

Abstract
Activation of connexin hemichannels is involved in the pathophysiology of disorders that include deafness, stroke, and cardiac infarct. This aspect makes hemichannels an attractive therapeutic target. Unfortunately, most available inhibitors are not selective or isoform specific, which hampers their translational application. The absence of a battery of useful inhibitors is due in part to the absence of simple screening assays for the discovery of hemichannel-active drugs. Here, we present an assay that we have recently developed to assess hemichannel function. The assay is based on the expression of functional human connexins in a genetically modified bacterial strain deficient in K+ uptake. These modified cells do not grow in low-K+ medium, but functional expression of connexin hemichannels allows K+ uptake and growth. This cell-growth-based assay is simple, robust, and easily scalable to high-throughput multi-well platforms.
Keywords: connexin, gap junction, high-throughput screening, ion channel, infarct, cerebrovascular accident, aminoglycoside
Introduction
Membrane proteins correspond to ~30 percent of genomes and are frequently expressed at low levels [1]. However, they are the targets of the majority of drugs currently on the market and drug transporting membrane proteins are also important targets to affect pharmacokinetics [2,3]. Ion channels constitute a subgroup of membrane proteins characterized by their conductance, gating, and selectivity. When open, ion channels form a hydrophilic pathway across the membrane through which ions flow at a high rate by electrodiffusion, determining their conductance, whereas the relative permeability of different ions, due to differences in charge and/or size, determines selectivity. Finally, factors such as voltage, ligands, and post-translational modifications produce conformational changes that open and close the channels (gating). Some ion channels are highly selective (e.g., voltage-gated K+ channels), while others are not. The latter group includes gap-junction channels (GJCs) and hemichannels (HCs) formed by connexins.
There are 21 human connexin isoforms, with lengths between 226 and 543 amino acids [4,5]. Connexins can oligomerize to form homomeric or heteromeric HCs (Figure 1) of varying permeability properties, regulation, and associations with other proteins [4,6]. Each connexin has four transmembrane helices (M1 to M4), two extracellular loops, and cytoplasmic hydrophilic regions (N- and C-terminal regions and intracellular loop) (Figure 1) [4-6]. Sequence analysis shows that the intracellular regions are poorly conserved, whereas the M1-extracellular loop 1-M2 sequence is well conserved, especially the M1 sequence [5]. Six connexins subunits assemble as hexamers to form HCs [6], and head-to-head docking of HCs from adjacent cells forms GJCs (Figure 1) [6].
Figure 1.

Connexin channels and hemichannels. Schematic representation of a connexin subunit (monomer), a hemichannel (hexamer), and a gap-junction channel (dodecamer). M1 to M4: transmembrane helices. Each monomer is depicted as a cylinder in the hemichannel and the gap-junction channel. This figure is reproduced from one originally published in the J Biol Chem [30], and is reproduced with permission from the American Society for Biochemistry and Molecular Biology.
Because of their large pore size, GJCs are permeable to small hydrophilic molecules of up to 400-800 Da (including many second messengers) [6]. Therefore, they not only mediate cell-to-cell electric coupling, but are involved in chemical coupling between neighboring cells. The general properties of GJCs and HCs in terms of pore size and selectivity are quite similar [6,7], with the pore formed by M1 and M2, and the narrowest region of the pore near the extracellular side of the membrane [4,8,9]. Since uncharged hydrophilic molecules can diffuse through GJCs and HCs, they are often referred to as channels, as opposed to ion channels.
Connexins in Health and Disease
HCs are mostly closed under normal conditions, but they play an important role in autocrine and paracrine signaling, by mediating the transmembrane fluxes of signaling molecules/metabolites such as ATP, NAD+, glutamate, glutathione, PGE2, and glucose [10]. Cx43, a 382-amino acid connexin, is expressed in parenchymal cells of a variety of organs, such as cardiac muscle, brain and kidney, as well as in capillary endothelial cells [11]. Connexins are abundantly expressed in the excitation-conduction system and in the contractile myocardium, and Cx43 GJCs mediate the cell-to-cell conduction of the electrical impulse generated by the sinoatrial node, essential for the coordinated contraction of the heart [12-16]. Heart disease is the most common cause of death in the U.S., and many of these deaths are caused by cardiac ischemia and arrhythmias. Cx43 GJCs and HCs have important roles in the damage of the heart muscle elicited by ischemia, and the genesis and maintenance of arrhythmias [14,15,17-20]. Cx43 also plays important roles in ventricular arrhythmias, including the most lethal one, ventricular fibrillation [12,14,15,17,19]. Cx43 HCs play a pathophysiological role in ischemic damage of the heart (myocardial infarction), brain (stroke), and kidneys (ischemic renal tubule necrosis) [17,18,21-26]. The activation of Cx43 HCs in cardiomyocytes, astrocytes, and renal proximal tubule cells under conditions that mimic ischemia contributes to the cell damage [17,18,21-26]. Under physiological conditions several factors maintain the HCs mostly closed. These include normal extracellular Ca2+ in the low millimolar range, the cell-negative membrane voltage, and phosphorylation by PKC [11,27-35]. However, HCs open in ischemia, even in the continuous presence of millimolar extracellular [Ca2+] [11,21-26,28,30-33,35]. The mechanism is not completely understood, but seems to involve changes in post-translational modifications (decreased phosphorylation due to ATP depletion and increased phosphatase activity, and/or changes in nitrosylation due to the oxidative stress), although the increase in cytosolic [Ca2+] can also play a role [11,18,22,24,25,33,35-41]. GJCs link the cytoplasm of adjacent cells, compartments of very similar composition, whereas plasma membrane HCs link the intracellular and extracellular fluids, which have very different compositions. Abnormal HCs opening can lead to losses of metabolites, Ca2+ influx with alterations of signaling and protease activation, equilibration of ionic gradients, and cell swelling. Therefore, abnormal HC opening can result or contribute to cell damage and/or death in a number of disorders (Figure 2).
Figure 2.
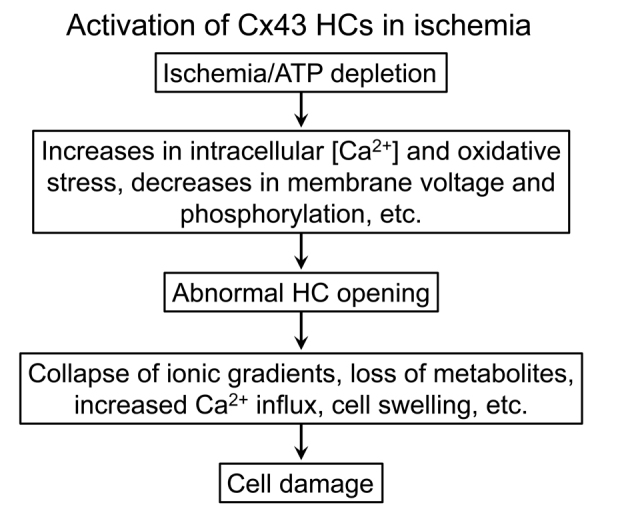
Activation of Cx43 hemichannels under ischemic conditions. Activation of Cx43 hemichannels (HCs) participates in the damage of cardiomyocytes, glia and renal tubule cells in ischemia. The pathways of activation have not been clearly defined, but there is evidence for all those indicated in the figure.
Connexin 26 (Cx26) has paramount importance in the inner ear. Hearing loss is very common and can occur at any time from infancy to old age [36,42-47]. Approximately 1/1,000 infants have profound hearing impairment, and ~50 percent of the cases are due to single gene mutations, mostly Cx26 mutations [36,42-46]. In addition, a role of Cx26 in non-genetic deafness is also likely [42,44,46,48,49]. Cx26, the main connexin in the inner ear, is smaller (226 amino acids) than Cx43 (382 amino acids) and their primary sequences display < 30 percent amino-acid identity. Cx26 has a very short C-terminal domain (< 20 residues vs. > 150 residues in Cx43). Based on their lower permeability to fluorescent dyes and metabolites, it seems that GJCs and HCs formed by Cx26 have a smaller apparent pore size than those formed by Cx43 [6].
In the inner ear, the cochlea houses the organ of Corti, a narrow spiral containing the hair cells that transduce sound into electrical impulses. The cochlear gap-junctional communication network is essential for hearing [45,46,50]. Cx26 mutations may cause deafness by reducing gap-junctional communication with decreased K+ recycling into the endolymph [50,51], a mechanism recently questioned [52], or by selectively reducing cell-to-cell permeability to signaling molecules such as inositol trisphosphate (IP3) [53-56]. Deafness due to “leaky” HCs has also been proposed [43,57,58]; in this case, cell damage, with the resulting deafness, would occur as a consequence of uncompensated water and solute fluxes (Figure 2). In addition, it has been shown that some deafness-associated mutants display increased Ca2+ permeability, and it has been speculated that the increased Ca2+ influx results in apoptosis and death of hair and supporting cells [59,60]. Although the detailed mechanisms of deafness are not definitively understood, Cx26 HC inhibitors are potential therapeutic leads for deafness mediated by leaky HCs, including HCs that display increased Ca2+ permeability [43,57-62].
There are many additional disorders where targeting connexin HCs may prove useful. These include HCs in the central nervous system, where under some abnormal conditions activated microglia can release massive amounts of glutamate through connexin HCs, which damages neural cells. This process could play a major role in the neuronal damage of a variety of neurodegenerative diseases, and targeting of HCs has been proposed for therapy [63-65]. Another potential use of HC inhibitors is for the inhibition of neovascularization in the treatment of cancer [65].
High-Throughput Assays to Assess Gap-junction Channel Function
Dye transfer experiments have been traditionally used to assess GJC functionality and have been recently exploited for high-throughput screening (HTS) assays aimed at discovering GJC inhibitors. These assays are based on the transport from donor to acceptor cells via GJCs. In one assay, donor cells were loaded with calcein, a fluorescent probe, and its transfer to acceptor cells was imaged using automated fluorescence microscopy [66]. Another assay was based on the permeability of GJCs to Ca2+ [67]. Donor cells co-expressing Cx43 and α1A adrenergic receptors were paired with acceptor cells co-expressing Cx43 and aequorin, a Ca2+-sensitive luminescent protein. Activation of α1A receptors led to Ca2+ waves that spread to the acceptor cells via GJCs and were detected by the increase in aequorin luminescence. A rather similar principle was used in another assay, where donor cells co-expressed a I- transporter and connexins, the acceptor cells co-expressed connexins, and a yellow fluorescent protein variant whose fluorescence emission is quenched by the anion [68]. In this assay, influx of I- into the donor cell is followed by diffusion through GJCs into the acceptor cell, where it quenches the yellow fluorescent protein emission. In all these assays, changes in the signal that reflect decrease donor/acceptor transport were used to identify new GJC inhibitors [66-68]. These assays have been proven useful, but have limitations such as the relative complexity of sample preparation and data analysis. They are also focused on GJCs and cannot be used to assess the function of HCs. There are many HC assays based on methodologies such as dye uptake and electrophysiology [6,10], but complexity and cost make their adaptation for HTS of large chemical libraries costly and difficult.
Expression of Human Connexin Hemichannels in Bacteria
Studies with isolated systems where experimental conditions are controlled are important to understand the bases of normal function and the molecular mechanisms of diseases. Recombinant connexins for research studies are generally expressed in mammalian cells, insect cells or frog oocytes. Until our recent report, the insect cell/baculovirus expression system was the only available system that yielded purified connexins in the amounts necessary for detailed biochemical and biophysical studies [8,31,69-73]. In our recent report, we developed and optimized an Escherichia coli-based expression/purification system that yields milligram amounts of functional human Cx26 HCs [74]. Bacteria were transformed with a plasmid containing E. coli-optimized DNA for the expression of human Cx26 with a C-terminal poly-His tag preceded by a protease cleavage site (TEV protease) to remove the tag after purification. Cx26 was purified by immobilized metal affinity chromatography based on the affinity of the Cx26 poly-His tag for Co2+, followed by size-exclusion chromatography. The highly-purified Cx26 formed very stable HCs in detergent [74]. The human Cx26 HCs purified from bacteria were structurally and functionally identical to those purified from insect cells and essentially all HCs reconstituted in unilamellar liposomes were functional [72,74]. Although differences between bacterial and mammalian post-translational modifications could be an issue, the problem is minimized because there is no evidence of direct regulation of Cx26 by post-translational modifications and connexins are not glycosylated. In contrast, phosphorylation of the C-terminal domain of Cx43 clearly regulates function [11,31,33,35,73], but it is possible to phosphorylate purified connexins in vitro [31,73]. The bacterial expression system has the potential to accelerate the pace of structural and functional connexin research.
An Assay for Hemichannel Function in Bacteria
Independently of the usefulness of the bacterial expression system, our report showed that it is possible to express functional human connexin HCs in E. coli [74]. We took advantage of that observation to develop a new cell-based assay to evaluate the function of human HCs expressed in bacteria. We used an E. coli strain (LB2003) with deletion of three K+ uptake systems (Kdp, Kup and Trk) [75,76]. LB2003 cells do not grow in low-[K+] medium, but grow under conditions where K+ influx and intracellular [K+] are expected to increase such as increasing [K+] in the growth medium or expressing recombinant K+-selective channels [75,77,78]. K+ is necessary for growth because of its involvement in many important cellular processes such as maintaining turgor pressure, activation of enzymes and intracellular pH regulation. We were able to achieve growth of LB2003 cells in low-[K+] medium by expressing human Cx26 (Figure 3), Cx43, or Cx46 [74,79]. On one hand, this phenomenon of growth recovery (growth complementation) was expected because HCs provide a pathway for K+ influx, as K+ channels do. On the other hand, HCs are “large” and poorly-selective channels that can also have deleterious effects on the cells (e.g., depolarization, alterations in metabolites homeostasis; see Figure 2). Since there is a favorable electrochemical driving force for K+ electrodiffusion across the E. coli inner membrane, it is expected that the increased K+ permeability elicited by HC expression will produce growth complementation by increasing K+ influx and steady-state cytosolic [K+]. In fact, our recent data support such a mechanism by showing that intracellular [K+] is increased by ~30 mM in LB2003 cells expressing Cx26 HCs that are grown in 4 mM [K+] [79].
Figure 3.

Growth complementation by expression of Cx26 HCs in LB2003 cells. (a) Schematic representation of the LB2003 cells. (b) Growth complementation by Cx26 shown as percent of the maximal growth in the Cx26-expressing cells. Data are means ± SEM. Adapted from J Biomol Screen [79] with permission from SAGE Publications. See [79] for details.
Two sets of results support the notion that growth complementation by connexin expression is the result of the presence of functional HCs: 1) Growth complementation was blocked by known HC inhibitors that included divalent cations, 2-aminoethoxydiphenyl borate, octanol, and aminoglycosides (Figure 4) [74,79]. Inhibitors of connexin-formed channels with affinities in the low-nM range are not available, but these compounds at the concentrations employed are known to inhibit HCs [80-83]. Although non-specific, divalent cations are well-known inhibitors of HCs [84]; 2) The sensitivity of growth complementation by Cx26 mutants to the inhibitory effect of Ca2+ was altered [79]. The Cx26 mutants G45E and D50N form functional HCs with low sensitivity to the inhibitory effect of extracellular [Ca2+] [59,61,62]. These mutants were able to produce growth complementation, but inhibition by external [Ca2+] was severely hampered (D50N) or abolished (G45E) in the cells expressing the mutants [79]. Our assay complements more complex methodologies that can provide far more detailed mechanistic information, such as permeability assays and electrophysiological studies [6,10].
Figure 4.

Effects of HC inhibitors on Cx26-dependent growth complementation. Reduction in growth complementation by connexin HC inhibitors in LB2003 cells expressing Cx26. Cells were grown without any additions (control, not shown), or in medium supplemented with 50 µM 2-aminoethoxydiphenyl borate (APB), 1 mM 1-octanol (Oc), 100 µM kanamycin A, 1 mM Ca2+ (Ca), 20 mM Mg2+ (Mg), or 100 µM Zn2+ (Zn). Data are means ± SEM. Adapted from J Biomol Screen [79] with permission from SAGE Publications. See [79] for details.
The Z’ factor is a commonly used statistical parameter to assess the quality of a HTS assay [85]. It is a dimensionless parameter that takes into account the day-to-day and well-to-well variability of the samples. It is calculated from the sample means (µ) and standard deviations (σ) as: Z’ = 1 – (3σs + 3σc)/ǀµs - µcǀ, where the subscripts s and c denote sample and control, respectively. An assay with Z’ ≥ 0.5 is best, whereas values between 0 < Z’ < 0.5 point to a marginal assay that requires optimization, and assays with Z’ < 0 are not suitable for HTS [85]. With a Z’ of 0.8, our assay has a great potential for HTS (Figure 5) [79].
Figure 5.

Evaluation of assay performance. Values are presented as means ± SD of data from multi-well plates from 276 (No Cx26 and Cx26) and 72 (Cx26 + kanamycin) measurements performed in four independent experiments. Growth was normalized to the mean of Cx26-expressing cells under control conditions. The HC inhibitor kanamycin was used at a concentration of 10 µM, which produces ~50 percent inhibition. The calculated Z’ factors for Cx26 vs. No Cx26 and Cx26 + kanamycin A vs. No Cx26 were 0.8 and 0.6, respectively. Adapted from J Biomol Screen [79] with permission from SAGE Publications. See [79] for details.
Because of its simplicity, low-cost, easy scalability, reproducibility, and sensitivity, the assay presented here should be useful for the discovery of new and better HC inhibitors [79]. However, it can miss inhibitors because of factors such as limited access to the periplasmic space due to the presence of the outer membrane, indirect effects that need interaction of inhibitors with proteins not present in bacteria, and partial HC inhibition that is insufficient to decrease cell [K+] to impair cell growth.
The HTS HC function assay presented here has the potential for screening large chemical libraries to discover new, effective, and specific HC inhibitors for research and therapy. To accomplish this aim, scaling up the assay from 96-well to 384- or 768-well plates will be desirable. Since Cx26 and Cx43/Cx46 are among the most dissimilar connexin isoforms [4-6], it seems likely that our assay can be used to assess the function of HCs formed by most or all connexin isoforms.
Acknowledgments
This work was supported by National Institutes of Health grants R01GM79629, 3R01GM079629-03S1 and R01GM097159, American Heart Association Texas Affiliate Inc. grant 14GRNT18750014, and Welch Foundation grant BI-1757.
Glossary
- Cx26
connexin 26
- Cx43
connexin 43
- GJC
gap-junction channel
- HC
hemichannel
- HTS
high-throughput screening
- OD600
absorbance measured at 600 nm
Author Contributions
Drs. Krishnan, Fiori, Cuello, and Altenberg co-wrote the review article.
References
- Krogh A, Larsson B, von Heijne G. et al. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1(9):727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- International Transporter Consortium. Giacomini KM, Huang SM, Tweedie DJ. et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9(3):215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen MS, Nygaard Axelsen L, Sorgen PL. et al. Gap junctions. Compr Physiol. 2012;2(3):1981–2035. doi: 10.1002/cphy.c110051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abascal F, Zardoya R. Evolutionary analyses of gap junction protein families. Biochim Biophys Acta. 2013;1828(1):4–14. doi: 10.1016/j.bbamem.2012.02.007. [DOI] [PubMed] [Google Scholar]
- Harris AL, Locke D. Permeability of connexin channels. In: Harris AL, Locke D, editors. Connexins: A guide. New York: Humana Press; 2009. pp. 165–206. [Google Scholar]
- Saez JC, Berthoud VM, Branes MC. et al. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83(4):1359–1400. doi: 10.1152/physrev.00007.2003. [DOI] [PubMed] [Google Scholar]
- Maeda S, Nakagawa S, Suga M. et al. Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature. 2009;458(7238):597–602. doi: 10.1038/nature07869. [DOI] [PubMed] [Google Scholar]
- Bennett BC, Purdy MD, Baker KA. et al. An electrostatic mechanism for Ca(2+)-mediated regulation of gap junction channels. Nat Commun. 2016;7:8770. doi: 10.1038/ncomms9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez JC, Leybaert L. Hunting for connexin hemichannels. FEBS lett. 2014;588(8):1205–1211. doi: 10.1016/j.febslet.2014.03.004. [DOI] [PubMed] [Google Scholar]
- Marquez-Rosado L, Solan JL, Dunn CA. et al. Connexin43 phosphorylation in brain, cardiac, endothelial and epithelial tissues. Biochim Biophys Acta. 2012;1818(8):1985–1992. doi: 10.1016/j.bbamem.2011.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontes MS, van Veen TA, de Bakker JM. et al. Functional consequences of abnormal Cx43 expression in the heart. Biochim Biophys Acta. 2012;1818(8):2020–2029. doi: 10.1016/j.bbamem.2011.07.039. [DOI] [PubMed] [Google Scholar]
- Duffy HS. The molecular mechanisms of gap junction remodeling. Heart Rhythm. 2012;9(8):1331–1334. doi: 10.1016/j.hrthm.2011.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Fishman GI. Designer gap junctions that prevent cardiac arrhythmias. Trends Cardiovasc Med. 2013;23(2):33–38. doi: 10.1016/j.tcm.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severs NJ. Connexins in the heart. In: Harris AL, Locke D, editors. Connexins: A guide. New York: Humana Press; 2009. pp. 435–455. [Google Scholar]
- Jalife J, Morley GE, Vaidya D. Connexins and impulse propagation in the mouse heart. J Cardiovasc Electrophysiol. 1999;10(12):1649–1663. doi: 10.1111/j.1540-8167.1999.tb00230.x. [DOI] [PubMed] [Google Scholar]
- De Vuyst E, Boengler K, Antoons G. et al. Pharmacological modulation of connexin-formed channels in cardiac pathophysiology. Br J Pharmacol. 2011;163(3):469–483. doi: 10.1111/j.1476-5381.2011.01244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura T, Miki T, Yano T. Role of the gap junction in ischemic preconditioning in the heart. Am J Physiol Heart Circ Physiol. 2010;298(4):H1115–H1125. doi: 10.1152/ajpheart.00879.2009. [DOI] [PubMed] [Google Scholar]
- Delmar M, Makita N. Cardiac connexins, mutations and arrhythmias. Curr Opin Cardiol. 2012;27(3):236–241. doi: 10.1097/HCO.0b013e328352220e. [DOI] [PubMed] [Google Scholar]
- Gollob MH. Cardiac connexins as candidate genes for idiopathic atrial fibrillation. Curr Opin Cardiol. 2006;21(3):155–158. doi: 10.1097/01.hco.0000221574.95383.6f. [DOI] [PubMed] [Google Scholar]
- Contreras JE, Sanchez HA, Eugenin EA. et al. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc Natl Acad Sci USA. 2002;99(1):495–500. doi: 10.1073/pnas.012589799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retamal MA, Schalper KA, Shoji KF. et al. Possible involvement of different connexin43 domains in plasma membrane permeabilization induced by ischemia-reperfusion. J Membr Biol. 2007;218(1-3):49–63. doi: 10.1007/s00232-007-9043-y. [DOI] [PubMed] [Google Scholar]
- Saez JC, Contreras JE, Bukauskas FF. et al. Gap junction hemichannels in astrocytes of the CNS. Acta Physiol Scand. 2003;179(1):9–22. doi: 10.1046/j.1365-201X.2003.01196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara L, Bao X, Bello-Reuss E. et al. Do connexin 43 gap-junctional hemichannels activate and cause cell damage during ATP depletion of renal-tubule cells? Acta Physiol Scand. 2003;179(1):33–38. doi: 10.1046/j.1365-201X.2003.01198.x. [DOI] [PubMed] [Google Scholar]
- Vergara L, Bao X, Cooper M. et al. Gap-junctional hemichannels are activated by ATP depletion in human renal proximal tubule cells. J Membr Biol. 2003;196(3):173–184. doi: 10.1007/s00232-003-0636-9. [DOI] [PubMed] [Google Scholar]
- John SA, Kondo R, Wang SY. et al. Connexin-43 hemichannels opened by metabolic inhibition. J Biol Chem. 1999;274(1):236–240. doi: 10.1074/jbc.274.1.236. [DOI] [PubMed] [Google Scholar]
- Bargiello T, Brink P. Voltage-gating mechanisms of connexin channels. In: Harris AL, Locke D, editors. Connexins: A Guide. New York: Humana Press; 2009. pp. 103–128. [Google Scholar]
- Li WE, Nagy JI. Connexin43 phosphorylation state and intercellular communication in cultured astrocytes following hypoxia and protein phosphatase inhibition. Eur J Neurosci. 2000;12(7):2644–2650. doi: 10.1046/j.1460-9568.2000.00162.x. [DOI] [PubMed] [Google Scholar]
- Chen Y, Deng Y, Bao X. et al. Mechanism of the defect in gap-junctional communication by expression of a connexin 26 mutant associated with dominant deafness. FASEB J. 2005;19(11):1516–1518. doi: 10.1096/fj.04-3491fje. [DOI] [PubMed] [Google Scholar]
- Bao X, Chen Y, Lee SH. et al. Membrane transport proteins with complete replacement of transmembrane helices with polyalanine sequences remain functional. J Biol Chem. 2005;280(10):8647–8650. doi: 10.1074/jbc.M413536200. [DOI] [PubMed] [Google Scholar]
- Bao X, Reuss L, Altenberg GA. Regulation of purified and reconstituted connexin 43 hemichannels by protein kinase C-mediated phosphorylation of Serine 368. J Biol Chem. 2004;279(19):20058–20066. doi: 10.1074/jbc.M311137200. [DOI] [PubMed] [Google Scholar]
- Bao X, Chen Y, Reuss L. et al. Functional expression in Xenopus oocytes of gap-junctional hemichannels formed by a cysteine-less connexin 43. J Biol Chem. 2004;279(11):9689–9692. doi: 10.1074/jbc.M311438200. [DOI] [PubMed] [Google Scholar]
- Bao X, Altenberg GA, Reuss L. Mechanism of regulation of the gap junction protein connexin 43 by protein kinase C-mediated phosphorylation. Am J Physiol Cell Physiol. 2004;286(3):C647–C654. doi: 10.1152/ajpcell.00295.2003. [DOI] [PubMed] [Google Scholar]
- Gomez-Hernandez JM, de Miguel M, Larrosa B. et al. Molecular basis of calcium regulation in connexin-32 hemichannels. Proc Natl Acad Sci USA. 2003;100(26):16030–16035. doi: 10.1073/pnas.2530348100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 2009;419(2):261–272. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird DW. The gap junction proteome and its relationship to disease. Trends Cell Biol. 2010;20(2):92–101. doi: 10.1016/j.tcb.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Berthoud VM, Beyer EC. Oxidative stress, lens gap junctions, and cataracts. Antioxid Redox Signal. 2009;11(2):339–353. doi: 10.1089/ars.2008.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Huang T, Zhu Y. et al. Connexin43 hemichannels contribute to cadmium-induced oxidative stress and cell injury. Antioxid Redox Signal. 2011;14(12):2427–2439. doi: 10.1089/ars.2010.3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decrock E, Vinken M, Bol M. et al. Calcium and connexin-based intercellular communication, a deadly catch? Cell Calcium. 2011;50(3):310–321. doi: 10.1016/j.ceca.2011.05.007. [DOI] [PubMed] [Google Scholar]
- De Vuyst E, Wang N, Decrock E. et al. Ca(2+) regulation of connexin 43 hemichannels in C6 glioma and glial cells. Cell Calcium. 2009;46(3):176–187. doi: 10.1016/j.ceca.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Orellana JA, Sanchez HA, Schalper KA. et al. Regulation of intercellular calcium signaling through calcium interactions with connexin-based channels. Adv Exp Med Biol. 2012;740:777–794. doi: 10.1007/978-94-007-2888-2_34. [DOI] [PubMed] [Google Scholar]
- Ravecca F, Berrettini S, Forli F. et al. Cx26 gene mutations in idiopathic progressive hearing loss. J Otolaryngol. 2005;34(2):126–134. doi: 10.2310/7070.2005.04017. [DOI] [PubMed] [Google Scholar]
- Lee JR, White TW. Connexin-26 mutations in deafness and skin disease. Expert Rev Mol Med. 2009;11:e35. doi: 10.1017/S1462399409001276. [DOI] [PubMed] [Google Scholar]
- Martinez AD, Acuna R, Figueroa V. et al. Gap-junction channels dysfunction in deafness and hearing loss. Antioxid Redox Signal. 2009;11(2):309–322. doi: 10.1089/ars.2008.2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel R, Forge A. Connexins in the inner ear. In: Harris AL, Locke D, editors. Connexins: A guide. New York: Humana Press; 2009. pp. 419–434. [Google Scholar]
- Sabag AD, Dagan O, Avraham KB. Connexins in hearing loss: a comprehensive overview. J Basic Clin Physiol Pharmacol. 2005;16(2-3):101–116. doi: 10.1515/jbcpp.2005.16.2-3.101. [DOI] [PubMed] [Google Scholar]
- Zoidl G, Dermietzel R. Gap junctions in inherited human disease. Pflugers Archiv : Eur J Physiol. 2010;460(2):451–466. doi: 10.1007/s00424-010-0789-1. [DOI] [PubMed] [Google Scholar]
- Gale JE, Piazza V, Ciubotaru CD. A mechanism for sensing noise damage in the inner ear. Curr Biol. 2004;14(6):526–529. doi: 10.1016/j.cub.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Orzan E, Murgia A. Connexin 26 deafness is not always congenital. Int J Pediatr Otorhinolaryngol. 2007;71(3):501–507. doi: 10.1016/j.ijporl.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Zhao HB, Kikuchi T, Ngezahayo A. et al. Gap junctions and cochlear homeostasis. J Membr Biol. 2006;209(2-3):177–186. doi: 10.1007/s00232-005-0832-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi T, Adams JC, Miyabe Y. et al. Potassium ion recycling pathway via gap junction systems in the mammalian cochlea and its interruption in hereditary nonsyndromic deafness. Med Electron Microsc. 2000;33(2):51–56. doi: 10.1007/s007950070001. [DOI] [PubMed] [Google Scholar]
- Patuzzi R. Ion flow in stria vascularis and the production and regulation of cochlear endolymph and the endolymphatic potential. Hear Res. 2011;277(1-2):4–19. doi: 10.1016/j.heares.2011.01.010. [DOI] [PubMed] [Google Scholar]
- Hernandez VH, Bortolozzi M, Pertegato V. et al. Unitary permeability of gap junction channels to second messengers measured by FRET microscopy. Nat Methods. 2007;4(4):353–358. doi: 10.1038/nmeth1031. [DOI] [PubMed] [Google Scholar]
- Gossman DG, Zhao HB. Hemichannel-mediated inositol 1,4,5-trisphosphate (IP3) release in the cochlea: a novel mechanism of IP3 intercellular signaling. Cell Commun Adhes. 2008;15(4):305–315. doi: 10.1080/15419060802357217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao HB. Connexin26 is responsible for anionic molecule permeability in the cochlea for intercellular signalling and metabolic communications. Eur J Neurosci. 2005;21(7):1859–1868. doi: 10.1111/j.1460-9568.2005.04031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzone R, Veronesi V, Gomes D. et al. Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS lett. 2003;533(1-3):79–88. doi: 10.1016/s0014-5793(02)03755-9. [DOI] [PubMed] [Google Scholar]
- Gerido DA, DeRosa AM, Richard G. et al. Aberrant hemichannel properties of Cx26 mutations causing skin disease and deafness. Am J Physiol Cell Physiol. 2007;293(1):C337–C345. doi: 10.1152/ajpcell.00626.2006. [DOI] [PubMed] [Google Scholar]
- Stong BC, Chang Q, Ahmad S. et al. A novel mechanism for connexin 26 mutation linked deafness: cell death caused by leaky gap junction hemichannels. The Laryngoscope. 2006;116(12):2205–2210. doi: 10.1097/01.mlg.0000241944.77192.d2. [DOI] [PubMed] [Google Scholar]
- Sanchez HA, Mese G, Srinivas M. et al. Differentially altered Ca2+ regulation and Ca2+ permeability in Cx26 hemichannels formed by the A40V and G45E mutations that cause keratitis ichthyosis deafness syndrome. J Gen Physiol. 2010;136(1):47–62. doi: 10.1085/jgp.201010433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrinoni A, Codispoti A, Serra V. et al. Connexin 26 (GJB2) mutations, causing KID Syndrome, are associated with cell death due to calcium gating deregulation. Biochem Biophys Res Commun. 2010;394(4):909–914. doi: 10.1016/j.bbrc.2010.03.073. [DOI] [PubMed] [Google Scholar]
- Lopez W, Gonzalez J, Liu Y. et al. Insights on the mechanisms of Ca(2+) regulation of connexin26 hemichannels revealed by human pathogenic mutations (D50N/Y). J Gen Physiol. 2013;142(1):23–35. doi: 10.1085/jgp.201210893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez HA, Villone K, Srinivas M. et al. The D50N mutation and syndromic deafness: altered Cx26 hemichannel properties caused by effects on the pore and intersubunit interactions. J Gen Physiol. 2013;142(1):3–22. doi: 10.1085/jgp.201310962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Suzumura A. Gap junctions and hemichannels composed of connexins: potential therapeutic targets for neurodegenerative diseases. Front Cell Neurosci. 2014;8:189. doi: 10.3389/fncel.2014.00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch M, Kielian T. Hemichannels in neurodegenerative diseases: is there a link to pathology? Front Cell Neurosci. 2014;8:242. doi: 10.3389/fncel.2014.00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, O'Carroll SJ, Henare K. et al. Connexin hemichannel induced vascular leak suggests a new paradigm for cancer therapy. FEBS lett. 2014;588(8):1365–1371. doi: 10.1016/j.febslet.2014.02.003. [DOI] [PubMed] [Google Scholar]
- Li Z, Yan Y, Powers EA. et al. Identification of gap junction blockers using automated fluorescence microscopy imaging. J Biomol Screen. 2003;8(5):489–499. doi: 10.1177/1087057103257309. [DOI] [PubMed] [Google Scholar]
- Haq N, Grose D, Ward E. et al. A high-throughput assay for connexin 43 (Cx43, GJA1) gap junctions using codon-optimized aequorin. Assay Drug Dev Tech. 2013;11(2):93–100. doi: 10.1089/adt.2012.469. [DOI] [PubMed] [Google Scholar]
- Lee JY, Choi EJ, v J. A new high-throughput screening-compatible gap junctional intercellular communication assay. BMC Biotech. 2015;15:90. doi: 10.1186/s12896-015-0211-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima A, Doi T, Mitsuoka K. et al. Roles of Met-34, Cys-64, and Arg-75 in the assembly of human connexin 26. Implication for key amino acid residues for channel formation and function. J Biol Chem. 2003;278(3):1807–1816. doi: 10.1074/jbc.M207713200. [DOI] [PubMed] [Google Scholar]
- Ambrosi C, Boassa D, Pranskevich J. et al. Analysis of four connexin26 mutant gap junctions and hemichannels reveals variations in hexamer stability. Biophys J. 2010;98(9):1809–1819. doi: 10.1016/j.bpj.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer KA. The gap junction proteins beta 1-connexin (connexin-32) and beta 2-connexin (connexin-26) can form heteromeric hemichannels. J Biol Chem. 1995;270(12):6768–6772. [PubMed] [Google Scholar]
- Fiori MC, Figueroa V, Zoghbi ME. et al. Permeation of calcium through purified connexin 26 hemichannels. J Biol Chem. 2012;287(48):40826–40834. doi: 10.1074/jbc.M112.383281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X, Lee SC, Reuss L. et al. Change in permeant size selectivity by phosphorylation of connexin 43 gap-junctional hemichannels by PKC. Proc Natl Acad Sci USA. 2007;104(12):4919–4924. doi: 10.1073/pnas.0603154104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiori MC, Krishnan S, Cortes DM. et al. Functional hemichannels formed by human connexin 26 expressed in bacteria. Biosci Rep. 2015;35(2) doi: 10.1042/BSR20140089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpe S, Bakker EP. Requirement of a large K+-uptake capacity and of extracytoplasmic protease activity for protamine resistance of Escherichia coli. Arch Microbiol. 1997;167(2/3):126–136. [PubMed] [Google Scholar]
- Buurman ET, McLaggan D, Naprstek J. et al. Multiple paths for nonphysiological transport of K+ in Escherichia coli. J Bacteriol. 2004;186(13):4238–4245. doi: 10.1128/JB.186.13.4238-4245.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmer J, Zeilinger C. MjK1, a K+ channel from M. jannaschii, mediates K+ uptake and K+ sensitivity in E. coli. FEBS lett. 2003;547(1-3):165–169. doi: 10.1016/s0014-5793(03)00706-3. [DOI] [PubMed] [Google Scholar]
- Ptak CP, Cuello LG, Perozo E. Electrostatic interaction of a K+ channel RCK domain with charged membrane surfaces. Biochemistry. 2005;44(1):62–71. doi: 10.1021/bi048390f. [DOI] [PubMed] [Google Scholar]
- Krishnan S FM, Whisenant TE, Cortes DM. et al. An E. coli-based assay to assess the function of recombinant human hemichannels. J Biomol Screen. 2016 doi: 10.1177/1087057116675321. [DOI] [PubMed] [Google Scholar]
- Verselis VK, Srinivas M. Connexin channel modulators and their mechanisms of action. Neuropharmacology. 2013;75:517–524. doi: 10.1016/j.neuropharm.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai D, del Corsso C, Srinivas M. et al. Block of specific gap junction channel subtypes by 2-aminoethoxydiphenyl borate (2-APB). J Pharmacol Exp Ther. 2006;319(3):1452–1458. doi: 10.1124/jpet.106.112045. [DOI] [PubMed] [Google Scholar]
- Burt JM, Spray DC. Volatile anesthetics block intercellular communication between neonatal rat myocardial cells. Circ Res. 1989;65(3):829–837. doi: 10.1161/01.res.65.3.829. [DOI] [PubMed] [Google Scholar]
- Figueroa VA, Retamal MA, Cea LA. et al. Extracellular gentamicin reduces the activity of connexin hemichannels and interferes with purinergic Ca(2+) signaling in HeLa cells. Front Cell Neurosci. 2014;8:265. doi: 10.3389/fncel.2014.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasciani I, Temperan A, Perez-Atencio LF. et al. Regulation of connexin hemichannel activity by membrane potential and the extracellular calcium in health and disease. Neuropharmacology. 2013;75:479–490. doi: 10.1016/j.neuropharm.2013.03.040. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]