

Abstract
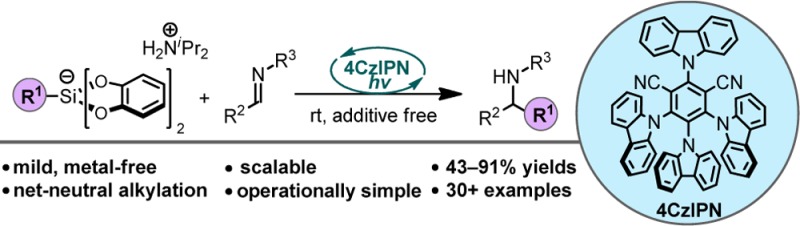
An operationally simple, mild, redox-neutral method for the photoredox alkylation of imines is reported. Utilizing an inexpensive organic photoredox catalyst, alkyl radicals are readily generated from the single-electron oxidation of ammonium alkyl bis(catecholato)silicates and are subsequently engaged in a C–C bond-forming reaction with imines. The process is highly selective, metal-free, and does not require a large excess of the alkylating reagent or the use of acidic additives.
Keywords: visible light, photocatalysis, imines, hypervalent silicon, radical alkylation
The resurgence of visible-light photoredox catalysis over the past decade has resulted in an influx of synthetically valuable methods for the construction of compounds whose synthesis would be challenging using two-electron reaction manifolds.1 Photoredox catalysts, by precisely controlling the rate of radical generation, facilitate selective reactions that would be extremely difficult using stoichiometric reagents where reactive radical species are present in noncatalytic quantities. These photoredox-mediated processes contrast with radical chain processes where unproductive redox quenching and undesired side reactions are often unavoidable. As a result, photoredox-mediated single electron transfer (SET) processes have led to a revival of radical-based chemistry and are already being adopted for the construction of complex molecules where traditional radical approaches are incompatible.1
Recently, our group and others have developed powerful methods for generating and engaging Csp3-centered radicals in a dual-catalytic cross-coupling system.2 The identification of benchtop-stable precursors from which radicals can be photocatalytically generated via SET oxidation or reduction is an underlying hurdle that must be overcome in the development of this type of catalysis. In the course of our investigations, our group and others have reported that alkyl bis(catecholato)silicates readily undergo SET oxidation under photoredox conditions and are viable radical precursors in photoredox/nickel dual catalysis.3 Ultimately, it has been shown that alkyl bis(catecholato)silicates serve as progenitors for both primary and secondary alkyl radicals that can be engaged in numerous processes including: (1) Csp2–Csp3 cross-couplings with aryl halides/(pseudo)halides and alkenyl halides,3,4 (2) the functionalization of azaborine isosteres,5 and (3) the synthesis of thioethers via H atom transfer.6 When compared with other radical precursors, alkyl bis(catecholato)silicates pose a number of advantages, specifically the ability to undergo homolysis under neutral conditions as well as the ability to be oxidized at relatively low and uniform oxidation potentials (Eox = 0.34 to 0.90 V).3 Despite having been discovered in 1964, these hypervalent silicate species have few known synthetic applications.7 Thus, in light of this deficiency and their numerous advantages as radical precursors, we initiated a program to explore their utility in radical-mediated processes.
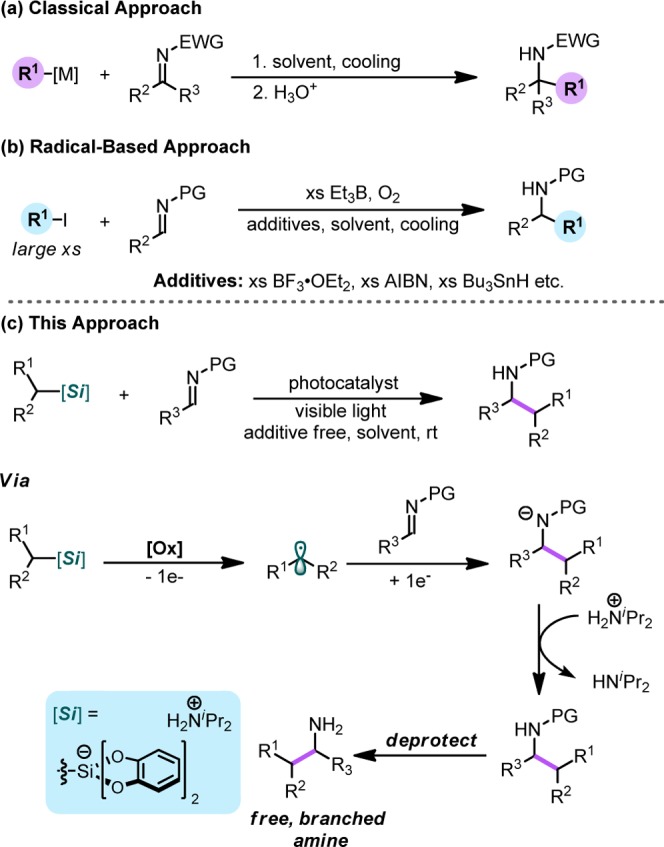
In the context of this program, we became interested in the possibility of using hypervalent silicon species to effect redox-neutral alkylation processes, such as the alkylation of imines. Imines are widely used as building blocks for preparing complex amine-containing, biologically active compounds and heterocyclic species.8 Classical strategies rely on preformed organometallic reagents to achieve efficient alkylation (Scheme 1a), but more recently, radical-based methods have emerged as versatile, functional-group-tolerant approaches for accessing complex amines.8,9 Classical radical-based methods generate radicals using a Et3B/O2 system or reductants (tin reagents or Zn).9 The source of alkyl radical is typically alkyl iodides (Scheme 1b).9 Photoredox catalysis has been instrumental in furthering this radical-based approach by enabling regulated radical generation under milder conditions.10 Recent reports in this vein have accomplished the radical alkylation process elegantly and even with some degree of stereocontrol.10a Although these methods furnish attractive diamines or amino alcohols, the focus of these strategies was on the installation of α-heteroatom-stabilized radicals.10a Thus, these previous methods do not provide a general solution to the radical alkylation of imines. We envisioned that alkyl bis(catecholato)silicates with alkylammonium counterions would be uniquely poised to fill this methodological gap because of their capacity to serve as efficient progenitors for a variety of radicals (1°, 2°, and benzylic) under mild, photocatalytic conditions. The radicals generated from these hypervalent silicon species can be coupled with imines, and the resulting nitrogen anion can then undergo protonation by the acidic ammonium cation to afford the desired alkylated product (Scheme 1). Moreover, unlike prior reports, which utilize expensive organometallic photocatalysts,10 organic photocatalysts such as 4CzIPN, 1, were anticipated to facilitate the SET events necessary for this process to occur.11
Scheme 1. Comparison of the Envisioned Photoredox-Mediated Radical Addition to Imines by Alkylsilicate Radical Precursors vs Standard Alkylation Approaches: (a) via Traditional Organometallic Reagents; (b) via Stoichiometric Generation of Radicals; (c) Alternate Photoredox-Based Approach Using Alkylsilicates.
On the basis of reported successes of N-sulfonyl aldimines as radical acceptors10a and our own experiences with these hypervalent silicon species as effective oxidative radical precursors, we attempted photoredox-mediated alkylation using the p-tolyl imine 2a and cyclohexylsilicate 3a with the organometallic photocatalyst Ru(bpy)3(PF6)2 in DMF. Although alkylation was observed, the reaction generally proceeded slowly, thus requiring higher photocatalyst loadings, longer reaction times, and/or photocatalysts with longer excited-state lifetimes (i.e., [Ir{dFCF3ppy}2(bpy)]PF6).1 Solvent optimization studies revealed that DMSO was the ideal solvent for this transformation.12 In this solvent, numerous photocatalysts, including organic photocatalysts (Table 1, entries 1 and 6), efficiently facilitated alkylation. Moreover, the stoichiometry of 3 could be lowered to just 1.1 equiv, speaking to the efficiency of the described reaction, which starkly contrasts with the stoichiometric radical-based methods where a large excess of the radical precursor is needed.9 Control experiments confirmed that this reaction required both irradiation and a photocatalyst to occur (see Supporting Information for details). Ultimately, we elected to use 4CzIPN (Table 1, entry 1), because it was the most efficient as well as less costly and more sustainable compared to its organometallic counterparts. We later found that we could halve the photocatalyst loading and still effect complete alkylation, albeit with longer reaction times (24 h).
Table 1. Screening of Select Photocatalystsa.

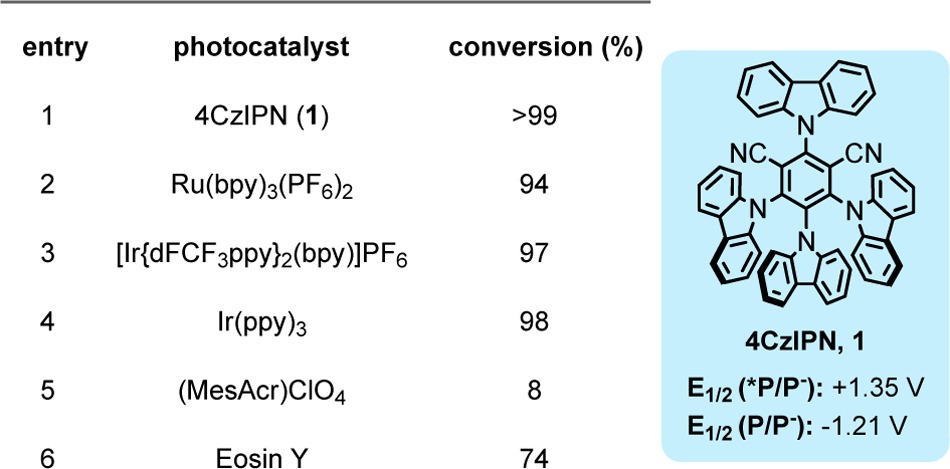
Conversion determined by GC/MS and/or HPLC analysis. The given potentials are vs the standard calomel electrode (SCE).
After determining suitable conditions, the scope of the alkylation process was next explored in the context of various N-sulfonyl aldimines using cyclohexylsilicate 3a (Table 2). N-Methyl-, phenyl-, p-toluenesulfonyl, and even N,N-dimethylsulfamidyl aldimines (2a–c, 2n) were all competent partners in this reaction, providing the desired products in good yields (4a–c, 4n). Alkylation of 2a could easily be scaled up (5 mmol, 10-fold increase) with neither yield nor reaction time being compromised. Modulation of the aryl ring with a variety of substituents was well-tolerated. Both substantially electron donating (2i–k) and electron-withdrawing (2f–h, 2l–n) groups provided products in good yields, regardless of the electronic nature of the aryl backbone. Of particular note, imine alkylation proceeded smoothly for a variety of halogen-substitutions, even for those that may be prone to radical SNAr-type reactions, such as difluoro imine 2g, thus providing a handle for further functionalization of the amine products (e.g., 4f, 4h, or 4n). In addition, these substrates and those containing electrophilic substituents (i.e., cyano- and ester-containing substrates 4l and 4m, respectively) would be incompatible with classical organometallic-based approaches. Although aryl N-sulfonyl imines were amenable to alkylation, attempts to use alkyl imines were not met with success (4s). Beyond N-sulfonyl-protected aryl imines, unprotected imines were also examined. Although N-alkyl imines gave no appreciable conversion, N-phenyl and N-pyridyl imines were competent toward alkylation (4o–4r). These systems were equally insensitive to electronic changes and underwent alkylation with ease. In addition, subsequent oxidation of secondary amine products was not observed. Unfortunately, this process could not be extended to ketimines (e.g., 4t).
Table 2. Evaluation of Imines Under Photoredox Conditions Using 3aa.
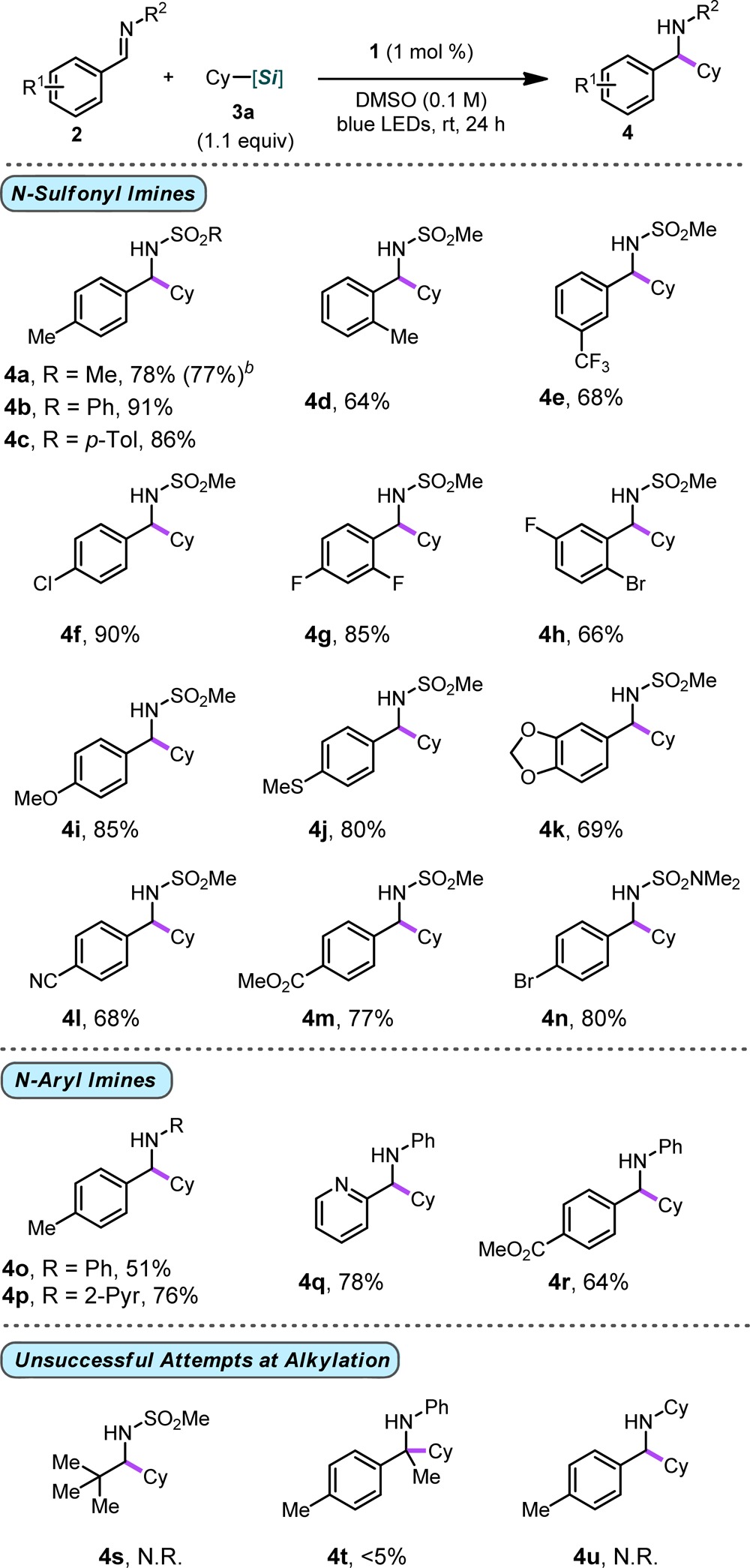
General reaction conditions: imine (1.0 equiv, 0.5 mmol), alkylsilicate (1.1 equiv), 4CzIPN (1 mol %), DMSO (0.1 M), rt, 24 h.
Yield in parentheses indicates yield on a 5 mmol scale.
The generality of this method was next explored in the context of various alkylsilicates (Table 3). Numerous alkyl groups could be installed on imines in varying electronic environments. We found that both classes of imines, N-aryl as well as N-sulfonyl, underwent alkylation in good yields. In these cases, the loading of the alkylsilicate and the photocatalyst needed to be raised to 1.3 equiv and 3 mol %, respectively.13 This is likely a consequence of off-cycle reactions consuming some of the alkylsilicate (i.e., homocoupling and H atom abstraction processes). Alkylsilicates that furnish stabilized (4v, 4w, and 4x) and nonstabilized (4y and 4z) primary radicals upon oxidation all performed well under the reaction conditions, providing the desired products in good yields. Additionally, protic groups as well as those that are prone to nucleophilic addition processes could be installed with ease using this approach (e.g., 4aa–4af). Importantly, this method retained its tolerance of various aryl substituents regardless of the alkylsilicate used.
Table 3. Photoredox Alkylation of Imines Using Various Alkylsilicatesa.
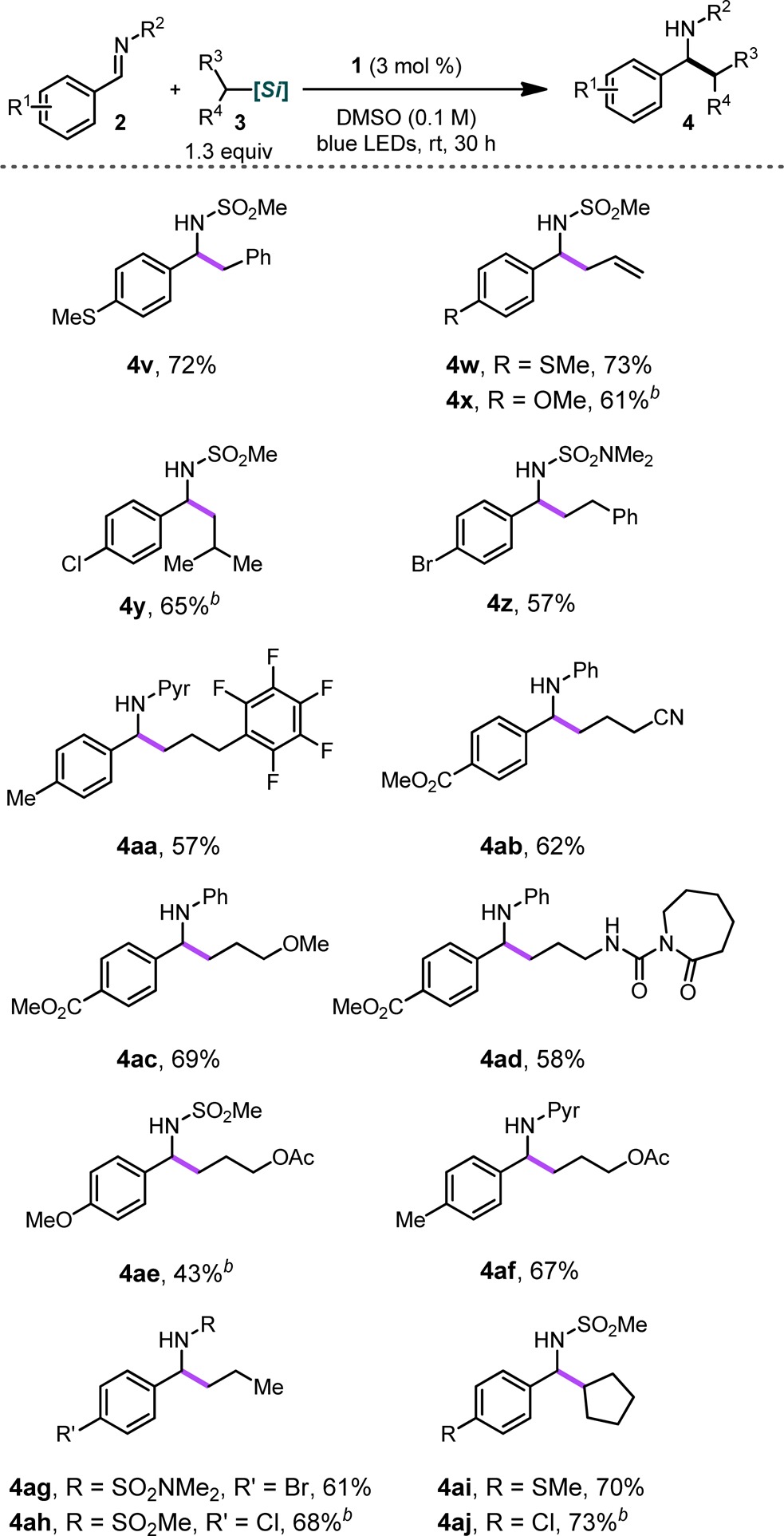
General reaction conditions: imine (1.0 equiv, 0.5 mmol), alkylsilicate (1.3 equiv), 4CzIPN (1 mol %), DMSO (0.1 M), rt, 24 h.
Reactions performed using 3 mol % Ru(bpy)3(PF6)2 instead of 4CzIPN.
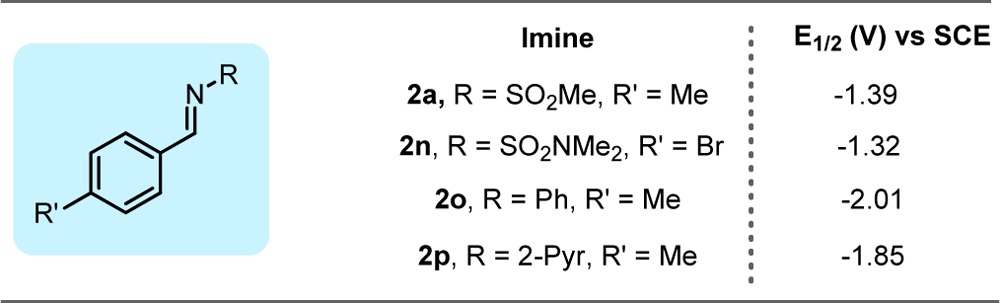
Mechanistically, we hypothesized that the described alkylation process can proceed through two plausible pathways: (a) SET reduction of the imine III to generate IVa, a possible “persistent” radical,14 followed by radical–radical coupling with II, generated from I upon SET oxidation (Figure 1a), or (b) addition of alkyl radical II to imine III, followed by reduction of the resulting N-centered radical IVb (Figure 1b). To probe the mechanism, electrochemical data were obtained for four classes of imines (Table 4) and compared to the redox potential of photocatalyst 1 (Table 1).11a On the basis of these data, Mechanism B is more probable for N-aryl imines, because of the unfavorable reduction of imine III to form IVa. Although this mechanism is also likely operative for N-sulfonyl imines, Mechanism A cannot be ruled out given the small difference between the reduced ground state of 4CzIPN and these imines (<0.2 V). Indeed, reduction of N-sulfonyl imines by photoredox catalysts to generate “persistent” radical anions has been suggested previously.10a
Figure 1.
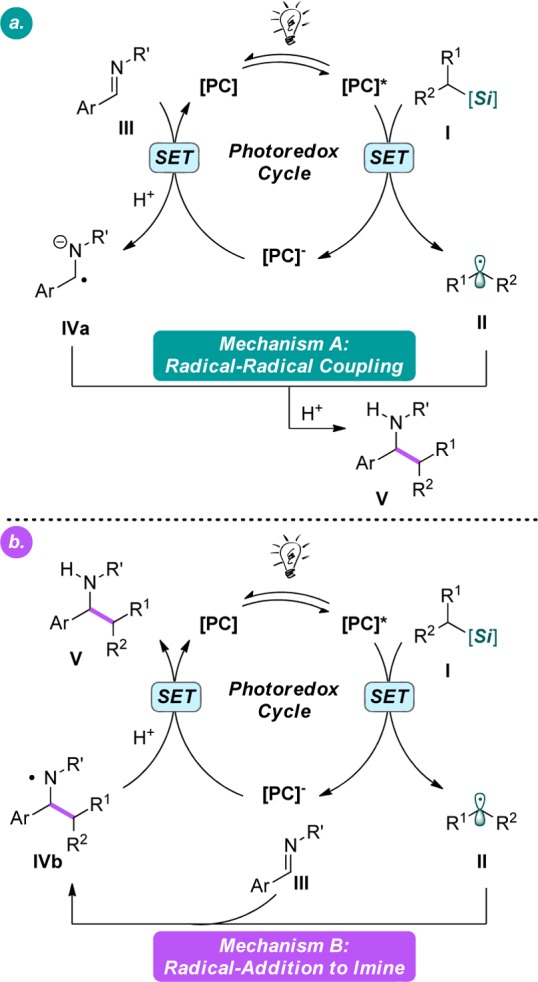
Postulated mechanistic pathways for imine alkylation.
Table 4. Measured E1/2 of Imines Using Cyclic Voltammetry.
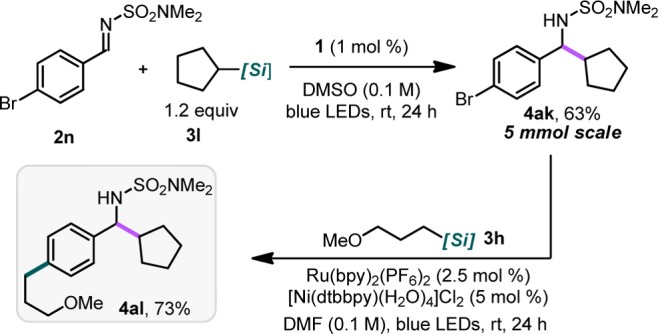
To demonstrate the broad utility of alkylsilicates for installation of Csp3 groups, a successive alkylation and cross-coupling sequence was executed using brominated imine 2n (Scheme 2). First, alkylation using imine 2n with 3l was performed on a 5 mmol scale to both reaffirm the scalability of the alkylation process and furnish 4ak. Subsequently the product (4ak) was subjected to the Ni/photoredox dual-catalytic conditions previously reported by our group,3a affording the elaborated cross-coupled product 4al in 73% yield.
Scheme 2. Successive Photoredox-Catalyzed Alkylation/Cross-Coupling Reactions Alkylsilicates.
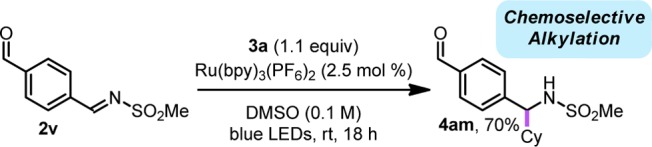
Finally, to probe the limits of the described method, selective alkylation of an aldehyde-containing imine was attempted. In this competition study, imine 2v underwent facile alkylation with complete chemoselectivity (Scheme 3). This result is in line with the enhanced electrophilic character of sulfonyl aldimines over their aldehyde congeners in DMSO.15 Thus, in this case, the radical is behaving in a similar manner to soft carbanions without the inherent sensitivity toward protic groups.
Scheme 3. Chemoselectivity in Photoredox Alkylation.
In summary, a simple, scalable, and efficient method for the alkylation of imines under photoredox conditions is described. The method avoids traditional, superstoichiometric radical initiators and utilizes an organic photoredox catalyst to mediate radical generation. A variety of protected and unprotected amines can be accessed via this alkylation strategy under mild, net-neutral conditions. This method is less restrictive in scope than many previously reported methods because it facilitates the installation of various alkyl groups, including those containing protic functional groups and those originating from nonstabilized radicals. Additionally, the resulting products can be further elaborated by employing the same radical progenitors used for the alkylation. Therefore, this synthetic method has the capacity to be combined with others to generate structural diversity rapidly.
Acknowledgments
We sincerely thank Mr. David Primer, Dr. Alvaro Gutierrez-Bonet, Mr. Geraint Davies, Dr. Simon Lang, Ms. Rebecca Wiles, and Dr. James Phelan of the University of Pennsylvania (UPenn) for useful discussions. We thank Mr. Xingpin Li (UPenn) for assistance in preparation of some alkylsilicates. We thank Dr. Rakesh Kohli (UPenn) and Dr. Charles W. Ross III (UPenn) for assistance in obtaining HRMS and Dr. Jun Gu (UPenn) for NMR advice.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.6b03665.
Experimental procedures and characterization data for all compounds (PDF)
The authors are grateful for the financial support provided by NIGMS (R01 GM113878). C.B.K. is grateful for an NIH NRSA postdoctoral fellowship (F32 GM117634).
The authors declare no competing financial interest.
Supplementary Material
References
- a Prier C. K.; Rankic D. A.; Macmillan D. W. C. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Narayanam J. M. R.; Stephenson C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. 10.1039/B913880N. [DOI] [PubMed] [Google Scholar]; c Romero N. A.; Nicewicz D. A. Chem. Rev. 2016, 116, 10075–10166. 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]; d Shaw M. H.; Twilton J.; Macmillan D. W. C. J. Org. Chem. 2016, 81, 6898–6926. 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For seminal reports, see:; a Tellis J. C.; Primer D. N.; Molander G. A. Science 2014, 345, 433–436. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zuo Z.; Ahneman D. T.; Chu L.; Terrett J. A.; Doyle A. G.; MacMillan D. W. C. Science 2014, 345, 437–440. 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]; For reviews, see:; c Tellis J. C.; Kelly C. B.; Primer D. N.; Jouffroy M.; Patel N. R.; Molander G. A. Acc. Chem. Res. 2016, 49, 1429–1439. 10.1021/acs.accounts.6b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Skubi K. L.; Blum T. R.; Yoon T. P. Chem. Rev. 2016, 116, 10035–10074. 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Gui Y.-Y.; Sun L.; Lu Z.-P.; Yu D.-G. Org. Chem. Front. 2016, 3, 522–526. 10.1039/C5QO00437C. [DOI] [Google Scholar]
- a Jouffroy M.; Primer D. N.; Molander G. A. J. Am. Chem. Soc. 2016, 138, 475–478. 10.1021/jacs.5b10963. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Corce V.; Chamoreau L.-M.; Derat E.; Goddard J.-P.; Ollivier C.; Fensterbank L. Angew. Chem., Int. Ed. 2015, 54, 11414–11418. 10.1002/anie.201504963. [DOI] [PubMed] [Google Scholar]
- a Patel N. R.; Kelly C. B.; Jouffroy M.; Molander G. A. Org. Lett. 2016, 18, 764–767. 10.1021/acs.orglett.6b00024. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lévêque C.; Chenneberg L.; Corcé V.; Goddard J.-P.; Ollivier C.; Fensterbank L. Org. Chem. Front. 2016, 3, 462–465. 10.1039/C6QO00014B. [DOI] [Google Scholar]
- Jouffroy M.; Davies G. H. M.; Molander G. A. Org. Lett. 2016, 18, 1606–1609. 10.1021/acs.orglett.6b00466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouffroy M.; Kelly C. B.; Molander G. A. Org. Lett. 2016, 18, 876–879. 10.1021/acs.orglett.6b00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Frye C. L. J. Am. Chem. Soc. 1964, 86, 3170–3171. 10.1021/ja01069a054. [DOI] [Google Scholar]; b Seganish W. M.; DeShong P. J. Org. Chem. 2004, 69, 1137–1143. 10.1021/jo035309q. [DOI] [PubMed] [Google Scholar]; c Matsuoka D.; Nishigaichi Y. Chem. Lett. 2015, 44, 163–165. 10.1246/cl.140940. [DOI] [Google Scholar]
- For reviews, see:; a Ferraris D. Tetrahedron 2007, 63, 9581–9597. 10.1016/j.tet.2007.06.043. [DOI] [Google Scholar]; b Martin S. Pure Appl. Chem. 2009, 81, 195–204. 10.1351/PAC-CON-08-07-03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For recent radical addition of imines using radicals generated from superstoichiometric radical precursors and Et3B, see:; a Ueda M.; Miyabe H.; Miyata O.; Naito T. Tetrahedron 2009, 65, 1321–1326. 10.1016/j.tet.2008.12.031. [DOI] [Google Scholar]; b Lee S.; Kim S. Tetrahedron Lett. 2009, 50, 3345–3348. 10.1016/j.tetlet.2009.02.136. [DOI] [Google Scholar]; c Miyabe H.; Ueda M.; Naito T. Chem. Commun. 2000, 2059–2060. 10.1039/b006574i. [DOI] [Google Scholar]; d Fernández-Salas J. A.; Rodríguez-Fernández M. M.; Maestro M. C.; García-Ruano J. L. Eur. J. Org. Chem. 2014, 2014, 5265–5272. 10.1002/ejoc.201402355. [DOI] [Google Scholar]; e Fujii S.; Konishi T.; Matsumoto Y.; Yamaoka Y.; Takasu K.; Yamada K. J. Org. Chem. 2014, 79, 8128–8133. 10.1021/jo501332j. [DOI] [PubMed] [Google Scholar]; f Yamada K.; Konishi T.; Nakano M.; Fujii S.; Cadou R.; Yamamoto Y.; Tomioka K. J. Org. Chem. 2012, 77, 1547–1553. 10.1021/jo2025042. [DOI] [PubMed] [Google Scholar]; g Fernández-Salas J. A.; Maestro M. C.; Rodríguez-Fernández M. M.; García-Ruano J. L.; Alonso I. Org. Lett. 2013, 15, 1658–1661. 10.1021/ol400439g. [DOI] [PubMed] [Google Scholar]; Miscellaneous methods:; h Friestad G. K.; Ji A.; Baltrusaitis J.; Korapala C. S.; Qin J. J. Org. Chem. 2012, 77, 3159–3180. 10.1021/jo2026349. [DOI] [PubMed] [Google Scholar]; For reviews on radical additions to imines see:; i Friestad G. K. Tetrahedron 2001, 57, 5461–5496. 10.1016/S0040-4020(01)00384-2. [DOI] [Google Scholar]; j Miyabe H.; Ueda M.; Naito T. Synlett 2004, 1140–1157. 10.1055/s-2004-822889. [DOI] [Google Scholar]
- a Uraguchi D.; Kinoshita N.; Kizu T.; Ooi T. J. Am. Chem. Soc. 2015, 137, 13768–13771. 10.1021/jacs.5b09329. [DOI] [PubMed] [Google Scholar]; b Fava E.; Millet A.; Nakajima M.; Loescher S.; Rueping M. Angew. Chem., Int. Ed. 2016, 55, 6776–6779. 10.1002/anie.201511235. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hsieh S.-Y.; Bode J. W. Org. Lett. 2016, 18, 2098–2101. 10.1021/acs.orglett.6b00722. [DOI] [PubMed] [Google Scholar]
- a Luo J.; Zhang J. ACS Catal. 2016, 6, 873–877. 10.1021/acscatal.5b02204. [DOI] [Google Scholar]; b Gutierrez-Bonet A.; Tellis J. C.; Matsui J. K.; Vara B. A.; Molander G. A. ACS Catal. 2016, 6, 8004–8008. 10.1021/acscatal.6b02786. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lévêque C.; Chenneberg L.; Corcé V.; Ollivier C.; Fensterbank L. Chem. Commun. 2016, 52, 9877–9880. 10.1039/C6CC04636C. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for further details on optimization.
- In certain cases, Ru(bpy)3(PF6)2 was used to demonstrate both the versatility of this alkylation protocol and simplify purification. At higher catalyst loadings, 4CzIPN can be difficult to separate from some of the alkylated products.
- a Studer A. Chem. - Eur. J. 2001, 7, 1159–1164. . [DOI] [PubMed] [Google Scholar]; b Fischer H. Chem. Rev. 2001, 101, 3581–3610. 10.1021/cr990124y. [DOI] [PubMed] [Google Scholar]; c Studer A. Chem. Soc. Rev. 2004, 033, 267–273. 10.1039/b307652k. [DOI] [PubMed] [Google Scholar]
- Appel R.; Mayr H. J. Am. Chem. Soc. 2011, 133, 8240–8251. 10.1021/ja200820m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.