

Abstract
Background/Aim: Proteomics based on high-resolution mass spectrometry (MS) is the tool of choice for the analysis of protein presence, modifications and interactions, with increasing emphasis on the examination of tumor tissues. Application of MS-based proteomics offers a detailed picture of tumor tissue characteristics, facilitating the appreciation of different tumor entities, whilst providing reliable and fast results for therapeutic marker targeting and prognostic factor assessment. Through use of the high analytical resolution of nano-high-pressure liquid chromatography (nanoHPLC) and the high resolution of an Orbitrap Elite mass spectrometer, the present study aimed to provide knowledge on the proteome of the generally unknown entity of pediatric ependymal tumors. Materials and Methods: Ten resected specimens of childhood ependymoma were analyzed through a one-dimensional (1D) nanoLC-MS/MS approach. Method optimization steps were undertaken for both the sample preparation/protein extraction procedure and LC parameters, aiming to achieve the highest possible identification rates. Results: Following method optimization, each nanoLC-MS/MS run resulted in identification of more than 5,000 proteins and more than 25,000 peptides for every analyzed sample, thus detailing the greater part of the ependymoma proteome. Identified proteins were found to spread throughout all known tumor categories regarding their molecular function and subcellular localization. Conclusion: Through the proposed nanoLC-MS/MS method herein we report, for the firs time, the ependymoma proteome database. A large number of similarities regarding proteome content are revealed compared to other two pediatric brain tumor entities; astrocytomas and medulloblastomas. Furthermore, through our approach, the majority of currently proposed markers for ependymoma (e.g. nucleolin, nestin, Ki67 and laminin subunit A2 ) as well as all major key players of the phosphoinositide 3-kinase pathway (seemingly implicated in ependymoma), were definitely detected.
Keywords: Ependymoma, pediatric brain tumors, central nervous system tumors, nanoLC-MS/MS, proteomics, method development
Pediatric brain tumors are a leading cause of cancer-related death in children (1). The application of high-throughput technologies (e.g. proteomics, and genomics) to the analysis of these tumor types has generated an abundance of molecular information, while in parallel, adequately continuing to provide knowledge on both the biological and clinical aspects of this devastating disease affecting children (2).
Ependymoma, the third most common tumor in children, is thought to arise from ependymal cells in the wall of the cerebral ventricles or the spinal canal and therefore occurs most frequently in the posterior fossa or the spinal cord (3,4). A variety of different sub-types of ependymomas have been identified, while the anaplastic variant seems to have the worse prognosis (5). Surgery remains the mainstay of treatment for ependymomas, while patients with posterior fossa ependymomas who have tumors amenable to gross total resection and are subsequently treated with radiotherapy, have a 70% or greater likelihood of long-term survival (6).
Due to the heterogeneity of the disease, its biological characteristics remain largely unknown and prognostic factors are basically based on clinical and histological criteria (i.e. age, extent of tumor resection, and histological grade). Therefore, biological, both genetic and proteomic, alterations that could be used to further characterize these tumors as well as identifying molecules that can be used as targets for therapy, need to be discovered.
Proteins, being the major conductors of genetic information and the molecules that can better reflect the functional status of the cell, are key targets in central nervous system (CNS) cancer research (7). This is why the elucidation of protein expression and their modifications is crucial in brain cancer biology, also aiding in discovery of predictors of cancer risk, detection of biomarkers for early diagnosis and identification of therapeutic targets (8). Proteomics, working together with genomics, may be able to redefine current ependymoma classifications and management protocols (9).
In our previous work, we reported on protein/proteomic signatures of pediatric astrocytomas and pediatric medulloblastomas, having had the opportunity to unravel parts of the molecular signature of these two distinct malignancy types, based on experiments utilizing two-dimensional gel-based protein separation and protein quantitation via image analysis and western blotting. It must, however, be underlined that the two-dimensional gel-based approach is biased to identify relatively high-abundant proteins and may miss on low-abundance regulatory molecules, including signal transducers, membrane proteins etc. Currently, gel-free approaches, namely liquid chromatography-tandem mass spectrometry (LC-MS/MS), are being widely used for clinical tissue and cell-line analyses (10).
The aim of the current study was two-fold: (i) to develop a sensitive and reproducible LC-MS/MS method for full in-depth proteomic screening of brain tumor tissues, and (ii) to build a high-confidence, in-depth proteome map from clinical cancer tissue specimens of ependymal origin, offering a first step towards their targeted protein validation in the clinical setting by the anticancer research community.
Materials and Methods
Tissue specimens. All samples were obtained from patients who underwent tumor resection surgery at Aghia Sophia Children’s Hospital, Athens, Greece. All procedures were in accordance with approved human subject guidelines, and were approved by the Ethical Committee of the Athenian University. After informed consent was obtained from the patients’ parents, samples were collected at initial diagnosis with no prior exposure to chemotherapy or radiation therapy.
All specimen analyzed in the study belonged to the cellular (WHO grade II) ependymoma subtype and all affected individuals were Caucasian (Table I). Resected tumors were stained with hematoxylin and eosin and examined by light microscopy.
Table I. Patient characteristics.

sCrea: Serum creatinine; T-bil: total bilirubin; ECOG PS: Eastern Coopeative Oncology Group Performance Status.
Sample preparation. Prior to analyses, all tissue samples were washed in in a sucrose buffer (HEPES, pH 7.5, 320 mM sucrose, 1 mM EDTA, 5 mM dithioerythreitol, 1 mg/ml protease inhibitors) for removal of excess blood from tissue. Next, tissues were powderized through grinding in liquid nitrogen. Further homogenization for disruption of all remaining intact protein structures was performed by tip-sonication for three cycles, 18 seach, under 38% amplification. The lysis buffer used was 7 M urea, 1.5 M Tris-HCl, 0.1 M sodium dodecyl sulfate. The homogenate was left at room temperature for 1 h and centrifuged at 12,700 × g for 30 min. De-salting was performed with Ultrafree-4 centrifugal filter unit (Millipore, Billerica, MA, USA). The protein content of the supernatant was determined using the Bradford quantification method. Protein extraction was sequentially performed by addition of 150 μl of extraction buffer to the sample solution. Finally, 150 μg of protein was further processed for peptide generation.
Peptide generation. Proteins were reduced, in solution, by incubation with 0.1 mM DTE in Tris-HCl pH 6.8 for 30 min at 36˚C. Proteins were then alkylated by the addition of 0.05 mM iodoacetamide for 30 min at room temperature in the dark. A trypsin solution (Roche, Hoffman-La-Roche, Basel, Switzerland) (final concentration 500 ng/μl) was added to the sample at a trypsin to protein ratio of 1:100, mixed by pipetting and digestion took place overnight at 36˚C. The following day, trypsinization was terminated by the addition of 5% acetic acid (vol/vol). Finally, peptide-containing solutions were vacuum-dried for 1 h (until complete dryness). Powder was re-constituted in 100 μl of water with 0.1% formic acid. After cleaning for impurities by filtering through a Millex® syringe-driven filter unit, nanoLC-MS/MS analysis followed.
One-dimensional nano-liquid chromatography (1D-nanoLC) separation. Peptides generated in the previous step were separated in an Ultimate3000 system nanoLC system (Dionex; Thermo Scientific, Bremen, Germany). Peptides were loaded onto a C-18 pre-column (100 μm inner diameter ×2 cm; 100 Å, 3-μm-bead-packed, Acclaim PepMap 100; Thermo Scientific) at 10 μl/min in 99.9% water with 0.1% formic acid. After 6 min of desalting, the pre-column was switched online with the analytical C-18 column (75 μm ×50 cm; 100 Å, 2-μm-bead-packed Acclaim PepMap RSLC; Thermo Scientific) that was equilibrated with mobile phase A (99.9% water with 0.1% formic acid). Elution time for all runs was 360 min for under a non-linear gradient of mobile phase B (99.9% acetonitrile with 0.1% formic acid) (Table II) at a constant 300 nl/min flow rate.
Table II. Nano-high-pressure liquid chromatography gradient steps followed during the analysis.
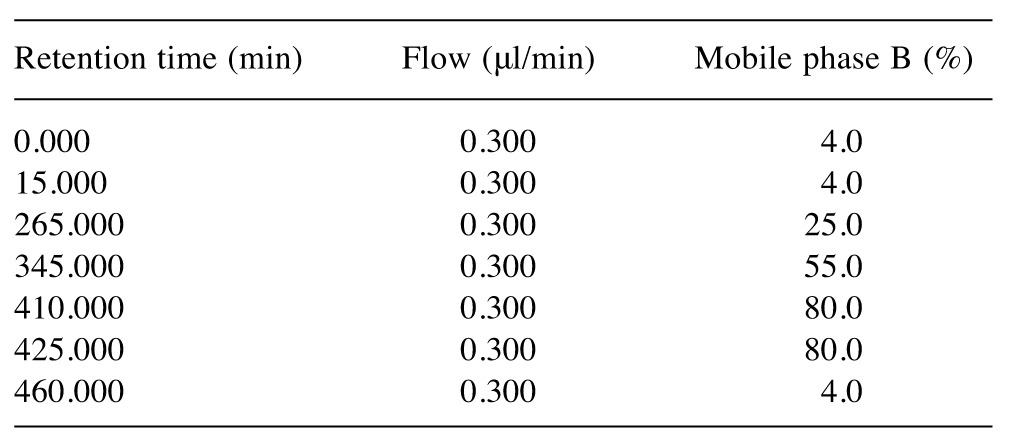
Mobile phase A: 99.9% acetonitrile, 0.1% H2O.
Orbitrap mass spectrometry. Mass spectra of eluted peptides were collected in an Orbitrap Elite mass spectrometer (Thermo Scientific) fitted with a nano-spray source, practically as previously described by our group (11,12). The instrument was run in a data-dependent acquisition mode with the XCalibur™ v.2.2 SP1.48 software (Themo Scientific). Full-scan data were acquired on the 300-2,000 m/z range under 60,000 resolution and a maximum injection time of 100 ms. Data-dependent tandem MS for the 20 most intense ions was performed with higher-energy collision dissociation fragmentation in the Orbitrap at a resolving power of 15,000 and a collision energy of 36. Resulting fragments were analyzed on the Orbitrap; MS/MS spectra were acquired with 15,000 resolving power and a maximum injection time of 120 ms. Measurements were performed using m/z 445.120025 as lock mass.
Data analysis. Raw data (each file consisting of an average of 72,000 spectra) were processed in Proteome Discoverer (version 1.4.0.388; Thermo Scientific), and searches were performed as described previously (11). Raw data were analyzed on the Homo sapiens UniProtKB/Swiss-Prot database (20,200 protein entries, reviewed 28-1-2016) using SequestHT under the following parameters: two maximum missed cleavages for trypsin; oxidation of methionine as variable modification; 10 ppm peptide mass tolerance; and 0.05 ppm fragment ion tolerance. Peptide spectral matches were validated using percolator based on q-values at 1% false discovery rate. Additional peptide filtering was performed based on Xcorr versus peptide charge values (percolator maximum Delta Cn was set at 0.05). Values of 2.2 for doubly-charged and 3.5 for triply-charged peptides were used. The minimum length of acceptable identified peptides was set as 6 amino acids.
Pathway analysis, functional clustering and classification. The Proteome Discoverer software was used to retrieve annotation information for identified proteins. Proteins were assigned their Gene Ontology (GO) terms and protein classification was performed based on these GO terms for biological process and subcellular localization. All identified proteins were analyzed; when more than one assignment was available, all the functional annotations were considered in the final results list. Theoretical molecular weight and pI were also used to classify proteins according to their physicochemical characteristics.
Functional analysis of the protein sets was performed using the Ingenuity Pathway Knowledge Base (Ingenuity Systems, Redwood City, CA, USA). Analysis resulted in identification of biological functions and diseases that were most significant to our protein data sets. Proteins showing highest expression in ependymoma samples (false discovery rate of 30%) were considered for the analysis. The focus genes from all protein lists were overlaid onto a global molecular network, developed from information contained in the Ingenuity Pathway Knowledge Base and this generated networks based on the connectivity of the individual proteins according to their relevance with the tissues under study.
Results
In the present study, we embarked on analyzing the protein expression patterns of pediatric ependymoma tissues. Ten selected resected specimens were subjected to 1D-nanoLC-MS/MS analysis and to strengthen the quality and reproducibility of protein profiling of each sample/tissue, analysis consisted of two technical replicates. The methodology followed in order to increase the number of identifications, and in parallel save on analysis time and quantity of material required, consisted of experimenting with several sample preparation procedures with regard to the protein extraction process, as well as testing in LC parameters.
Initially, tissues were prepared under two different approaches, the first consisting of tissue homogenization in liquid nitrogen followed by protein extraction in 0.1 M Tris-HCl, supplemented with 4% SDS and 0.1 M DTE (buffer A) and the second consisting of homogenization in Wheaton grind homogenizer followed by extraction in 8 M urea in ammonium bicarbonate (50 mM) (buffer B) (Figure 1). Better results with regard to total protein identification numbers were achieved when the procedure including dissemination in liquid nitrogen and buffer A was followed.
Figure 1. Deep proteome nano-high-performance liquid chromatography–tandem mass spectrometric (LC-MS/MS) analysis flowchart. Pediatric ependymoma resections were lysed under two different experimental approaches and proteins were extracted and digested by trypsin. Generated peptides were analyzed in Orbitrap Elite mass spectrometer. Method optimization steps to increase final protein identification rates were performed in sample lysis and LC parameters.

Protocol optimization with regard to LC parameters included testing with a three-step non-linear gradient (Table II) against a classical linear 2-35% mobile phase B gradient. Results indicated that an even distribution across the chromatographic run was achieved in the first, tailored method, compared to MS1 ions detected using linear gradients. Therefore, chromatographic profile design of the gradient offered a nearly constant number of unique peptides identified per minute of retention time. All optimized methods followed in the present study are described in detail in the Materials and Methods section.
After reaching a consensus with regard to optimizing the experimental procedure, all tissues were analyzed under the same protocol. The number of proteins identified in each tissue sample varied, with a mean of 4,000±150 protein groups and a mean of 25,000±550 peptides identified per sample. Only proteins (protein groups) identified/present in all samples were included in the final reference ependymoma protein database, consisting of 4,157 entries. An abstract of the final database showing protein coverage, unique peptides identified, peptide spectral matches, molecular weight and pI of identified proteins is given in Figure 2.
Figure 2. Final protein database of pediatric ependymoma. Only proteins (protein groups) identified/present in all samples (n=10) were included in the final reference ependymoma protein database, consisting of 4,157 entries. An Excel file form (an exemplary abstract is shown for reasons of convenience and comprehension), is presented including accession numbers of identified proteins (according to UniProt), name and description, sequence coverage, number of unique peptides, number of protein’s amino acid residues (AAs), protein molecular weight (MW) and protein isoelectric point (pI).

Furthermore, an investigation of the mRNA–miRNA interaction was carried out for each gene encoding the identified proteins. For this purpose, DIANA-TarBase v7.0, a database indexing more than half a million experimentally supported mRNA–miRNA interactions was used (13,14). DIANA-TarBase v7.0 enables users to easily identify positive or negative experimental results, the utilized experimental methodology, experimental conditions including cell/tissue type and treatment. As a result, all experimentally validated miRNA:gene interactions for each human gene encoding the identified proteins is demonstrated in Table III.
Table III. List of experimentally validated mRNA–miRNA interactions as obtained by DIANA-TarBase. The official names of the human genes encoding for every protein of Figure 2 are demonstrated in the first column, whereas all human microRNAs that are experimentally validated to interact with their mRNA transcripts are shown in the second column. The prediction score for each mRNA–miRNA interaction is presented in parentheses next to each microRNA.

Regarding GO terms, ependymoma proteins were grouped according to their biological process as well as localization. The majority of identified proteins (29%) are involved in cell metabolic processes, while a significant number (22.5%) are engaged in cell structure, 16.5% in localization procedures, 12% have biological regulatory roles, while others are connected to biogenesis (7%), development and stimulatory or immune responses (4%), or exert multiple functions (3%). Less frequently represented are proteins with biological adhesion (2%), proteins associated with apoptosis (2%) and cell reproduction (1%), as well as those associated with locomotion (1%) (Figure 3A).
Figure 3. Classification of ependymoma proteins according to their biological process (A) and subcellular localization (B). The distribution frequencies with regard to the specified categories within the given charts are indicated as a percentage of the total number of protein entries. Physicochemical characteristics were also utilized to further classify the ependymoma proteins according to their pI (C).
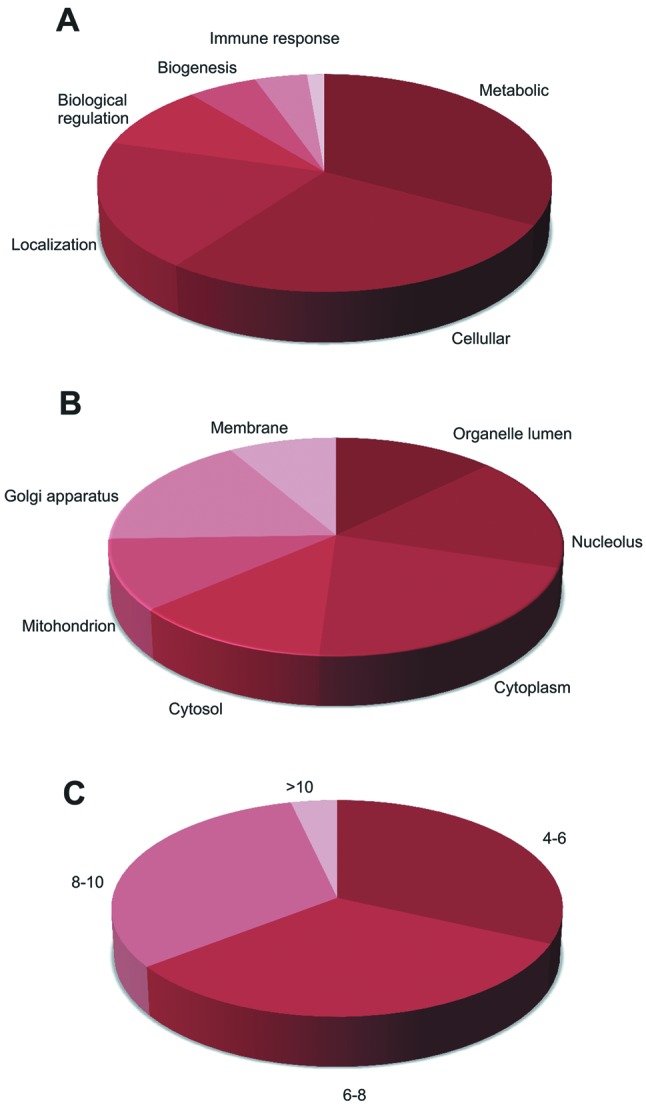
In terms of their cellular distribution, a large proportion of proteins were found to be localized in organelle lumen (12.54%) and a greater number of proteins (14.8%) were components of the nucleolus. The majority of pediatric ependymoma proteins were found to be cytoplasmic (18.66%), while identified proteins were also present in other subcellular compartments such as the cytosol (13%), the mitochondrion (10%) and golgi apparatus (14%), and 7% were membranous. Ten percent of identified proteins were extracellular, as well as comprising proteins of the endoplasmic reticulum (Figure 3B).
Ependymoma proteins were further clustered according to their biophysical characteristics; the assessed theoretical pI values predicted an even protein accumulation in both the acidic region (pI 4-7) and pI 8-10 region (Figure 3C).
The ependymoma protein database was further analyzed through the Ingenuity Pathway Knowledge Base. By this functional tool, protein biological functions and/ diseases that are most significant to the datasets are identified. The rank product list, representing proteins present in the ependymoma analyzed samples, generated the following top networks: (i) Cellular function and maintenance, post-translational modification, protein folding (Ingenuity Pathway analysis internal score, 60); (ii) dermatological diseases and conditions, inflammatory disease, cancer (score, 41); and (iii) cancer, hematological disease, reproductive system disease (score, 39). Cancer, dermatological diseases and gastrointestinal diseases are the main diseases connected to the list. The top canonical pathways associated with high significance are the 14-3-3-mediated signaling (p-value 3.25e-16) and the protein ubiquitination pathway (p-value 1.2e-11).
Several networks, implicating the analyzed molecules, showing protein–protein interactions, were generated by the software. A very interesting network implicating identified molecules significant in cell death of ependymal tissues is presented in Figure 4.
Figure 4. Signaling pathway network mined for proteins identified in pediatric ependymoma. Construction of the diagram was performed by the Ingenuity Pathway Analysis software, as described in the Materials and Methods section. Gray nodes indicate focus genes/proteins of ependymoma. Solid lines represent direct interactions between proteins. Direct interactions are defined as those where two proteins directly contact each other, with no intermediate step and usually comprise chemical modifications.

Discussion
In the present study, by employing a modified cutting-edge one-dimensional shotgun proteomic approach, we aimed to unveil for the first time the proteome of a pediatric brain tumor entity, generally unknown regarding its molecular-proteomic biological traits, pediatric ependymoma.
With regard to proteome optimization steps employed in the present study, aiming at reaching greater protein coverage without compromising on speed of analysis and sensitivity, we herein propose a universal sample preparation method for for nanoLC-MS/MS analysis of pediatric brain tissues, which can, under certain conditions, be applied to all experiments on brain tissue specimens, whether malignant or not. Optimization steps were assessed on two parameters: (i) sample preparation by two different extraction buffers, and (ii) LC gradient conditions.
Under the concentrations provided, the proposed lysis buffer (buffer A) in combination with tissue severance in liquid nitrogen was optimal in attaining maximal dissociation of protein complexes, with adequate protein extraction, as well as solubility of the extracted material.
Regarding experimentation on LC parameters, it is well known that for complex peptide mixtures, conventional linear gradients produce an unequal spread of peptides, and peptide distributions deviate considerably from uniformity, with larger numbers of peptides eluting in the middle of the run, and relatively few peptides eluting in the beginning and towards the end of the gradient time. In the present study, we propose a three-step non-linear gradient that seems to provide an even distribution of all eluted ions across the chromatographic run compared to MS1 ions produced using linear gradients.
Under a constant flow rate of 300 nl/min, the improved chromatographic parameters were set to begin at 4% acetonitrile increasing linearly to 25% over 250 min, then to 55% over 80 min, and finally to 80% over 30 min. We also experimented on elution time duration and reached the conclusion that a clear and linear correlation between the chromatographic peak capacity in peptide elution (15) was achieved at 360 min of total elution time.
By following the above experimental protocol modifications regarding protein extraction and chromatographic parameters, we developed an LC-MS/MS method able to deliver more than 5,000 protein entries belonging to 4,155 protein groups and 25,345 peptides, thus mapping in-depth for the first time the greater part of the proteome of this pediatric-type human brain malignancy.
Previously, we had undertaken steps in studying the other two major pediatric brain tumor malignancies: pediatric astrocytoma (16) and pediatric medulloblastoma (15), by means of proteomics technologies. The current series of analyzed samples sheds light on similarities in protein patterns throughout the panel of the three pediatric brain tumor malignancies. It is disappointing that we cannot yet discuss in-depth differences of protein content (in a quantitative manner) between the three types, since the proteomic technologies employed for astrocytomas and medulloblastomas far lacked the forward/analytical character of the nanoLC-MS/MS procedure described herein.
Regarding the biological traits of the material under study, all major components of the mitogen-activated protein kinase pathway that had been reported to be de-regulated in grade 1 tumors (astrocytomas) (Q13418, Q04759, P12931, P63104, Q04917, P27348, P28482) by us and others (16-18), are present in the final list of ependymomas, further confirming the intermediate character of this type of malignancy (5).
Furthermore, regarding expression of heat-shock molecules, similarities were found in heat-shock protein 90 (P08238), 60-kDa heat-shock protein (P10809) and 70-kDa heat-shock protein (P34932), which were found to be equally expressed in in low-grade gliomas (astrocytomas) and ependymomas, consistent with the general scheme that heat shock proteins are expressed as a response to various stress stimuli, including cancer. However, heat-shock protein 71-kDa (P11142), and heat-shock protein beta (P04792) and 90-alpha (P07900) were exclusively expressed in ependymomas.
Interestingly, both prohibitin (P35232), prohibitin 2 (Q99623), glial fibrillary acidic protein (GFAP; P14136) and vimentin (P08670) were among the proteins expressed in ependymomal tissues, similarly to astrocytomas. Prohibitin, a pleiotropic protein overexpressed in several tumor types, has been implicated in the regulation of cell proliferation, invasive migration and survival, and the ERK pathway (19). Positivity of tumors for both immunohistochemical markers vimentin and GFAP revealed a similar character of ependymoma to that of astrocytoma, revealing a high requirement for chaperoning activity inside tumor cells, enhanced by the presence of heat-shock proteins (20,21).
Expression of the galectin protein family is in tight correlation with human gliomas and relevant to the modulation of invasion of tumor astrocytes into brain parenchyma (22). Galectins are glycan-binding proteins highly expressed in several tumor types (including brain neoplasms) and have been correlated with adverse prognosis of certain tumor types; strong expression of galectin-3 has been found in astrocytomas and medulloblastomas (23). In our study, galectins 1, 3, 8, 9 and 9b were found to be present in our analyzed series of patients. Due to their unique structure, galectins can oligomerize forming lattices upon binding to multivalent oligossacharides and influence several pathological events, such as tumorigenesis, invasion and metastasis (24)
Peroxiredoxins I-VI have recently been shown to have a role in the tumorigenesis of astrocytic brain tumors (25,26). The presence of all perixoredoxin molecules, except for peroxideroxin-3, was noted in our studied ependymoma samples, in contrast to Haapasalo et al. who reported absence of perixiredoxin 4 (26). Peroxiredodin presence advocates the presence of defense in oxidative damage, to surpass the effect of oxidative stress taking place in ependymoma tissues.
A similar outline was depicted for similarities and differences regarding ependymal tissues and medullostoma. Coronin 1A, 1B, 1C and 2B were identified as components of epeyndymoma tissues. Coronin 1B (Q9BR76) is known to be implicated in contractility at adherens junctions and therefore influences apoptotic cell extrusion (27). Furthermore, coronin 1A (P31146), a newly identified p53 transcriptional target and a homotrimeric F-actin binding protein, important for cell migration and brain morphogenesis, whose expression has been implicated in diffuse glioma malignancies (28,29) was found to be part of the ependymomal proteome; this specific molecule was also present in our previous analysis of medulloblastoma tissues (17).
Furthermore, drebrin (Q16643), an actin-binding protein involved in the regulation of actin filament organization, playing a significant role in cell motility and having been implicated in glioma cell invasiveness (28), has equally been found to be expressed in medulloblastoma and ependymoma tissues. Both drebrin and synaptophysin (P08247) are neuronal synaptic markers playing roles in microglial activation prior to any visible signs of neuronal cell death, including neuronal cleaved caspase-3 activation (31).
Similarly to medulloblastoma, programmed cell death 6-interacting protein (Q8WUM4), a key regulator of tumor growth and angiogenesis implicated in the PI3K pathway, protein lyric (Q86UE4), a protein that activates the nuclear factor-κΒ transcription factor and promotes anchorage-independent growth of immortalized melanocytes and astrocytes and secreted protein acidic and rich in cysteine (SPARC) (P09486), an inducer of neuronal differentiation as a cell defense mechanism against tumor transformation, were all found to be present in ependymoma tissues (32-34).
Overall, all putative biological markers proposed through genomic studies/approaches identified in larger cohorts of samples including nucleolin (P19338) (35), Ki-67 (P46013) (36), nestin (P48681) (37) and laminin subunit A2 (P24043) (38) were identified as being present in the our analyzed samples, thus further confirming the analytical power of the method of analysis proposed herein.
The PI3K pathway is one of the most commonly activated pathways in cancer, including glioma and medulloblastoma cancer types (17,39,40). PI3Ks are lipid kinases that transduce signals from growth factors and cytokines influencing diverse biological functions, including cellular proliferation, survival, and motility. The PI3K pathway has been reported to be activated in a large number of ependymoma cases examined by polymerase chain reaction and immunohistochemical analyses (41). It is of great importance that the nanoLC-MS/MS method developed in the present study allows for definite identification of key molecules of the specific pathway, thus allowing for further confirmation (in a quantitative manner) of deregulation of the specific molecules once compared to tissues that are non-malignant (manuscript in preparation). More specifically, proteins integrin (P05106), MAPK1 (P28482), MEK1/2 (Q2750), p7056K (P23443), NF-κB (P19838) and B-cell lymphoma 2-like protein (Q9BXK5) were all present in the final protein list of ependymomas, reported in our present study.
Whether future molecular pathophysiology experiments lead research of this specific brain malignancy towards the PI3K pathway as a potential therapeutic target and potential prognostic marker for ependymoma, or towards any other relevant pathway or isolated molecule or panel of molecules, one fact is certain: proteomics, and more specifically, enhanced in-depth nanoLC-MS/MS analysis, will provide the tools for assessing results of all molecular approaches in an easily accessible and highly reproducible manner.
Acknowledgements
The Authors would like to thank all patients and their families without whom advance in pediatric brain tumor research would not be possible.
References
- 1.Firme MR, Marra MA. The molecular landscape of pediatric brain tumors in the next-generation sequencing era. Curr Neurol Neurosci Rep. 2014;14(9):474. doi: 10.1007/s11910-014-0474-4. [DOI] [PubMed] [Google Scholar]
- 2.Anagnostopoulos AK, Tsangaris GT. Proteomics of pediatric brain tumors. Exp Reviews Proteomics. 2014;11(5):641–648. doi: 10.1586/14789450.2014.939633. [DOI] [PubMed] [Google Scholar]
- 3.de Bont JM, den Boer ML, Kros JM, Passier MM, Reddingius RE, Smitt PA, Luider TM, Pieters R. Identification of novel biomarkers in pediatric primitive neuroectodermal tumors and ependymomas by proteome-wide analysis. J Neuropathol Exp Neurol. 2007;66(6):505–516. doi: 10.1097/01.jnen.0000240475.35414.c3. [DOI] [PubMed] [Google Scholar]
- 4.Merchant TE, Fouladi M. Ependymoma: New therapeutic approaches including radiation and chemotherapy. J Neurooncol. 2005;75:287–299. doi: 10.1007/s11060-005-6753-9. [DOI] [PubMed] [Google Scholar]
- 5.de Bont JM, Packer RJ, Michiels EM, den Boer ML, Pieters R. Biological background of pediatric medulloblastoma and ependymoma: a review from a translational research perspective. Neuro Oncol. 2008;10(6):1040–1060. doi: 10.1215/15228517-2008-059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nieder C, Andratschke NH, Grosu AL. Re-irradiation for recurrent primary brain tumors. Anticancer Res. 2016;36(10):4985–4995. doi: 10.21873/anticanres.11067. [DOI] [PubMed] [Google Scholar]
- 7.Da Costa GG, Gomig TH, Kaviski R, Santos Sousa K, Kukolj C, De Lima RS, De Andrade Urban C, Cavalli IJ, Ribeiro EM. Comparative proteomics of tumor and paired normal breast tissue highlights potential biomarkers in breast cancer. Cancer Genomics Proteomics. 2015;12(5):251–261. [PubMed] [Google Scholar]
- 8.Weidle UH, Birzele F, Kollmorgen G, Rüger R. Dissection of the process of brain metastasis reveals targets and mechanisms for molecular-based intervention. Cancer Genomics Proteomics. 2016;13(4):245–258. [PubMed] [Google Scholar]
- 9.Anagnostopoulos AK, Vougas K, Kolialexi A, Mavrou A, Foundoulakis M, Tsangaris GT. The protein profile of the human immature T-cell line CCRF-CEM. Cancer Genomics Proteomics. 2005;2 (4):271–300. [PubMed] [Google Scholar]
- 10.Polisetty RV, Gautam P, Sharma R, Harsha HC, Nair SC, Gupta MK, Uppin MS, Challa S, Puligopu AK, Ankathi P, Purohit AK, Chandak GR, Pandey A, Sirdeshmukh R. LC-MS/MS analysis of differentially expressed glioblastoma membrane proteome reveals altered calcium signaling and other protein groups of regulatory functions. Mol Cell Proteomics. 2012;11(6):M111.013565. doi: 10.1074/mcp.M111.013565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anagnostopoulos AK, Stravopodis DJ, Tsangaris GT. Yield of 6,000 proteins by 1D nLC-MS/MS without pre-fractionation. J Chromatogr B Analyt Technol Biomed LifeSci. 2016:pii: S1570-0232(16)30656-0.. doi: 10.1016/j.jchromb.2016.08.031. [DOI] [PubMed] [Google Scholar]
- 12.Velentzas AD, Anagnostopoulos AK, Velentzas PD, Mpakou VE, Sagioglou NE, Tsioka MM, Katarachia S, Manta AK, Konstantakou EG, Papassideri IS, Tsangaris GT, Stravopodis DJ. Global proteomic profiling of Drosophila ovary: a high-resolution, unbiased, accurate and multifaceted analysis. Cancer Genomics Proteomics. 2015;12(6):369–384. [PubMed] [Google Scholar]
- 13.Paraskevopoulou MD, Vlachos IS, Hatzigeorgiou AG. DIANA-TarBase and DIANA Suite Tools: Studying experimentally supported microRNA targets. Curr Protoc Bioinformatics. 2016;55:12.14.1–12.14.18. doi: 10.1002/cpbi.12. [DOI] [PubMed] [Google Scholar]
- 14.Vlachos IS, Paraskevopoulou MD, Karagkouni D, Georgakilas G, Vergoulis T, Kanellos I, Anastasopoulos IL, Maniou S, Karathanou K, Kalfakakou D, Fevgas A, Dalamagas T, Hatzigeorgiou AG. DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 2015;43(Database issue):p. D153–159. doi: 10.1093/nar/gku1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thakur SS, Geiger T, Chatterjee B, Bandilla P, Fröhlich F, Cox J, Mann M. Deep and highly sensitive proteome coverage by LC-MS/MS without prefractionation. Mol Cell Proteomics. 2011;10(8):M110.003699. doi: 10.1074/mcp.M110.003699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anagnostopoulos AK, Dimas KS, Papathanassiou C, Braoudaki M, Anastasiadou E, Vougas K, Karamolegou K, Kontos H, Prodromou N, Tzortzatou-Stathopoulou F, Tsangaris GT. Proteomics studies of childhood pilocytic astrocytoma. J Proteome Res. 2011;10(5):2555–2565. doi: 10.1021/pr200024m. [DOI] [PubMed] [Google Scholar]
- 17.Anagnostopoulos AK, Papathanassiou C, Karamolegou K, Anastasiadou E, Dimas KS, Kontos H, Koutsopoulos A, Prodromou N, Tzortzatou-Stathopoulou F, Tsangaris GT. Proteomic studies of pediatric medulloblastoma tumors with 17p deletion. J Proteome Res. 2015;14(2):1076–1088. doi: 10.1021/pr501219f. [DOI] [PubMed] [Google Scholar]
- 18.Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N, Toedt G, Wittmann A, Kratz C, Olbrich H, Ahmadi R, Thieme B, Joos S, Radlwimmer B, Kulozik A, Pietsch T, Herold-Mende C, Gnekow A, Reifenberger G, Korshunov A, Scheurlen W, Omran H, Lichter P. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest. 2008;118(5):1739–1749. doi: 10.1172/JCI33656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao Y, Liang H, Zhang F, Luan Z, Zhao S, Wang XA, Liu S, Bao R, Shu Y, Ma Q, Zhu J, Liu Y. Prohibitin overexpression predicts poor prognosis and promotes cell proliferation and invasion through ERK pathway activation in gallbladder cancer. J Exp Clin Cancer Res. 2016;35:68. doi: 10.1186/s13046-016-0346-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernando G, Paul F, Laura J, Alejandra AM, Gabriela M, Alberto PL. Is the WNT/β catenin signalling pathway activated in seminoma? An immunohistochemical study. J Cancer Res Ther. 2016;12(2):1075–1079. doi: 10.4103/0973-1482.147392. [DOI] [PubMed] [Google Scholar]
- 21.Palm T, Figarella-Branger D, Chapon F, Lacroix C, Gray F, Scaravilli F, Ellison DW, Salmon I, Vikkula M, Godfraind C. Expression profiling of ependymomas unravels localization and tumor grade-specific tumorigenesis. Cancer. 2009;115(17):3955–3968. doi: 10.1002/cncr.24476. [DOI] [PubMed] [Google Scholar]
- 22.Rorive S, Belot N, Decaestecker C, Lefranc F, Gordower L, Micik S, Maurage CA, Kaltner H, Ruchoux MM, Danguy A, Gabius HJ, Salmon I, Kiss R, Camby I. Galectin-1 is highly expressed in human gliomas with relevance for modulation of invasion of tumor astrocytes into the brain parenchyma. Glia. 2001;3:241–255. doi: 10.1002/1098-1136(200103)33:3<241::aid-glia1023>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 23.Borges CB, Bernardes ES, Latorraca EF, Becker AP, Neder L, Chammas R, Roque-Barreira MC, Machado HR, de Oliveira RS. Galectin-3 expression: a useful tool in the differential diagnosis of posterior fossa tumors in children. Childs Nerv Syst. 2011;27(2):253–257. doi: 10.1007/s00381-010-1262-3. [DOI] [PubMed] [Google Scholar]
- 24.Pereira JX, Azeredo MC, Martins FS, Chammas R, Oliveira FL, Santos SN, Bernardes ES, El-Cheikh MC. The deficiency of galectin-3 in stromal cells leads to enhanced tumor growth and bone marrow metastasis. BMC Cancer. 2016;16:636. doi: 10.1186/s12885-016-2679-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jarvela S, Rantala I, Rodriguez A, Kallio H, Parkkila S, Kinnula VL, Soini Y, Haapasalo H. Specific expression profile and prognostic significance of peroxiredoxins in grade II-IV astrocytic brain tumors. BMC Cancer. 2010;22:100–104. doi: 10.1186/1471-2407-10-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haapasalo T, Nordfors K, Järvelä S, Kok E, Sallinen P, Kinnula VL, Haapasalo HK, Soini Y. Peroxiredoxins and their expression in ependymomas. J Clin Pathol. 2013;66(1):12–17. doi: 10.1136/jclinpath-2012-201048. [DOI] [PubMed] [Google Scholar]
- 27.Michael M, Meiring JC, Acharya BR, Matthews DR, Verma S, Han SP, Hill MM, Parton RG, Gomez GA, Yap AS. Coronin 1B reorganizes the architecture of F-actin networks for contractility at steady-state and apoptotic adherens junctions. Dev Cell. 2016;37(1):58–71. doi: 10.1016/j.devcel.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 28.Di Giovanni S, Knights CD, Rao M, Yakovlev A, Beers J, Catania J, Avantaggiati ML, Faden AI. The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J. 2006;17:4084–4096. doi: 10.1038/sj.emboj.7601292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thal D, Xavier CP, Rosentreter A, Linder S, Friedrichs B, Waha A, Pietsch T, Stumpf M, Noegel A, Clemen C. Expression of coronin-3 (coronin-1C) in diffuse gliomas is related to malignancy. J Pathol. 2008;214(4):415–424. doi: 10.1002/path.2308. [DOI] [PubMed] [Google Scholar]
- 30.Terakawa Y, Agnihotri S, Golbourn B, Nadi M, Sabha N, Smith CA, Croul SE, Rutka JT. The role of drebrin in glioma migration and invasion. Exp Cell Res. 2013;319(4):517–528. doi: 10.1016/j.yexcr.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 31.Jebelli J, Hooper C, Pocock JM. Microglial p53 activation is detrimental to neuronal synapses during activation-induced inflammation: Implications for neurodegeneration. Neurosci Lett. 2014;583:92–97. doi: 10.1016/j.neulet.2014.08.049. [DOI] [PubMed] [Google Scholar]
- 32.Rho SB, Song YJ, Lim MC Lee SH, Kim B, Park SY. Programmed cell death 6 (PDCD6) inhibits angiogenesis through PI3K/mTOR/p70S6K pathway by interacting of VEGFR-2. Cell Signal. 2012;24(1):131–139. doi: 10.1016/j.cellsig.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 33.Emdad L, Sarkar D, Su ZZ, Randolph A, Boukerche H, Valerie K, Fisher PB. Activation of the nuclear factor kappa B pathway by astrocyte elevated gene-1: implications for tumor progression and metastasis. Cancer Res. 2006;66(3):1509–1516. doi: 10.1158/0008-5472.CAN-05-3029. [DOI] [PubMed] [Google Scholar]
- 34.Tai IT, Dai M, Owen DA, Chen LB. Genome-wide expression analysis of therapy-resistant tumors reveals SPARC as a novel target for cancer therapy. J Clin Invest. 2005;115:1492–502. doi: 10.1172/JCI23002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ridley L, Rahman R, Brundler MA, Ellison D, Lowe J, Robson K Prebble E, Luckett I, Gilbertson RJ, Parkes S, Rand V, Coyle B, Grundy RG. Multifactorial analysis of predictors of outcome in pediatric intracranial ependymoma. Neuro Oncol. 2008;10:675–689. doi: 10.1215/15228517-2008-036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bennetto L, Foreman N, Harding B, Hayward R, Ironside J, Love S, Ellison D. Ki-67 immunolabelling index is a prognostic indicator in childhood posterior fossa ependymomas. Neuropathol Appl Neurobiol. 1998;24:434–440. doi: 10.1046/j.1365-2990.1998.00143.x. [DOI] [PubMed] [Google Scholar]
- 37.Milde T, Hielscher T, Witt H, Kool M, Mack SC, Deubzer HE Oehme I, Lodrini M, Benner A, Taylor MD, von Deimling A, Kulozik AE, Pfister SM, Witt O, Korshunov A. Nestin expression identifies ependymoma patients with poor outcome. Brain Pathol. 2012;22:848–860. doi: 10.1111/j.1750-3639.2012.00600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Witt H, Mack SC, Ryzhova M, Bender S, Sill M, Isserlin R Benner A, Hielscher T, Milde T, Remke M, Jones DT, Northcott PA, Garzia L, Bertrand KC, Wittmann A, Yao Y, Roberts SS, Massimi L, Van Meter T, Weiss WA, Gupta N, Grajkowska W, Lach B, Cho YJ, von Deimling A, Kulozik AE, Witt O, Bader GD, Hawkins CE, Tabori U, Guha A, Rutka JT, Lichter P, Korshunov A, Taylor MD, Pfister SM. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell. 2011;20:143–157. doi: 10.1016/j.ccr.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chakravarti A, Zhai G, Suzuki Y, Sarkesh S, Black PM, Muzikansky A, Loeffler JS. The prognostic significance of phosphatidylinositol 3-kinase pathway activation in human gliomas. J Clin Oncol. 2004;22:1926–1933. doi: 10.1200/JCO.2004.07.193. [DOI] [PubMed] [Google Scholar]
- 40.Hartmann W, Digon-Sontgerath B, Koch A, Waha A, Endl E, Dani I Denkhaus D, Goodyer CG, Sörensen N, Wiestler OD, Pietsch T. Phosphatidylinositol 30-kinase/AKT signaling is activated in medulloblastoma cell proliferation and is associated with reduced expression of PTEN. Clin Cancer Res. 2006;12:3019–3027. doi: 10.1158/1078-0432.CCR-05-2187. [DOI] [PubMed] [Google Scholar]
- 41.Rogers HA, Mayne C, Chapman RJ, Kilday JP, Coyle B, Grundy RG. PI3K pathway activation provides a novel therapeutic target for pediatric ependymoma and is an independent marker of progression-free survival. Clin Cancer Res. 2013;19(23):6450–6460. doi: 10.1158/1078-0432.CCR-13-0222. [DOI] [PubMed] [Google Scholar]