

Abstract
Formal syntheses of tetracyclic terpenoids frondosin B and liphagal are described. Both synthetic routes rely on the use of platinum-catalyzed α,β-unsaturated carbene formation for the key C–C bond forming transformations. The successful route toward frondosin B utilizes a formal (4 + 3) cycloaddition, while the liphagal synthesis features the vinylogous addition of an enol nucleophile as a key step. Both synthetic routes are discussed, revealing insights into structural requirements in the catalytic α,β-unsaturated carbene reaction manifold.
Graphical Abstract
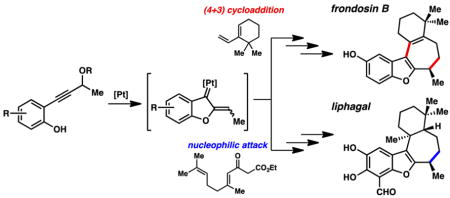
Frondosin B and liphagal are tetracyclic terpenoids that share a common carbon skeleton, and both possess promising and distinct bioactivity.1 Frondosin B was reported to be an antagonist toward interleukin-8 receptors, which are involved in inflammatory disorders such as rheumatoid arthritis.1a,2 Liphagal has been found to have human phosphatidylinositol-3-kinase (PI3K) α-selective inhibitory activity;1b,3 compounds that modulate the PI3K signaling pathway may have therapeutic potential in the treatment of autoimmune disorders, cancer, and cardiovascular disease. Their interesting architectures coupled with their potential to serve as leads in the search for therapeutics have inspired numerous groups to undertake total syntheses of both frondosin B4 and liphagal.1b,5
We and others have been investigating the formation and application of transient α,β-unsaturated Pt carbenes generated via Pt catalysis.6 These intermediates are capable of reacting in a variety of ways, including cycloadditions,6a–d 1,2-migrations,6e–g and interception by nucleophiles.6h–j As such they can construct a diverse spectrum of products, portending considerable utility in synthesis. It was therefore somewhat surprising that methods associated with the generation of α,β-unsaturated Pt carbenes had in fact not been used en route to a natural product at the outset of this work. Nonetheless, we anticipated that both frondosin B and liphagal could be prepared using this chemistry, ideally utilizing Pt catalysis to rapidly construct the B and C rings of these molecules (Figure 1). Specifically, we planned to employ a general strategy joining a carbene originating from precursor phenol 3 with diene 5. The resulting tetracyclic products, when appropriately decorated, could then be transformed to frondosin B via alkene isomerization (6 → 1, R′ = H), while alkene reduction would complete the synthesis of liphagal (6 → 2, R′ = Me).
Figure 1.

Generalized Pt-carbene approach to frondosin B/liphagal molecular core.
This strategy raised some pertinent questions. Mainly, how would the respective dienes react with the carbene intermediate? Dienes featuring enhanced nucleophilicity (largely silyl enol ethers) have been demonstrated to undergo net (4 + 3) cycloadditions.6b,c,7 On the other hand, we had observed intramolecular ene-type reactivity with electron-rich alkene nucleophiles.6h,8 We anticipated that we could potentially advance several possible adducts to the tetracyclic motif, but the uncertainty of which product(s) would form would undoubtedly impact our strategy. Other more subtle questions (e.g., steric characteristics of specific diene, electronic nature of phenol precursor) may also need to be examined.
We began our effort targeting the structurally simpler frondosin B, preparing the two components of the key cycloaddition reaction (Scheme 1). The phenol component (3a) was synthesized by a four-step sequence beginning with p-methoxyphenol (7). The phenol moiety was first converted to a methoxymethyl acetal. Ortho-iodination9 followed by hydrolysis afforded iodophenol 9. Sonogashira coupling10 with benzyl ether 1011 produced the target alkyne for carbene generation (3a). A known two-step sequence converted 2,2-dimethylcyclohexanone (11) to the requisite diene.12
Scheme 1.

Frondosin B: Syntheses of Reactants Alkyne 3a and Diene 5a
With phenol 3a and diene 5a in hand, we investigated the key carbene reaction (Scheme 2). To our delight, treating the two components with catalytic [(C2H4)2PtCl2]2 (Zeise’s dimer) in 1,4-dioxane at 100 °C led to observable formation of tetracycle 6aa. From this result it was clear that electron-neutral diene 5a was a competent cycloaddition partner and that the (4 + 3) process6b,c was the predominant pathway. Interestingly, a notable amount of structural isomer 12 was also produced; this compound presumably arises from a differential bond migration onto the Pt-carbene species after an initial (4 + 2) process.13 Both of these compounds were formed as apparently single diastereomers, indicating these carbene-based cycloadditions can be highly stereoselective. The addition of ligand appeared to somewhat diminish the formation of the alternate product (12), and also improved the yield to a measurable degree. We evaluated different ligand systems and found that inclusion of Feringa’s phosphor-amidite14 provided the best results, with a 74% isolated yield of chromatographically inseparable tetracycles 6aa and 12, in a 2.5:1 ratio.15 These compounds could be separated after the acid-mediated isomerization4i to conjugated benzofurans 13 and 14.16 The syntheses of tetracycle 6aa, and subsequently tetracycle 13, represent a formal total synthesis of frondosin B; demethylation affords the natural product.4a,b,g,i,k
Scheme 2.

Pt-Catalyzed (4 + 3) Cycloaddition toward Frondosin B
Having established the capacity of the (4 + 3) reaction to efficiently generate this tetracyclic skeleton in the formal synthesis of frondosin B, we turned our attention to the more highly substituted liphagal (Scheme 3). Here, we chose sesamol (15) as a starting point for the phenol component because its use would ultimately lead to late stage intermediates that should be viable precursors to the natural product. To this end, silyl protection of 15 followed by TFA-catalyzed iodination17 gave aryl iodide 17. Sonogashira coupling10 with alkyne 10 followed by removal of the TBS group provided phenol 3b. A known procedure allowed us to prepare diene 5b in one step from β-cyclocitral (19).18 Unfortunately, our attempts to perform the cycloaddition between phenol 3b and diene 5b failed to give detectable amounts of the desired product (6bb). The reason for this reaction failure was not immediately clear; both the diene and phenol components had been changed from the frondosin B cycloaddition.
Scheme 3.

Cycloaddition Approach to Liphagal
To diagnose the origin of the failure of this cycloaddition, we systematically paired each of the components from the successful process with those of the unsuccessful one (Figure 2). Reacting the methyl-containing diene (5b) with less electron-rich phenol 3a failed to give the predicted cycloadduct, instead forming compound 20, the electrophilic substitution product via diene attack on the carbene. Evidently, in this case the presence of the methyl group was responsible for the inability to complete the cyclization event, but the exact role that it may play depends on the mechanistic nature of the cycloaddition itself. In the case that the cycloaddition prefers a concerted pathway, the methyl group would disfavor the reactive s-cis conformation of the diene necessary for the cycloaddition (Scheme 4). The inability of the diene to achieve this conformation would then force the reaction to follow an asynchronous pathway, a mechanism that may be favored regardless.19 An allylic cation generated by nucleophilic attack of diene 5b on carbene 4a could be unable to undergo cyclization because of the steric congestion of the reacting centers. This cation would then simply undergo loss of a proton to generate diene 20.
Figure 2.

Alkyne/diene reaction combinations in Pt carbene catalysis.
Scheme 4.

Failed Cycloaddition–Electrophilic Substitution on Diene 5b
Curiously, pairing the simpler (and previously successful) diene 5a with sesamol-derived phenol 3b also failed to give a cycloaddition product, this time leading only to decomposition. This seems to indicate that the electron-rich nature of this phenol was also detrimental toward affording the desired reactivity. We hypothesized that electron density from the benzene ring could be delocalized into the Pt carbene, attenuating its electrophilicity (Figure 3).20 The cycloaddition route to liphagal therefore appeared to suffer from problems arising from both components, but this realization did not necessarily imply that phenol 3b would be unsuccessful in all manifolds of α,β-unsaturated Pt carbene chemistry.
Figure 3.

Electronic rationale for poor reactivity of phenol 3b.
Depending on to what extent delocalization reduces the electrophilicity of putative carbene intermediate 4b, we thought it may still react with stronger nucleophiles. In fact, we recently reported that enols derived from β-dicarbonyl compounds are competent nucleophiles in this chemistry.6j With the correct substitution pattern, a β-dicarbonyl compound may lead to nucleophilic addition products that could intercept the Andersen biomimetic approach1b downstream. Along those lines, our revised route to liphagal is illustrated in Figure 4. An acid catalyzed cyclization of an unconjugated diene similar to known cases1b,5a,b would give the tetracyclic core of the natural product. This intermediate (23) could be produced from ketoester 24 via decarboxylation and reduction. We would prepare ketoester 24 from alkyne 3b and dicarbonyl compound 25 using our previously reported method.
Figure 4.

Revised synthetic approach toward liphagal.
Toward this end, we prepared the pronucleophile component (25) via the Roskamp reaction21 from geranial (26, Scheme 5). We were pleased to find that this β-ketoester approach was significantly more effective than the previous attempts using a diene. We were able to produce alkylation adduct 24 in 76% yield using the more reactive enol nucleophile,22 whereas, before, reactions with phenol partner 3b had given only decomposition as discussed above. From this stage, a decarboxylation/reduction/acetylation sequence afforded acetate 28. Mild allylic reduction23 yielded a polyene that, after treatment with chlorosulfonic acid, gave tetracycle 22 in modest yield, with this species constituting a formal synthesis of liphagal.5f
Scheme 5.

Completed Formal Total Synthesis of Liphagal Using Pt-Carbene Formation in Combination with Stabilized Nucleophile
In summary, we were pleased that our efforts not only culminated in the total syntheses of frondosin B and liphagal but also provided important insights into unsaturated Pt-carbene catalysis. The (4+3) cycloaddition chemistry was easily applicable to the synthesis of frondosin B, and our finding that phosphoramidite ligands improve the carbene-based cycloaddition suggests future opportunities in catalytic reaction design. The analogous cycloaddition was unsuccessful in the liphagal context, but gratifyingly the enol interception strategy we developed provided a fruitful solution. We anticipate that the studies herein may prove beneficial to the inclusion of these methods in future synthetic strategies toward complex molecules. Further studies of α,β-unsaturated Pt carbenes are ongoing, and their results will be reported in due course.
Supplementary Material
Acknowledgments
The National Institutes of Health (NIGMS, R01GM110560) is gratefully acknowledged.
Footnotes
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.6b03682.
Experimental procedures, compound characterization data and spectra (PDF)
ORCID
Eric M. Ferreira: 0000-0001-9412-0713
The authors declare no competing financial interest.
References
- 1.(a) Patil AD, Freyer AJ, Killmer L, Offen P, Carte B, Jurewicz AJ, Johnson RK. Tetrahedron. 1997;53:5047. [Google Scholar]; (b) Marion F, Williams DE, Patrick BO, Hollander I, Mallon R, Kim SC, Roll DM, Feldberg L, Van Soest R, Andersen RJ. Org Lett. 2006;8:321. doi: 10.1021/ol052744t. [DOI] [PubMed] [Google Scholar]
- 2.For a recent review on the frondosins, see: VanHeyst MD, Wright DL. Eur J Org Chem. 2015;2015:1387.
- 3.Pereira AR, Strangman WK, Marion F, Feldberg L, Roll D, Mallon R, Hollander I, Andersen RJ. J Med Chem. 2010;53:8523. doi: 10.1021/jm100531u. [DOI] [PubMed] [Google Scholar]
- 4.(a) Inoue M, Carson MW, Frontier AJ, Danishefsky SJ. J Am Chem Soc. 2001;123:1878. doi: 10.1021/ja0021060. [DOI] [PubMed] [Google Scholar]; (b) Hughes CC, Trauner D. Angew Chem, Int Ed. 2002;41:1569. doi: 10.1002/1521-3773(20020503)41:9<1569::aid-anie1569>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]; (c) Kerr DJ, Willis AC, Flynn BL. Org Lett. 2004;6:457. doi: 10.1021/ol035822q. [DOI] [PubMed] [Google Scholar]; (d) Li X, Ovaska TV. Org Lett. 2007;9:3837. doi: 10.1021/ol701633z. [DOI] [PubMed] [Google Scholar]; (e) Olson JP, Davies HML. Org Lett. 2008;10:573. doi: 10.1021/ol702844g. [DOI] [PubMed] [Google Scholar]; (f) Mehta G, Likhite NS. Tetrahedron Lett. 2008;49:7113. [Google Scholar]; (g) Reiter M, Torssell S, Lee S, MacMillan DWC. Chem Sci. 2010;1:37. doi: 10.1039/C0SC00204F. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Garayalde D, Krüger K, Nevado C. Angew Chem, Int Ed. 2011;50:911. doi: 10.1002/anie.201006105. [DOI] [PubMed] [Google Scholar]; (i) Zhang J, Li L, Wang Y, Wang W, Xue J, Li Y. Org Lett. 2012;14:4528. doi: 10.1021/ol3020013. [DOI] [PubMed] [Google Scholar]; (j) Laplace DR, Verbraeken B, Van Hecke K, Winne JM. Chem - Eur J. 2014;20:253. doi: 10.1002/chem.201303273. [DOI] [PubMed] [Google Scholar]; (k) Oblak EZ, VanHeyst MD, Li J, Wiemer AJ, Wright DL. J Am Chem Soc. 2014;136:4309. doi: 10.1021/ja413106t. [DOI] [PubMed] [Google Scholar]
- 5.(a) Mehta G, Likhite NS, Kumar CSA. Tetrahedron Lett. 2009;50:5260. [Google Scholar]; (b) Deore V, Lohar MK, Mundada R, Roychowdhury A, Vishwakarma R, Kumar S. Synth Commun. 2010;41:177. [Google Scholar]; (c) George JH, Baldwin JE, Adlington RM. Org Lett. 2010;12:2394. doi: 10.1021/ol100756z. [DOI] [PubMed] [Google Scholar]; (d) Alvarez-Manzaneda E, Chahboun R, Alvarez E, Cano MJ, Haidour A, Alvarez-Manzaneda R. Org Lett. 2010;12:4450. doi: 10.1021/ol101173w. [DOI] [PubMed] [Google Scholar]; (e) Day JJ, McFadden RM, Virgil SC, Kolding H, Alleva JL, Stoltz BM. Angew Chem, Int Ed. 2011;50:6814. doi: 10.1002/anie.201101842. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Kamishima T, Kikuchi T, Narita K, Katoh T. Eur J Org Chem. 2014;2014:3443. [Google Scholar]
- 6.(a) Saito K, Sogou H, Suga T, Kusama H, Iwasawa N. J Am Chem Soc. 2011;133:689. doi: 10.1021/ja108586d. [DOI] [PubMed] [Google Scholar]; (b) Shu D, Song W, Li X, Tang W. Angew Chem, Int Ed. 2013;52:3237. doi: 10.1002/anie.201209266. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kusama H, Sogo H, Saito K, Suga T, Iwasawa N. Synlett. 2013;24:1364. [Google Scholar]; (d) Yang W, Wang T, Yu Y, Shi S, Zhang T, Hashmi ASK. Adv Synth Catal. 2013;355:1523. [Google Scholar]; (e) Allegretti PA, Ferreira EM. Org Lett. 2011;13:5924. doi: 10.1021/ol202649j. [DOI] [PubMed] [Google Scholar]; (f) Allegretti PA, Ferreira EM. Chem Sci. 2013;4:1053. [Google Scholar]; (g) Kwon Y, Kim I, Kim S. Org Lett. 2014;16:4936. doi: 10.1021/ol502465e. [DOI] [PubMed] [Google Scholar]; (h) Allegretti PA, Ferreira EM. J Am Chem Soc. 2013;135:17266. doi: 10.1021/ja408957a. [DOI] [PubMed] [Google Scholar]; (i) Shu D, Winston-McPherson GN, Song W, Tang W. Org Lett. 2013;15:4162. doi: 10.1021/ol4018408. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Allegretti PA, Huynh K, Ozumerzifon TJ, Ferreira EM. Org Lett. 2016;18:64. doi: 10.1021/acs.orglett.5b03246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xue/Li and Winne also reported comparable (4 + 3) cycloaddition approaches in their syntheses of frondosin B and 5-epi-liphagal, utilizing a presumed benzofuranyl cationic species as their three-atom component. See refs 4i and 4j.
- 8.Tang also reported the competency of two less nucleophilic dienes: cyclopentadiene and cyclohexadiene. See ref 6b.
- 9.(a) Townsend CA, Bloom LM. Tetrahedron Lett. 1981;22:3923. [Google Scholar]; (b) Gschwend HW, Rodriguez HR. Org React. 1979;26:1. [Google Scholar]
- 10.Chinchilla R, Nájera C. Chem Rev. 2007;107:874. doi: 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]
- 11.Ether 10 was synthesized by standard alkylation using 3-butyn-2-ol and BnBr. See the Supporting Information for details.
- 12.Tanis SP, Abdallah YM. Synth Commun. 1986;16:251. [Google Scholar]
- 13.A similar observation of differential bond migrations was made by Iwasawa. See ref 6c.
- 14.Teichert JF, Feringa BL. Angew Chem, Int Ed. 2010;49:2486. doi: 10.1002/anie.200904948. [DOI] [PubMed] [Google Scholar]
- 15.(a) The intermediacy of a furanyl cation via benzofuran formation followed by ether ionization was ruled out as a mechanistic pathway. The benzofuranyl Bn ether from intramolecular hydroalkoxylation of 3a was unreactive under the catalytic conditions. (b) Although the phosphoramidite ligand is chiral, enantioinduction in the cycloaddition was modest (<15% ee).
- 16.Benzofuran 13 was isolated as a 2.5:1 mixture of conjugated olefin isomers. These isomers can be purified after demethylation. See refs 4a, g, j.
- 17.Liron F, Fontana F, Zirimwabagabo JO, Prestat G, Rajabi J, La Rosa C, Poli G. Org Lett. 2009;11:4378. doi: 10.1021/ol9017326. [DOI] [PubMed] [Google Scholar]
- 18.Winne JM, Catak S, Waroquier M, Van Speybroeck V. Angew Chem, Int Ed. 2011;50:11990. doi: 10.1002/anie.201104930. [DOI] [PubMed] [Google Scholar]
- 19.Both Tang and Iwasawa discuss mechanistic considerations of the α,β-unsaturated carbene-based (4 + 3) cycloaddition. Multiple possible pathways are suggested, many of which implicate an asynchronous pathway, whose reactivity would be facilitated by the use of siloxydienes. See refs 6b, c.
- 20.An alternative possibility is that the electron-rich arene is acting as a competing nucleophile, leading to complex oligomerization of phenol 3b.
- 21.Holmquist CR, Roskamp EJ. J Org Chem. 1989;54:3258. [Google Scholar]
- 22.In our experience, carbene interception will occur at lower temperature ranges (23–50 °C) for more reactive nucleophiles (e.g., enol ethers), while less reactive nucleophiles (e.g., electron-neutral dienes) tend to require more elevated temperatures (80–100 °C) for efficient reaction times.
- 23.(a) am Ende CW, Zhou Z, Parker KA. J Am Chem Soc. 2013;135:582. doi: 10.1021/ja3108577. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Trost BM, Shen HC, Dong L, Surivet JP, Sylvain C. J Am Chem Soc. 2004;126:11966. doi: 10.1021/ja048078t. [DOI] [PubMed] [Google Scholar]; (c) Tsuji J, Mandai T. Synthesis. 1996;1996:1. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.