. [DOI] [PubMed] [Google Scholar]
- a Mestel R.Drugs from the Sea. Discover Magazine 1999. [Google Scholar]; b Rayl A. J. S.Oceans. Medicine Chests of the Future? The Scientist 1999. [Google Scholar]
- a Colin P. L.; Arneson C.. Tropical Pacific Invertebrates; Coral Reef Press: Beverly Hills, CA, 1995. [Google Scholar]; b Allen G. R.; Steene R.. Indo-Pacific Coral Reef Field Guide; Tropical Reef Research: Singapore, 1996. [Google Scholar]; c Gosliner T. M.; Behrens D. W.; Williams G. C.. Coral Reef Animals of the Indo-Pacific; Sea Challengers: Monterey, CA, 1996. [Google Scholar]
- Manes L. V.; Bakus G. J.; Crews P. Tetrahedron Lett. 1984, 25, 931–934. 10.1016/S0040-4039(01)80065-4. [DOI] [Google Scholar]
- Crews P.; Manes L.; Boehler M. Tetrahedron Lett. 1986, 27, 2797–800. 10.1016/S0040-4039(00)84645-6. [DOI] [Google Scholar]
- a Rodriguez J.; Nieto R. M.; Crews P. J. Nat. Prod. 1993, 56, 2034–2040. 10.1021/np50102a002. [DOI] [PubMed] [Google Scholar]; b Rudi A.; Kashman Y. J. Nat. Prod. 1994, 57, 829–832. 10.1021/np50108a023. [DOI] [PubMed] [Google Scholar]; c Searle P. A.; Richter R. K.; Molinksi T. F. J. Org. Chem. 1996, 66, 4073–4079. 10.1021/jo952261a. [DOI] [PubMed] [Google Scholar]; d Groweiss A.; Newcomer J. J.; O’Keefe B. R.; Blackman A.; Boyd M. R. J. Nat. Prod. 1999, 62, 1691–1693. 10.1021/np9902688. [DOI] [Google Scholar]
- D’Auria M. V.; Giannini C.; Minale L.; Zampella A.; Debitus C.; Frostin M. J. Nat. Prod. 1997, 60, 814–816. 10.1021/np970050q. [DOI] [Google Scholar]
- Fernandez R.; Dherbomez M.; Letourneux Y.; Nabil M.; Verbist J. F.; Biard J. F. J. Nat. Prod. 1999, 62, 678–680. 10.1021/np980330l. [DOI] [PubMed] [Google Scholar]
- Pettit G. R.; Hogan F.; Xu J.-P.; Tan R.; Nogawa T.; Cichacz Z.; Pettit R. K.; Du J.; Ye Q.-H.; Cragg G. C.; Herald C. L.; Howard M. S.; Goswami A.; Searcy J.; Tackett L.; Doubek D. L.; Willimas L.; Hooper J. H.; Schmidt J. M.; Chapuis J.-C.; Tackett D. N.; Craciumescu F. J. Nat. Prod. 2008, 71, 438–444. 10.1021/np700738k. [DOI] [PubMed] [Google Scholar]
- Ovenden S. P. B.; Nielson J. L.; Liptro C. H.; Willis R. H.; Tapiolas D. M.; Wright A. D.; Motti C. A. Mar. Drugs 2011, 9, 2469–2478. 10.3390/md9112469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kinder F. R.; Bair K. W.; Bontempo J.; Crews P.; Czuchta A. M.; Nemzek R.; Thale Z.; Vattay A.; Versace R. W.; Weltcheck S.; Wood A.; Zabludoff S. D.; Phillips P. E. Proc. Am. Assoc. Cancer Res. 2000, 41, 600. [Google Scholar]; b Phillips P. E.; Bair K. W.; Bontempo J.; Crews P.; Czuchta A. M.; Kinder F. R.; Vattay A.; Versace R. W.; Wang B.; Wood A.; Zabludoff S. Proc. Am. Assoc. Cancer Res. 2000, 41, 59. [Google Scholar]
- Kinder F. R.; Verscace R. W.; Bair K. W.; Bontempo J. M.; Casarz D.; Chen S.; Crews P.; Czuchta A. M.; Jagoe C. T.; Mou Y.; Nemzek R.; Phillips P. E.; Tran L. D.; Wang R. M.; Weltchek S.; Zabludoff S. J. Med. Chem. 2001, 44, 3692–3699. 10.1021/jm010188c. [DOI] [PubMed] [Google Scholar]
- Edwards D. J.; Gerwick W. H. J. Am. Chem. Soc. 2004, 126, 11432–11433. 10.1021/ja047876g. [DOI] [PubMed] [Google Scholar]
- Song L.; Barona-Gomez F.; Corre C.; Xiang L.; Udwary D. W.; Ausitn M. B.; Noel J. P.; Moore B. S.; Challis G. L. J. Am. Chem. Soc. 2006, 128, 14754–14755. 10.1021/ja065247w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel S. C.; Hoffman H.; Zhang J.; Debusshe L.; Haag-Richter S.; Kurz M.; Nardi F.; Lukat P.; Kochems I.; Tietgen H.; Schummer D.; Nicolas J.-P.; Calvet L.; Czepczor V.; Vrignaud P.; Muhlenweg A.; Pelzer S.; Muller R.; Bronstrup M. Angew. Chem., Int. Ed. 2015, 54, 15560–15564. 10.1002/anie.201508277. [DOI] [PubMed] [Google Scholar]
- Hoffmann H.; Haag-Richter S.; Kurz M.; Tietgen H.. WO2005/044803, 2005.
- Johnson T. A.; Sohn J.; Vaske Y. M.; White K. N.; Cohen T. L.; Vervoort H. C.; Tenney K.; Valeriote F. A.; Bjeldanes L. F.; Crews P. Bioorg. Med. Chem. 2012, 20, 4348–4355. 10.1016/j.bmc.2012.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wilson M. C.; Mori T.; Ruckert C.; Uria A. R.; Helf M. J.; Takada K.; Gernert C.; Steffens U. A. E.; Heycke N.; Schmitt S.; Rinke C.; Helfrich E. J. N.; Brachmann A. O.; Gurgui C.; Wakimoto T.; Kracht M.; Crusemann M.; Hentschel U.; Abe I.; Matsunaga S.; Kalinowski J.; Takeyama H.; Piel J. Nature 2014, 506, 58–62. 10.1038/nature12959. [DOI] [PubMed] [Google Scholar]; b Hentschel U.; Piel J.; Degnan S. M.; Taylor M. Nat. Rev. Microbiol. 2012, 10, 641–654. 10.1038/nrmicro2839. [DOI] [PubMed] [Google Scholar]
- Liu Q.-J.; Li H.; Chen S. P.; Zhou G.-C. Chin. Chem. Lett. 2011, 22, 505–507. 10.1016/j.cclet.2010.11.023. [DOI] [Google Scholar]
- Zhang W.; Liang Q.; Li H.; Meng X.; Li Z. Tetrahedron 2013, 69, 664–672. 10.1016/j.tet.2012.11.004. [DOI] [Google Scholar]
- Martin-Galvez F.; Garcia-Ruiz C.; Sanchez-Ruiz A.; Valeriote F. A.; Sarabia F. ChemMedChem 2013, 8, 819–831. 10.1002/cmdc.201300033. [DOI] [PubMed] [Google Scholar]
- Kinder F. R., unpublished.
- Tai W.-Y.; Zhang R.-T.; Ma Y.-M.; Gu M.; Liu G.; Li J.; Nan F.-J. ChemMedChem 2011, 6, 1555–1558. 10.1002/cmdc.201100164. [DOI] [PubMed] [Google Scholar]
- Liu G.; Ma Y. M.; Tai W. Y.; Xie C. M.; Li Y. L.; Li J.; Nan F. J. ChemMedChem 2008, 3, 74–78. 10.1002/cmdc.200700214. [DOI] [PubMed] [Google Scholar]
- Banwell M. G.; McRae K. J. J. Org. Chem. 2001, 66, 6768–6774. 10.1021/jo0159486. [DOI] [PubMed] [Google Scholar]
- Zhang F. R.; Shridhar B.; Gabelli S.; Chen X. C.; Miller M. S.; Nacev B. A.; Cheng Y. l.; Meyers D. J.; Tenney K.; Shim J. S.; Crews P.; Amzel L. M.; Ma D.; Liu J. O. J. Med. Chem. 2013, 56, 3996–4016. 10.1021/jm400227z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudin J. B.; Hareau G.; Julia S. A.; Ruel O. Tetrahedron Lett. 1991, 32, 1175–1178. 10.1016/S0040-4039(00)92037-9. [DOI] [Google Scholar]
- Okazoe T.; Takai K.; Utmoto K. J. Am. Chem. Soc. 1987, 109, 951–953. 10.1021/ja00237a081. [DOI] [Google Scholar]
- Towbin H.; Bair K. W.; DeCaprio A. J.; Eck M. J.; Kim S.; Kinder F. R.; Morollo A.; Mueller D. R.; Schindler P.; Zabludoff S.; Phillips P. E. J. Biol. Chem. 2003, 278, 52964–52971. 10.1074/jbc.M309039200. [DOI] [PubMed] [Google Scholar]
- Hu X.; Dang Y.; Tenney K.; Crews P.; Tsai C. W.; Sixt K. M.; Cole P. A.; Liu J. O. Chem. Biol. 2007, 14, 764–774. 10.1016/j.chembiol.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; LaMontagne K.; Sabio M.; Sharma S.; Versace R. W.; Yusuff R. W.; Phillips P. E. Cancer Res. 2004, 64, 2984–2987. 10.1158/0008-5472.CAN-04-0019. [DOI] [PubMed] [Google Scholar]
- Lu J.-P.; Yuan X.-H.; Yuan H.; Wang W.-L.; Wan B.; Franzblau S. G.; Ye Q.-Z. ChemMedChem 2011, 6, 1041–1048. 10.1002/cmdc.201100003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.-P.; Yuan X.-H.; Ye Q.-Z. Eur. J. Med. Chem. 2012, 47, 479–484. 10.1016/j.ejmech.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W.; Lu J.-P.; Ye Q.-Z. J. Med. Chem. 2012, 55, 8021–8027. 10.1021/jm3008695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y.; Shen S.; Verma I. M. Cancer Immunol. Res. 2014, 2, 823–831. 10.1158/2326-6066.CIR-14-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin S.-Q.; Wang J.-J.; Zhang C.-M.; Liu Z.-P. Curr. Med. Chem. 2012, 19, 1021–1035. 10.2174/092986712799320709. [DOI] [PubMed] [Google Scholar]
- Zhang P. T.; Yang X.; Zhang F.-R.; Gabelli S. B.; Wang R. X.; Zhang Y. H.; Bhat S.; Chen X. C.; Furlani M.; Amzel L. M.; Liu J. O.; Ma D. Bioorg. Med. Chem. 2013, 21, 2600–2617. 10.1016/j.bmc.2013.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X.; Addlagatta A.; Lu J.; Matthews B. W.; Liu J. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 18148–18153. 10.1073/pnas.0608389103. [DOI] [PMC free article] [PubMed] [Google Scholar]