

Abstract
Hydrogels are of growing interest for the delivery of therapeutics to specific sites in the body. For use as a delivery vehicle, hydrophilic precursors are usually laden with bioactive moieties and then directly injected to the site of interest for in situ gel formation and controlled release dictated by precursor design. Hydrogels formed by thiol–ene click reactions are attractive for local controlled release of therapeutics owing to their rapid reaction rate and efficiency under mild aqueous conditions, enabling in situ formation of gels with tunable properties often responsive to environmental cues. Herein, we will review the wide range of applications for thiol–ene hydrogels, from the prolonged release of anti-inflammatory drugs in the spine to the release of protein-based therapeutics in response to cell-secreted enzymes, with a focus on their clinical relevance. We will also provide a brief overview of thiol–ene click chemistry and discuss the available alkene chemistries pertinent to macromolecule functionalization and hydrogel formation. These chemistries include functional groups susceptible to Michael type reactions relevant for injection and radically-mediated reactions for greater temporal control of formation at sites of interest using light. Additionally, mechanisms for the encapsulation and controlled release of therapeutic cargoes are reviewed, including i) tuning the mesh size of the hydrogel initially and temporally for cargo entrapment and release and ii) covalent tethering of the cargo with degradable linkers or affinity binding sequences to mediate release. Finally, myriad thiol–ene hydrogels and their specific applications also are discussed to give a sampling of the current and future utilization of this chemistry for delivery of therapeutics, such as small molecule drugs, peptides, and biologics.
Keywords: Hydrogels, thiol, ene, click chemistry, drug delivery, controlled release
1. Introduction
Significant advancements have been made in the last decade to develop new therapeutics with the potential to improve the treatment of a variety of diseases, from small molecular weight hydrophilic and hydrophobic drugs to larger peptides and biologics (e.g., therapeutic proteins and antibodies). In particular, biologics have been a major area of growth for the pharmaceutical industry with the worldwide sales of biologics exceeding $92 billion in 2009.1 Despite this increasing demand, costs of drug development and production remain high.2-3 Delivery of therapeutics at a controlled rate to a targeted site affords opportunities to both improve treatment efficacy and reduce total treatment costs. However, successful development of drug carriers with appropriate therapeutic retention and release characteristics for clinical use remains a challenge and an area of active research. Tremendous progress has been made in the design of novel drug carriers, including liposomes,4-5 nanoparticles,6-8 polymersomes,9-11 dendrimers,12-13, microparticles,14-15 and hydrogels,16-17 with improved efficacy, prolonged drug action in vivo, reduced drug toxicity, and decreased drug-associated costs. Among these drug carriers, hydrogels have emerged as promising delivery vehicles, especially for biologics, owing to their high cargo loading efficiency and their ability to retain cargo bioactivity.18
Hydrogels, or hydrophilic polymer networks that imbibe and retain large amounts of water, have been fabricated for controlled release applications using a range of natural and synthetic polymers as their base building blocks. Due to their inherent biocompatibility and bioactivity, natural polymers, such as hyaluronic acid,19 chitosan,20 heparin,21 silk,22 and alginate,23 often provide synergistic interactions with cargo molecules and with cells in vivo. For example, Elia and coworkers recently demonstrated the delivery of small molecular weight drugs (cortisone and hydrocortisone) using hyaluronic acid-silk based hydrogels.24 On the other hand, synthetic biocompatible polymers, including poly(ethylene glycol) (PEG) and poly(vinyl alcohol) (PVA), generally offer greater flexibility for chemical modification, improved tunability over mechanical properties, facile incorporation of degradable functional groups, and limited batch-to-batch variation.18 For example, Sinko and coworkers investigated the use of PEG hydrogels for ocular drug delivery to injured rabbit corneas; sustained delivery of doxycycline over 7 days using these hydrogels resulted in accelerated wound healing compared to direct doses of doxycycline.25
Control over the formation of these hydrogels is essential for delivery applications since the connectivity (e.g., crosslink density) and mechanical properties of the network dictate mass transport and therapeutic release. For example, increasing the crosslink density decreases the distance between crosslinks in the hydrogel (i.e., mesh size), often resulting in a decreased initial burst of cargo.26-27 Yang and coworkers demonstrated mesh size-dependent release of the model protein bovine serum albumin (BSA) from PEG-based hydrogels with mesh sizes ranging from 4 nm to 14 nm.26 For hydrogel compositions with a mesh size below 6 nm, only about ∼15% of encapsulated BSA was released. In contrast, for the hydrogel with a mesh size of approximately 15 nm, ∼100% of encapsulated BSA was released within 2 hours.
Hydrogels have been formed by i) physical crosslinking (i.e., non-covalent interactions), including ionic, electrostatic, or hydrophobic interactions between the polymeric macromers, or ii) chemical crosslinking by reactive functional groups to form covalent linkages.28 Physically crosslinked hydrogels offer advantages for injectable formulations, including dynamic crosslinks for gel dissolution and therapeutic release, shear thinning for injection, and in situ formation without initiators or catalysts.29 However, covalently crosslinked hydrogels provide better control over crosslink density and allow easier incorporation of labile functional groups for stimuli-responsive degradability of and release from the delivery vehicle.30 Amongst covalent crosslinking chemistries, click reactions, including copper-free azide–alkyne cycloadditions, Diels-Alder, thiol–ene, and oxime reactions, are attractive for therapeutic delivery applications due to their fast reaction kinetics under mild conditions, permitting rapid formation in situ in the presence of cargo molecules and tissues.28, 31-32 In particular, thiol–ene click reactions have been broadly applied for the design of drug delivery carriers.33-35
In this review, we highlight recent work utilizing thiol–ene click reactions for controlled drug delivery applications. Drug incorporation and release mechanisms (section 2) and thiol–ene reactions for hydrogel formation (section 3) are discussed along with recent examples of material chemistries used for therapeutic delivery. We subsequently overview how thiol–ene hydrogels have been used for delivery of small molecular weight drugs, therapeutic peptides, and proteins (section 4) and examine future directions in the field (section 5).
2. Drug incorporation and release mechanisms
Strategies for incorporation of therapeutics into hydrogels generally fall into three categories: (1) encapsulation, where therapeutics are entrapped within the crosslinks of the polymer network (Fig. 1A); (2) tethering, where drugs of interest are covalently bound to the polymer network (Fig. 1B); and (3) affinity binding, where hydrophobic, ionic, or peptide interactions are utilized to retain therapeutics within the hydrogel network (Fig. 1C).36 To design effective drug delivery vehicles, these strategies are combined with appropriate release mechanisms to suit the application of interest; several examples are shown in the right hand column of Fig. 1. The release of therapeutics entrapped within or tethered to the matrix can be controlled by i) Fickian diffusion, ii) degradation of tethered linkages in response to relevant biological stimuli, or iii) combinations of both. With affinity binding, a ligand is added to the hydrogel with affinity for therapeutic(s) of interest. In this way, release is controlled by the reversible binding of the ligand to the therapeutic in combination with diffusion of the free species or matrix degradation.37 In this section, we will overview therapeutic delivery strategies specifically for thiol–ene hydrogel systems with relevant examples.
Figure 1. Therapeutic loading and release mechanisms from hydrogel-based delivery vehicles.
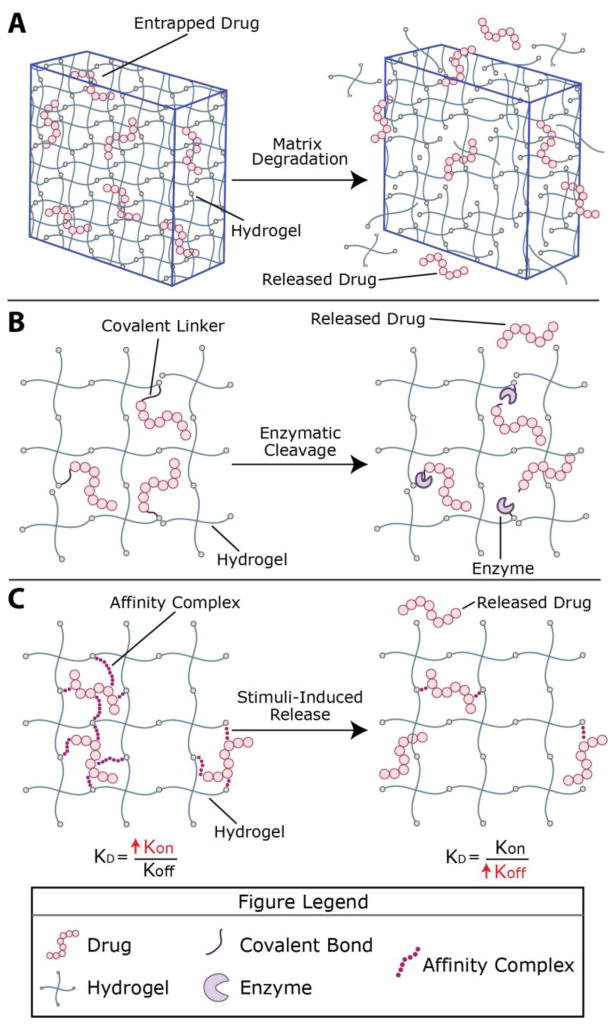
Hydrogels, including those formed by thiol–ene click chemistry, can be formed in vitro or in vivo for therapeutic delivery applications. Stoichiometric reaction between functional groups on multiarm poly(ethylene glycol) (PEG) macromers functionalized with specific alkenes or thiols, respectively, has been reported, producing thiol–ene hydrogels with nearly ideal network structure (shown here); these are of growing interest for delivery applications toward providing a well-defined and predictable mesh size. However, macromolecules of varied length and functionality, both of synthetic and natural origin and decorated with various alkenes or thiols, broadly have been utilized for the formation of thiol–ene hydrogels. Engineering of material structure and chemistry provide handles for controlling release profiles. A) For example, therapeutics frequently have been encapsulated within the hydrogel network, where the cargo is entrapped if the average pore size (e.g., mesh size) of the hydrogel is smaller than the drug; degradation of the network, which increases mesh size, controls release. B) Small molecule drugs or peptides, which can be difficult to entrap within hydrophilic highly-swollen hydrogels, have been tethered to the network and released upon tether cleavage (or complete network degradation); here, cleavage by a cell-secreted enzyme is depicted. C) Ligands for a therapeutic of interest also have been incorporated within hydrogels for controlling retention and release by affinity binding; reversible binding of the ligand dictated by kon/koff determines the fraction of bound/free therapeutic, where diffusion of the free species (or matrix degradation) controls release. These therapeutic loading and release mechanisms can be used in different combinations than those depicted here and have been used in thiol–ene hydrogels.
For the majority of applications, hydrogels are loaded with therapeutics of interest by physical encapsulation (Fig. 1A), mitigating the need to chemically modify the bioactive cargo, which can impact FDA approval.38 Loading strategies include mixing the therapeutic into the precursor solution prior to forming the hydrogel39 or diffusing therapeutics into the hydrogel after crosslinking.40 In addition, smaller therapeutics can be encapsulated within nano- and micro-gels (i.e., hydrogel particles on the size scale of nanometers or microns). These nano- or micro-gels are incorporated within hydrogels to restrict uncontrolled drug diffusion out of the bulk hydrogel immediately after formation.41 Diffusion dictated release occurs when the network mesh size is tuned to slightly larger than the diameter of the therapeutic of interest, benefiting patients by maintaining a potent therapeutic concentration in vivo over the course of hours or days. Hydrogels with inherent or incorporated degradability have been used for applications that require increased control of release over time or at a specific location, generally exploiting light-mediated, enzymatic, or hydrolytic degradation to release the drug of interest. Approaches for imparting degradability are discussed further in section 3.4.42 In addition to degradation, stimuli-responsive hydrogels whose swelling changes in response to a microenvironment stimulus have been used for the controlled release of entrapped therapeutics; increased swelling within the desired microenvironment leads to increased mesh size and consequently release. A recent review by He et al. highlights pH and temperature responsive drug delivery from a variety of materials, including stimuli-responsive thiol–ene hydrogels.43
Often, covalent conjugation (Fig. 1B) is required when the size of the therapeutic is too small to allow entrapment within the hydrogel or when diffusion of the therapeutic from the hydrogel is too rapid for a desired application. The therapeutic typically is modified for covalent attachment to the hydrogel and released by cleavage of the covalent linkage or by complete hydrogel degradation. With linker cleavage (e.g., enzymatic cleavage of a peptide linker), tethered molecules are released without altering the greater network structure.44 When therapeutics are incorporated into the polymer backbone, rather than as a pendant group, the matrix itself is degraded to facilitate release. Van Hove et al. applied the latter approach for the release of peptide-based drugs that promote angiogenesis: the drugs were flanked by enzymatically-degradable peptides that crosslinked the hydrogel (IPES↓LRAG, which degrades in response to specific matrix metalloproteinases [MMPs]), and the drug was released upon MMP-mediated hydrogel degradation.45
The third major class of techniques for therapeutic incorporation within thiol–ene hydrogels is affinity binding (Fig. 1C). Hydrophobic interactions, hydrogen bonding, and ionic inclusion complexes all have been used to retain therapeutics in hydrogel networks.46-47 Cleaving hydrophobic groups from the network decreases the ability of a gel to retain hydrophobic drugs, and ionic networks have been used to release therapeutics in response to changes in pH. For example, Arslan et al. designed a PEG-based hydrogel in which cyclodextrin-containing crosslinkers form an inclusion complex with the hydrophobic drug puerarin, which is traditionally used for treating glaucoma.48 The amount of puerarin loaded into each hydrogel was maximized by decreasing the molecular weight of PEG, which made the polymer and resulting hydrogel less hydrophilic, and by increasing the concentration of cyclodextrin, which has a hydrophobic interior cavity that entraps and releases hydrophobic drugs.
3. Thiol–ene reactions for hydrogel formation
Broadly, thiol–ene click chemistry refers to the reaction of thiol-containing compounds with alkenes, or ‘enes’, with close to one hundred percent conversion. Interest in thiol–ene chemistry for hydrogel design has arisen from several advantageous attributes for biological applications: i) thiol–ene reactions often proceed rapidly under mild conditions (e.g., water and buffers) that are compatible with cells and other biological molecules; ii) thiol–ene reactions have well-defined and well-characterized reaction mechanisms and products; and iii) the process of introducing thiols and alkenes to macromolecules for hydrogel synthesis is relatively simple compared to the introduction of other functional groups (e.g., strained cyclooctynes).49-50 In this section, we review some of the key aspects of thiol–ene reactions for their use in designing hydrogels for therapeutic release: the thiol–ene reaction mechanisms, the introduction of thiol groups onto macromolecules to generate macromers, and the characteristics of alkenes that can be used for thiol–ene chemistry to form hydrogels. This section concludes with a brief discussion of engineering thiol–ene hydrogels to control their degradation and release profiles.
3.1 Thiol–ene reaction mechanism
Reactions of thiols with alkenes typically occur by two different mechanisms: thiol-Michael type reactions and radically-mediated thiol–ene reactions. Selection of the reactive functional groups and solution conditions, as discussed in section 3.2, dictates the reaction mechanism and important characteristics relevant for consideration when designing hydrogels for therapeutic delivery (e.g., reaction initiation, time, and byproducts). While the reaction mechanisms are briefly discussed here, interested readers are referred to recent articles on thiol–ene chemistry for a comprehensive review.33-34, 49, 51
The thiol-Michael type reaction typically proceeds at room temperature without interference from water protons, making it amenable to hydrogel synthesis, which usually takes place in aqueous solutions. Generally, in thiol-Michael type reactions, a base (B) abstracts a proton from a thiol, forming a thiolate anion that acts as a nucleophile (Fig. 2A). The thiolate anion attacks the electrophilic β-carbon of an alkene to form a carbon-centered anion intermediate, which subsequently abstracts a proton from a conjugate acid (BH+) to generate the thiol-Michael addition product, regenerating the original base. The reaction continues, usually rapidly, until one of the reactive functional groups is consumed. Owing to the mechanism, these reactions typically are free of byproducts and can occur with no or low amounts of catalyst, making thiol-Michael reactions attractive for injectable hydrogel applications.
Figure 2. Thiol–ene reaction mechanisms.
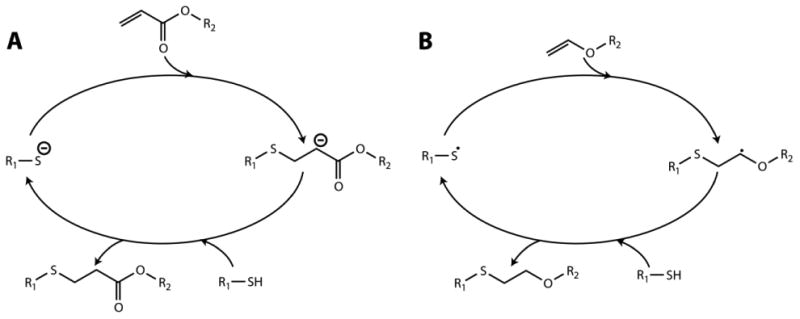
Thiol–ene reactions take place by A) Michael-type or B) radically-mediated mechanisms based on alkene selection. Michael-type reactions occur upon mixing of the reactive macromers, where the rate of reaction can be increased by addition of a base catalyst or increasing solution pH, making them well suited for injectable hydrogel formation. Radically-mediated thiol–ene reactions are initiated with the application of light and a photoinitiator, providing control over when and where hydrogels form.
In radically-mediated thiol–ene chemistry, the reaction also proceeds under mild aqueous conditions but requires an initiator to generate radicals that initiate the reaction between thiol and select alkene functional groups (Fig. 2B). Radical generation can be achieved using a thermal, oxidation-reduction, or photochemical process based on initiator selection.34 After initiation, the generated radical attacks a thiol to form a thiyl radical, which acts as an electrophile. Subsequently, the thiyl radical attacks the C=C double bond of the alkene, forming a thiol–ene addition product and generating a new thiyl radical. The propagation process is highly efficient resulting in a reaction of one thiol with one alkene. Unlike the traditional free radical chain polymerization (e.g., acrylates or methacrylates), radically-mediated thiol–ene reactions are relatively oxygen-insensitive.33
Photochemical processes are particularly attractive for designing thiol–ene hydrogels for biomedical applications as they afford control of where, when, and how fast the polymerization occurs by controlling the application of light.52 Photoinitiated thiol–ene reactions have been utilized to prepare various drug carriers, including microgels and nanogels. Photoinitiators such as Irgacure-2959,53 lithium acylphosphinate (LAP),54 and eosin-Y55 have been used extensively; initiator-free photopolymerization also has been reported but is less common.56 Thiol–ene photocrosslinking reactions typically have been carried out in the presence of biological cargoes (e.g., proteins, cells) with low cytocompatible doses of light (e.g., ∼5 to 10 mW/cm2 of long wavelength UV light at 365 nm for ∼2-5 minutes). Visible light light-mediated polymerizations of thiol–ene hydrogels also have been reported (e.g., 70,000 lux at 400 – 700 nm for ∼30 seconds).55 For translation to the clinic, both the cytocompatiblity and the depth of light penetration must be considered. While hydrogels may be be formed in situ during surgical procedures without concern of light penetration,57 formation of hydrogels by irradiation through tissues currently is limited to superficial depths by the scattering of light by tissues (e.g., penetration depths of ∼0.5 mm at 365 nm to 500 nm through skin).58-60 Toward increasing the range of wavelengths available for photopolymerizations in water, Grutzmacher and coworkers recently reported the synthesis of highly efficient, water-soluble visible light photoinitiators using monoacylphosphineoxide and bisacylphosphineoxide salts that are promising for biological applications based on their response to low doses of blue light (465 nm LED for <30 min).61-62 In addition, cyclic benzylidene ketone-based initiators that are water soluble and responsive to two-photon near infrared light (780 nm, ∼60 to 400 mW, 100 fs pulse) may prove useful for thiol–ene photopolymerizations in biomedical applications requiring increased light penetration in situ.63
3.2 Introduction of thiol functional groups on macromolecules
Thiol–ene hydrogels are prepared by covalent crosslinking of hydrophilic polymers containing reactive thiols and alkenes; these reactive macromolecules also are referred as macromolecular monomers or macromers (Fig. 3A). Thiol functional groups (also known as sulfhydryls) are generally introduced on a macromer by reactions with functional groups commonly found on natural and synthetic polymers, including amines, carboxylic acid groups, aldehydes, ketones, or reduced disulfides (Fig. 3B). The reactivity of the thiol can vary depending on the functional group(s) proximate to it, which affects the pKa.34 While a brief overview of incorporation of thiol groups on macromers is provided here, commonly used synthetic macromers, such as poly(ethylene glycol) functionalized with thiols, are commercially available.
Figure 3. Thiol–ene hydrogels.
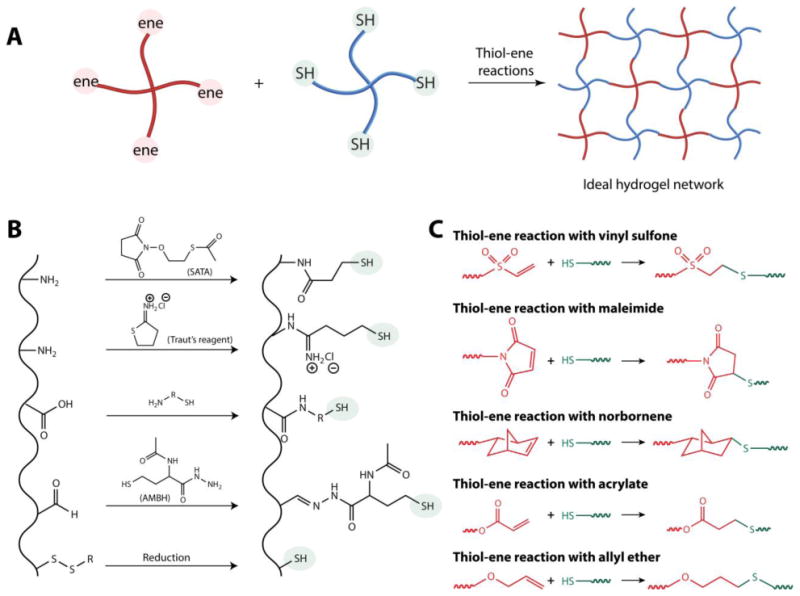
A) Hydrogels are formed by thiol–ene reactions of alkene (‘ene’) and thiol groups on multifunctional macromers. B) Thiol functional groups have been introduced on macromolecules to generate macromers by reacting (top to bottom) i) a pendant amine with N-succinimidyl S-acetylthioacetate (SATA, aqueous condition in a non-amine containing buffer) or ii) Traut's reagent (2-Iminothiolane, aqueous buffer range pH 7-10); iii) carboxylic acid using carbodiimide chemistry; iv) aldehyde and ketones with 2-acetamido-4-mercaptobutyric acid hydrazide (AMBH, under pH 7.4); or v) by reducing disulfide linkages using dithiothreitol (DTT), dithioerythritol (DTE), or tris(2-carboxyethyl)phosphine (TCEP) under aqueous conditions. C) Thiol-functionalized macromers have been reacted with various alkene-functionalized macromers to form hydrogels for therapeutic delivery applications, including (top to bottom) i) vinyl sulfone (base catalyzed), ii) maleimides (base catalyzed), iii) norbornenes (radically-mediated), iv) acrylate groups (radically-mediated), and v) allyl ethers (radically-mediated).
For polymers and proteins containing primary amine groups, thiols have been introduced by reacting primary amines using 2-iminothiolane (Traut's reagent), N-succinimididyl S-acerylthioacetate, or succinimidyl acetyl-thiopropropionate.64 For example, Stevens and coworkers thiolated primary amines on a latent form of recombinant transforming growth factor (TGF-β1) using 2-iminothiolane and subsequently reacted the thiolated growth factor with acrylate-functionalized PEG by radically-mediated thiol–ene reaction (see 3.3.4 for mechanistic details).65 The resulting hydrogel contained the latent form of TGF-β1, resulting in increased metabolic activity and protein deposition of encapsulated chondrocytes compared to a negative control while maintaining the bioactivity and stability of the released TGF-β1 over a 34 day time period.
Using carbodiimide chemistry, thiol groups have been introduced on a polymer or a protein by reaction of carboxylic acid groups with cystamine or similar reagents that contain a primary amine and a thiol. For example, Revzin and coworkers incorporated thiol functional groups on heparin using carbodiimide chemistry to modify 40% of the available carboxylic acid groups.66 Heparin is a highly-charged polysaccharide often used in hydrogels to mimic heparan sulfate that is found within the extracellular matrix and binds various proteins (e.g., growth factors or cytokines), mediating their release.21 Here, hydrogel microspheres crosslinked with the thiolated-heparin were formed by reaction of the thiol functional groups with acrylate end-functionalized PEG. The hydrogels containing heparin retained hepatocyte growth factor (HGF) over approximately 5 days, whereas control hydrogels without heparin contained minimal levels of HGF. This example highlights that thiolation and subsequent incorporation of macromolecules such as heparin can be useful in designing thiol–ene hydrogels for therapeutic delivery.
In addition, aldehyde or ketone groups on macromolecules have been modified using acetamido-4-mercaptobutyric acid hydrazide, a hydrazide derivative, to form reactive thiol functional groups.64 For macromolecules containing disulfide linkages, disulfide groups can be reduced to introduce free thiol groups for reaction with alkenes.64 For biomacromolecules, though, reduction of disulfides also may result in changes to native structure and reduced bioactivity.
While thiol–ene polymerization conditions are typically chosen to minimize side reactions, disulfide formation still can present a challenge in the consistent formation of thiol–ene hydrogels: thiol-functionalized macromers can react with each other to form disulfide linkages making them inaccessible for subsequent reaction with alkenes.67 Additionally, thiols on macromers can react with various functional groups that are present on biologics (i.e., off-target reactions leading to oxidation of cysteine residues on proteins).68 Uncontrolled reactions can contribute to loss of bioactivity and degradation of therapeutic peptides and proteins and hence generally are undesirable. Even so, this approach has been utilized to conjugate biologics to polymeric drug carriers.69-70
3.3 Types of alkenes used for thiol–ene hydrogel formation
3.3.1 Vinyl Sulfone
Vinyl sulfones are electron poor alkenes that react with thiols in slightly alkaline conditions (e.g., pH ∼ 8) to give stable β-thiosulfonyl linkages, known as thioether bonds (Fig. 3C).64 Vinyl sulfone groups generally are stable for extended periods of time under aqueous conditions near neutral pH. Vinyl sulfone groups have been introduced on macromers by reaction with free amine and hydroxyl functional groups under basic pH.71-72 For example, in seminal work, Lutolf and Hubbell reported the reaction of hydroxyl end-functionalized PEG with divinyl sulfone in the presence of sodium hydride to synthesize vinyl sulfone end-functionalized PEGs for use in hydrogel formation.71
Hydrogels prepared using thiol-vinyl sulfone crosslinking are attractive for drug delivery applications and as tissue engineering platforms owing to their mild reaction conditions.72-73 Hubbell and coworkers have demonstrated applications of vinyl sulfone-based hydrogels for therapeutic delivery, including the delivery of bone morphogenetic protein 2 (BMP-2) for bone regeneration.74-75 Building upon this, Peng et al. recently reported the synthesis of vinyl sulfone-functionalized dextran hydrogels for delivery of BMP-2 in vivo.73 Vinyl sulfone propionic acid was reacted with hydroxyl groups of dextran via esterification in a one-pot synthesis to yield a series of macromers with varying degrees of substitution (DS = ∼2 to ∼12.5). In vivo studies were performed in mice where hydrogels loaded with recombinant human bone morphogenetic protein-2 (rhBMP-2) were implanted in muscle pouch. Hydrogels prepared from macromers with higher degrees of substitution (DS = 12.5) exhibited slower degradation and higher ectopic bone formation (318 mg) compared to macromers with lower degrees of substitution (DS = 9, ectopic bone formation = 226 mg). This finding can be correlated to slower degradation of these thiol–ene hydrogels, resulting in sustained release of rhBMP-2 for bone formation.
3.3.2 Maleimides
Maleimides are electron poor alkenes and readily react with thiols by Michael-type additions to form succinimide thioether linkages (Fig. 3C). Maleimide groups often are incorporated onto macromolecules by carbodiimide chemistry (e.g., reaction of activated carboxylic acids with amines to introduce maleimide groups). Thiol-maleimide reactions offer a number of advantages: (1) at neutral pH, maleimides react with high selectivity for thiols; (2) thiol-maleimide reactions occur rapidly under physiological conditions; and (3) the thiol-maleimide linkage formed with aryl thiols can undergo retro-Michael reaction under reducing conditions for controlled degradation and release applications.76-77 However, it is important to note that maleimide groups undergo ring hydrolysis under aqueous conditions, yielding maleamic acid that is not reactive with thiols.64 Solution pH, temperature, neighboring functional groups, and hydroxyl ion concentration affect the rate of ring hydrolysis (k = 500 to 1600 M-1 s-1).78 While maleimide ring hydrolysis after formation of succinimide thioether linkages will not significantly change the properties of an existing hydrogel, ring hydrolysis in the precursor solution before hydrogel preparation can significantly increase network defects; such defects typically increase mesh size and reduce network retention of loaded therapeutics, affecting release characteristics.64 In addition, since unreacted small-molecule maleimides can be cytotoxic, thorough purification of maleimide-functionalized macromers after synthesis is needed.79
Thiol-maleimide reactions have successfully been used to design various cell-compatible hydrogels for tissue engineering and drug delivery applications.80-83 For example, Fu and Kao reported semi-interpenetrating thiol-maleimide hydrogels formed in situ for controlled release.81 Thiol and maleimide end-functionalized PEG were reacted in the presence of gelatin at 37 °C to form a semi-interpenetrating network. A model macromolecule, fluorescein isothiocyanate-labeled dextran (Mw ∼ 40,000 g/mol), was encapsulated in the network during hydrogel formation by mixing with the precursor solutions. In the absence of gelatin, 80% of the dextran cargo was released after 1 hour, whereas the incorporation of gelatin slowed release of the cargo molecule to 25 hours (100% release). The difference in the release profiles was attributed to reduced mesh size as a result of additional physical entanglements caused by gelatin incorporation.
3.3.3 Norbornene
Norbornene is a strained cyclohexane ring with a methylene bridge that reacts with thiols in the presence of free radicals in a 1:1 ratio, suggesting limited to no norbornene homopolymerization under gelation conditions (Fig. 3C).84 For hydrogel formation, the thiol-norbornene reaction has been carried out in minutes using low cytocompatible doses of long wavelength ultraviolet light with the photoinitiators Irgacure-2959 and LAP (10 mW/cm2 at 365 nm)84 or using visible light with the photoinitiator eosin-Y (70000 Lux at 400 – 700 nm).55, 85 Since norbornene groups are relatively stable, high conversions have been achieved when stoichiometric amounts of reactive functional groups are used.86 The rate of reaction has been controlled by varying the light intensity, wavelength, and the concentration of photoinitiator,53, 84, 87-88 as has been similarly done for other radically-mediated thiol–ene reactions noted below (3.3.4). For a comprehensive review of hydrogels prepared using thiol-norbornene reactions, readers are referred to a recent review by Lin and coworkers.88 Broadly, the ability to crosslink hydrogels with light using radically-mediated thiol–ene reactions (e.g., thiol-norbornene, thiol-acrylate) provides control over hydrogel formation at positions and times of interest (i.e., spatiotemporal control), which is attractive for the formation of hydrogels in complex geometries, including tissue defects (e.g., critically-sized bone defects), and for stabilizing hydrogel microparticles.89-90
3.3.4 Other alkenes
Acryloyl groups (H2C=CH–C(=O)–) contain double bonds that are electron poor in nature and react with thiols to form stable thioether linkages (Fig. 3C) via a step-growth mechanism under basic conditions or in the presence of free radicals. However, in the presence of free radicals, acryloyl groups also react with each other via free radical chain polymerization resulting in an overall mixed mode polymerization: thiyl radicals react with acrylates (1:1) by step growth polymerization while acrylates react with each other by a chain growth polymerization.91-92 These reactions of thiols with acryloyl groups have been used to design drug delivery carriers.93-95 Vermonden and coworkers reported the reaction of thiols and methacrylates to form thermosensitive hydrogels for controlled delivery of the model therapeutic peptide bradykinin (BK).95 Briefly, a thermoresponsive macromer functionalized with acrylates (ABA-triblock polymer containing N-isopropylacrylamide) was reacted with thiolated hyaluronic acid under basic conditions to form hydrogels with different polymer concentrations and different mesh sizes. First order release kinetics were observed in vitro, indicating diffusion-mediated release of the encapsulated BK, and the rate of release was controlled by the network mesh size. In addition to acryloyl groups, thiols can also react with vinyl ethers, allyl ethers, and allyloxycarbonyls by radically-mediated thiol–ene reactions with a range of reported rates (Fig. 3C); to date, such hydrogels primarily have been used for tissue engineering applications.96-98
Unsaturated double bonds within maleate or fumarate readily react with thiol functional groups under basic pH by a Michael-type reaction; such reactions of thiols with maleates or fumarates recently have been utilized for drug delivery.99-100 For example, Shen and coworkers recently reported the use of thiol–ene hydrogels for the delivery of camptothecin, a hydrophobic anti-cancer drug.99 Poly[oligo(ethylene glycol) mercaptosuccinate] was reacted with poly[oligo(ethylene glycol) maleate] to form hydrogel films (thickness ∼10 to 35 nm). Camptothecin was conjugated to the polymer backbone with hydrolytically cleavable ester linkages for controlled drug release over short periods of time (∼24 hours).
3.4 Administration of thiol–ene hydrogels in vivo
Thiol–ene hydrogels can be administered in vivo by i) intramuscular101 or subcutaneous injection102-103 of reactive precursor solutions that form a gel in situ, ii) application of precursor solutions at desired sites during surgical procedures followed by in situ gelation,57 or iii) surgical implantation of pre-formed gels.104 For in situ formation, liquid precursor solutions need to polymerize rapidly under physiologically relevant conditions at the site of interest (e.g., < 1 min): if the rate of reaction is too slow, precursor solutions containing therapeutic drugs may diffuse into surrounding tissues, resulting in poor hydrogel formation and potential immunogenic response to unreacted monomers.105 However, if the rate of reaction is too rapid, premature gel formation may occur within the syringe for thiol-Michael type reactions, resulting in an uneven hydrogel distribution within the tissue and large network defects that produce undesirable release profiles. One approach to mitigating this issue is using a double-barrel syringe to control reactive precursor mixing and achieve rapid and consistent in situ polymerizations.102-103
3.5 Degradation of thiol–ene hydrogels relevant for controlled release of therapeutics
Degradation of thiol–ene hydrogels is advantageous for therapeutic delivery applications, allowing tunable release of entrapped cargo controlled based on the rate of network degradation. In addition, degradation of hydrogel-based drug carriers after the useful lifetime of the hydrogel in the body avoids the need for removal by surgery. Degradation has been achieved by incorporating degradable groups, such as water-degradable ester linkages,106-107 enzymatically-degradable linkages (especially peptides),108 and photodegradable linkages,109 into the polymer backbone, crosslinks, or pendant groups of the hydrogel (Fig. 1).28 The number and type of degradable linkages and the local environment surrounding the degradable moiety alter the rate of degradation, providing handles for controlling the release rate of the therapeutic. For example, the rate of degradation via ester hydrolysis in thiol–acrylate hydrogels is influenced by the number of carbons between the sulfide and ester groups (e.g., k = 0.08 days-1 for one carbon atom and 0.02 days-1 for two carbon atoms) and the pH of the surrounding buffer (e.g., k = 0.07 days-1 at pH 7.4 and k = 0.28 days-1 at pH 8).110
Incorporation of enzymatically degradable peptide crosslinkers is a versatile way to control hydrogel degradation in a stimuli-responsive manner. For example, Patterson and Hubbell evaluated the rate of cleavage of a variety of different matrix metalloproteinase (MMP)-sensitive peptides (including VPMS↓MRGG from combinatorial screening, GPQG↓IAGQ from collagen, and faster-degrading variant GPQG↓IWGQ) and subsequently studied the use of select crosslinkers for controlling the degradation of thiol-vinyl sulfone PEG-based hydrogels in response to MMP-1 and MMP-2.44 The rate of hydrogel degradation in response to one or both MMPs was found to be dependent on the sequence of the peptide crosslinker, where sequences were identified to achieve hydrogel degradation times ranging from 1 to 10 days when incubated with MMP-1 and MMP-2. These peptide sequences also have been incorporated within hydrogels formed by variety of thiol–ene reactions, including thiol-norbornene, -methacrylate, and -maleimide, where gel degradation times have been tuned from days to weeks based on sequence selection and network connectivity.80, 84, 111 Aryl-thiol based succinimide thioether linkages formed using thiol-maleimide chemistry can undergo degradation via retro-Michael type reactions in thiol-rich reducing microenvironments, such as that provided by glutathione found in elevated levels within tumors.77, 112 This approach has been used to tune the delivery of the anti-coagulant heparin over a range of time scales, from 3 days to 49 days, depending on the reactivity of the thiol and the strength of the reducing microenvironment.82 For a comprehensive review of degradable and stimuli-responsive hydrogels for biological applications, readers are referred to a recent review by Kharkar et al.28
4. Delivery of bioactive cargo in thiol–ene hydrogels
4.1 Small molecular drug and therapeutic peptide delivery
Thiol–ene hydrogels have been developed to control the release of small molecular drugs by rational design and control of hydrogel mesh size.108, 113-114 However, controlled delivery of small cargo molecules using thiol–ene hydrogels faces two major challenges: maintaining precise control over the mesh size during equilibrium swelling and incorporating hydrophobic drugs within highly hydrophilic hydrogels. In this section, we will highlight recent advances in material chemistry for the delivery of small molecular weight drugs and therapeutic peptides using thiol–ene hydrogels.
Swelling of the hydrogels has been controlled by careful selection of macromers. For example, the hydrophilicity of PEG is increased by increasing the number of repeating units of ethylene glycol (e.g., increasing chain length). However, to control the swelling of hydrogels and thus control hydrogel mesh size, an optimum balance between hydrophilic and hydrophobic content is needed. By selecting low molecular weight macromer precursors, hydrogel syneresis can be achieved, a process by which water is lost after formation reducing the hydrogel mesh size. With this approach, Langer and coworkers reported the synthesis of PEG-based thiol–ene hydrogels that can undergo syneresis in physiological conditions, reducing gel volume by ∼40%. They utilized these materials for the controlled release of methylprednisolone sodium succinate (MPSS), a glucocorticoid prodrug with anti-inflammatory and immunosuppressant properties.113 Hydrogels were prepared using acrylate end-functionalized PEG and a commercially available trifunctional thiol macromer (ethoxylated trimethylolpropane tri-3-mercaptopropionate). Incorporation of MPSS within the hydrogel suppressed the rate of hydrolysis of the ester linkage between methylprednisolone and succinate groups (k = 3.12 ± 0.10 × 10−3 h−1 encapsulated as compared to k = 5.98 ± 0.50 × 10−3 h−1 in PBS), demonstrating how hydrogels can increase therapeutic stability. Sustained release of MPSS was achieved over ∼20 days independent of the initial drug loading concentration, providing an opportunity to change the drug concentration based on individual patients' needs without changing the drug carrier volume.
Incorporation of small molecular hydrophobic drugs within hydrogels presents additional challenges owing to their limited water solubility. Introduction of hydrophobic units within the hydrogel network have been used to improve drug-loading efficiency and influence the swelling behavior of the hydrogels. For example, Harth and coworkers reported polycarbonate-based thiol–ene hydrogels for sustained delivery of paclitaxel, a chemotherapeutic drug used to treat ovarian and breast cancer.114 To incorporate the hydrophobic drug and optimize its residence time in the hydrogel-based drug carrier, polycarbonate was incorporated within the hydrogel as an additional hydrophobic backbone component. A three component system (polycarbonate, poly(ethylene oxide), and semi-branched polyglycidols) was used to optimize the balance between hydrophilicity and hydrophobicity, controlling hydrogel swelling behavior and drug release kinetics. Hydrogels that undergo ester hydrolysis were formed using light-mediated thiol–ene reactions by reacting allyl-functionalized polycarbonates with linear thiol-functionalized poly(ethylene oxide). Paclitaxel was encapsulated within the network during hydrogel formation with high loading efficiency (>98%), and release of ∼7% to ∼30% of the drug was observed over 7 days depending on initial hydrogel swelling.
As noted in section 2, control over release kinetics also can be achieved by covalently conjugating the drug molecule to the hydrogel backbone. The rate of release of cargo molecules in such cases depends on the rate of degradation of the covalent linkage between the drug and the network. Covalently-linked drug molecules can have preprogrammed drug release kinetics (e.g., different numbers of hydrolytically degradable ester linkages with known rates of hydrolysis)115-116 or can cleave in response to the local microenvironment; both approaches have been used in drug delivery systems. For example, Anseth and coworkers reported the use of thiol–ene click hydrogels for cell-mediated delivery of dexamethasone (Dex), an anti-inflammatory and immunosuppressant steroid drug (Fig. 4A).108 Dex was covalently linked to a peptide crosslinker (KCGPQG↓IAGQCK), which was susceptible to enzymatic cleavage by MMPs secreted by cells within tissues. The hydrogels were formed by reacting norbornene end-functionalized PEG with the thiol groups of MMP-degradable peptide crosslinkers, and the release of Dex was controlled by cleavage of tethers. Since Dex is known to promote osteogenic differentiation of human mesenchymal stem cells (hMSCs), released Dex bioactivity was monitored by measuring hMSC alkaline phosphatase (ALP) activity, a marker of early osteogenic differentiation.117 hMSCs encapsulated within hydrogels containing Dex-conjugated MMP-degradable peptide showed a significant increase in ALP activity compared to hydrogels containing Dex conjugated to a non-degradable linker, demonstrating both the release and bioactivity of Dex. While such an approach can be translated to deliver other small molecules using thiol–ene hydrogels, the biological activity of drugs or released drug fragments may change substantially after conjugation and release from the network.118 For each drug of interest, tests must be run to ensure the conjugated and released drug retains its desired biological activity.
Fig. 4. Controlled release of small molecular weight drugs and therapeutic peptides.
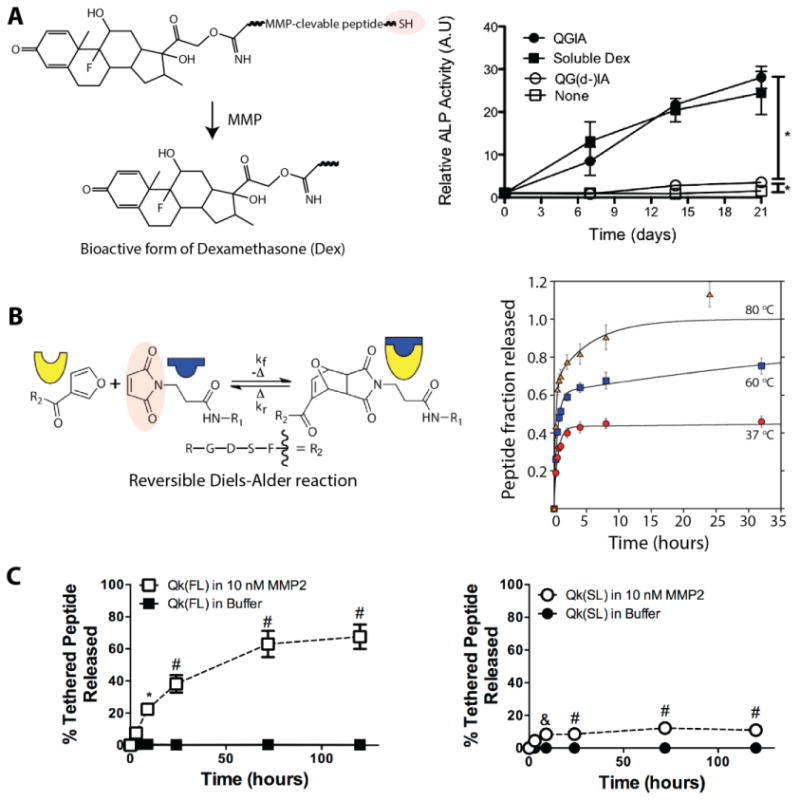
A) Dexamethasone (Dex) was covalently conjugated to the backbone of the hydrogel network using a thiol-functionalized MMP-sensitive peptide linker (KCGPQG↓IAGQCK). In the presence of cell-secreted MMP, the dexamethasone fragment (closed circle) was released and bioactive as demonstrated by enhanced alkaline phosphate (ALP) activity of encapsulated stem cells compared to the negative control (open circle, peptide sequence with substitution of D-isoleucine [QG(d-)I] making the linker MMP-insensitive and non-degradable). Adapted from Yang et al.108 with permission from Elsevier publishing. Copyright (2012). B) Furan-functionalized peptide (integrin-binding RGDS) was incorporated in thiol-maleimide hydrogels by a Diels-Alder reaction with excess maleimides. The release of peptide was controlled by a retro Diels-Alder reaction, which increases in rate with elevated temperature as demonstrated by the increasing fraction of peptide released at 37, 60, and 80 °C. Adapted from Koehler et al.123 with permission from ACS publishing. Copyright (2013). C) Toward promoting angiogenesis, a vascular endothelial growth factor mimetic peptide (Qk) was tethered to a non-degradable thiol–ene hydrogel using an MMP-cleavage linker, and temporal control of Qk release was achieved in vitro and in vivo by changing the MMP2-susceptible peptide tether (FL = Qk-PES↓LRAG-C-G, SL = Qk-VPLS↓LYSG-C-G). * p < 0.05; & p < 0.01; and # p < 0.0001 compared to buffer alone for each respective time point. Adapted from van Hove et al.124 with permission from Wiley publishing. Copyright (2015).
In the last decade, with significant advances in solid-phase synthesis and recombinant DNA technology, FDA-approved, peptide-based drugs have gained substantial importance for the treatment of human diseases.119-120 For example, peptide formulations such as cilengitide, taltirelin hydrate, and ziconotide acetate are in clinical trials for treating diseases associated with the central nervous system (i.e., spinocerebellar degeneration, ataxia, and severe chronic pain).121 Thiol–ene hydrogels have been used to deliver therapeutic peptides owing to their ability to maintain the bioactivity of cargo peptides and the ease of peptide encapsulation by facile crosslinking chemistries.122-123 Recently, Wang et al. demonstrated the release of a fluorescently-labeled library of model peptide drugs using thiol–ene hydrogels.122 Hydrogels were formed using a combination of a thiol-containing protein (ubiquitin-like domain tetramer of SATB1 with cysteine residues) and PEG-maleimide via a Michael-type crosslinking reaction. Controlled release of model peptide drugs (∼50% to 80% release) was achieved within approximately 24 hours, depending on the protein-peptide affinity interactions (i.e., a higher binding affinity peptide released more slowly). In another example, Koehler et al. reported the release of a covalently-linked peptide (integrin-binding RGDS) by reversible Diels-Alder reactions (Fig. 4B).123 PEG-based hydrogels were formed using thiol-maleimide chemisty, and temperature sensitive Diels-Alder linkages were used to conjugate the bioactive peptide functionalized with furan groups by reaction with excess maleimide groups. The release rate of peptide was tuned by the incorporation of different numbers of maleimide tethering sites (varied by incorporation of the monofunctional thiol) in the hydrogels: ∼45% to 60% of peptide was released by 35 hours depending on the number of maleimide tethering sites, and the percent release could be further varied with changes in local temperature (i.e., ∼40% cargo released at 37°C and ∼70% cargo released at 60°C). In another example, Benoit and coworkers reported thiol–ene hydrogels to control release of Qk, a proangiogenic peptide mimic of vascular endothelial growth factor using tissue-specific enzymatic activity.124 Qk was tethered to the hydrogel using enzymatically cleavable linkers, and variation in kcat/KM provided temporal control over release kinetics (i.e., ∼70% release was achieved using the sequence [Qk-PES↓LRAG-C-G] whereas only ∼15% release was achieved using the sequence [Qk-VPLS↓LYSG-C-G], Fig. 4C). These examples, along with those mentioned in sections 2 and 3, highlight approaches and methods for the design of thiol–ene hydrogels to deliver peptide-based therapeutics.
4.2 Therapeutic protein delivery
Protein therapeutics are in development to treat cancer, autoimmune diseases, protein deficiencies, and infectious diseases.125 The complexity of proteins allows them to complete tasks that small molecules cannot easily achieve, such as catalyzing an enzymatic reaction or inhibiting a biological process in a specific manner.126 For example, the antibody rituximab binds specifically to CD20, a cell-surface glycoprotein on B-cells, and has been approved to treat non-Hodgkin's lymphoma.127 However, their complexity leads to additional challenges, as protein structure must be maintained and proteolytic degradation must be avoided until the protein achieves the desired therapeutic effect either locally or systemically.126 Controlled release of therapeutic proteins from hydrogels offers the potential to maintain potent concentrations over extended periods of time and to limit premature degradation before the therapeutic reaches its desired target.128
As with small molecule drugs, the mesh size of hydrogel-based delivery vehicles is an important consideration; however, the mesh sizes of typical synthetic hydrogels and the size of therapeutic proteins are often on the same scale (approximately 1-10 nm). As a result, the protein often is encapsulated within the network and released by diffusion. If the protein size is smaller than the mesh size of the hydrogel, the protein can diffuse out of the hydrogel, usually at a slower rate than protein diffusion through water (and slower than uninhibited diffusion throughout the body). If the protein size is larger than the mesh size of the hydrogel, the protein is trapped until released by hydrogel degradation or a stimulus-triggered change in mesh size. Both approaches have proven useful depending on the desired time scale for therapeutic release as noted in examples below and discussed more broadly within a relevant review.129 In many cases, model proteins of different sizes, such as lysozyme (14 kDa) or BSA (66 kDa), are used. While these are useful and inexpensive models for protein release experiments, it is important to note that the bioactivity of therapeutic proteins might be compromised during loading or release from hydrogel delivery vehicles,130 and the compatibility of the hydrogel with a specific therapeutic protein of interest needs to be verified for each application.
PEG-based thiol–ene hydrogels are some of the most common types of hydrogels used in controlled release of therapeutic proteins. For example, Buwalda et al. crosslinked 8-arm PEG-poly(L-lactide)-acrylate block copolymers with multifunctional PEG-thiols by Michael addition for encapsulation and release of the model proteins lysozyme and albumin.131 Protein diffusion out of the hydrogel was slowed relative to diffusion in water and took place on time scales of days to weeks, demonstrating controlled release. Zustiak and Leach used an entirely PEG-based network formed by thiol-vinyl sulfone chemistry with hydrolytically degradable ester bonds for controlled protein release.39 Lysozyme encapsulated in the hydrogel diffused out within 18 hours, but the largest protein encapsulated, immunoglobulin, remained in the gel until complete hydrogel degradation at one week (Fig. 5A). In both of these cases, the diffusion rate was tuned by changing the number of ester bonds present in the network, affecting the degradation rate of the hydrogel and protein release.
Fig. 5. Controlled release of therapeutic proteins.
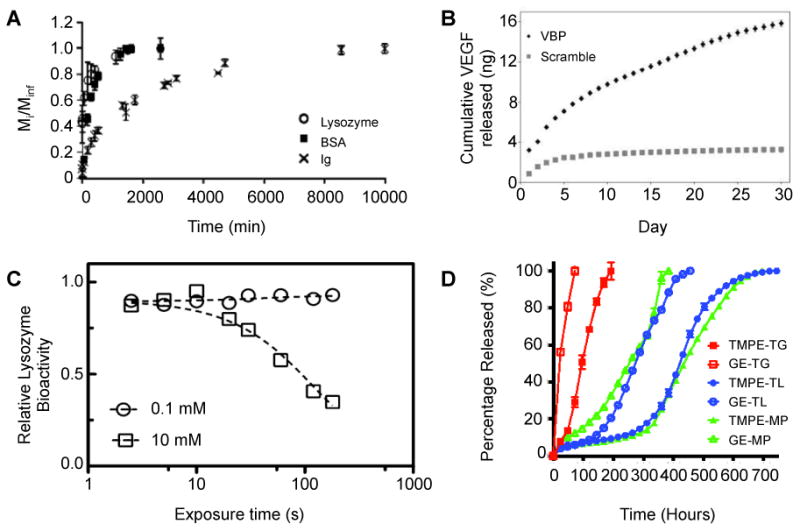
A) Proteins of various sizes were released from PEG-based hydrolytically-degradable thiol–vinyl sulfone hydrogels. Lysozyme, the smallest protein, was released in less than 24 hours, whereas Ig, the largest protein, was released upon complete gel degradation. Adapted from Zustiak and Leach39 with permission from Wiley. B) The growth factor VEGF was loaded into microspheres containing a peptide-based ligand derived from VEGF receptor 2 (VBP). VEGF was bound and released from the microspheres containing the VEGF receptor mimic, whereas microspheres containing a scrambled inert sequence exhibited a low level of release corresponding with random diffusion of VEGF into and out of the microspheres. Adapted from Impellitteri et al.133 with permission from Elsevier. C) Lysozyme was exposed to free radicals in conditions mimicking that of photoinitiated thiol–ene hydrogel polymerization. Higher radical concentrations would result from higher photoinitiator concentrations (10 mM), as compared to lower photoinitiator concentrations (0.1 mM), and from increased exposure time to light (plotted on x-axis). Increasing radical exposure decreased the bioactivity of lysozyme, indicating the importance of minimizing protein exposure to free radicals during gel formation. Bioactivity was increased upon introduction of reactive thiol and norbornene functional groups, supporting the protective effect of thiol–ene chemistry during hydrogel formation relative to acrylate homopolymerization (data not shown). Adapted from McCall and Anseth135 with permission from ACS Publishing. D) FITC-labeled ovalbumin was released from thiol-functionalized ethoxylated polyol ester/PEG-diacrylate based hydrogels of different compositions. By changing the ratio of the macromers within the hydrogel, the rate of release and the overall release profiles were tuned over a wide range of timescales. Adapted from Langer and coworkers42 with permission from Wiley.
While Michael-type reactions have enabled successful encapsulation and release of model proteins, gel formation in these systems begins upon mixing of all components. In contrast, photopolymerization allows for spatial and temporal control of polymerization, enabling the use of photolithography and micromolding for the formation of macroscale hydrogel geometries and microscale particles.132 Impellitteri et al. used a photopolymerized PEG-based thiol-norbornene reaction to encapsulate vascular endothelial growth factor (VEGF) in hydrogel microspheres containing a peptide mimic of the VEGF receptor (RTELNVGIDFNWEYP), serving as an affinity binding sequence that mediated VEGF release.133-134 The microspheres were formed in a water-in-water emulsion: one phase contained 4-arm PEG-norbornene, PEG-dithiol, and 0.5% w/w of the photoinitiator Irgacure 2959; the other phase contained 40 kDa dextran. The PEG macromers were crosslinked into stable microspheres by irradiation (low dose of UV light, 4 mW/cm2 for 8 minutes). Released VEGF was shown to be bioactive by its enhancement of human umbilical vein endothelial cell proliferation in vitro. (Fig. 5B).
McCall and Anseth compared the efficacy of photoinitiated thiol-norbornene or acrylate only reactions for the encapsulation of a model protein lysozyme within PEG-based hydrogels.135 At equal functional group concentrations (40 mM acrylate or 40 mM norbornene with 40 mM thiol) and the same photoinitiator concentration (1 mM LAP, 10 mW/cm2 at 365 nm for <5 min), the rapid thiol-norbornene system maintained a higher level of lysozyme activity after encapsulation and release than the slower (oxygen-inhibited) acrylate-based chain-growth system. This result was attributed to the protective effect of thiol–ene chemistry during hydrogel formation, which may limit the overall protein exposure to damaging free radicals in the gel-forming solution compared to acrylate homopolymerization (Fig. 5C).
Hydrogel degradation rates can be engineered to respond to microenvironment conditions (e.g., enzymes, reducing conditions, pH), allowing triggered release of a protein therapeutic, as discussed in section 3.5. For example, PEG-based hydrogels formed by thiol-norbornene photopolymerization with cysteine-functionalized, enzyme-sensitive peptide crosslinks were developed and used for protein release by Anseth and coworkers.136 The crosslinks contained a sequence (CGAAPV↓RGGGGC) that degrades in the presence of human neutrophil elastase (HNE), an enzyme upregulated at the site of inflammatory disease and injury.137 The model protein BSA was released from these hydrogels upon the application of human neutrophil elastase.136 Notably, no protein release was observed in the absence of the enzyme, demonstrating that the protein release was controlled by the degradation of the hydrogel in the presence of HNE.
In an alternative approach to localized protein release, Kharkar et al. formed PEG-based hydrogels sensitive to glutathione, which is elevated in tumors, using thiol-maleimide chemistry.83, 109 Hydrogels encapsulating BSA were formed with PEG-maleimide and PEG functionalized with different thiols that influenced hydrogel degradability. Approximately 40% of BSA was released from all compositions as a result of initial swelling; however, when the thiol-containing macromer contained an electron-withdrawing aryl group (PEG-4-mercaptophenylacetic acid), an additional 50% of the BSA was released in the presence of 10 mM glutathione over the course of 7 days. Negligible additional release was observed over 7 days for the compositions that were glutathione-insensitive, collectively demonstrating promise for tailoring protein release within tumor microenvironments.
While PEG-based materials are often commercially available and a number of them are FDA-approved,138 there is a limit to the tunability one can achieve simply by varying the end groups and functionality of PEG-based monomers. Langer and coworkers synthesized a library of thiol-functionalized ethoxylated polyol esters and reacted them with PEG-diacrylate to form a library of hydrogels with highly tailorable rates of degradation.42 By changing the thiol composition of the hydrogel from 100% thioglycolate-functionalized ethoxylated polyol ester to 100% thiolactate-functionalized ethoxylated polyol ester, the time for complete hydrogel degradation was varied from ∼ 12 days to ∼ 38 days. The release of many model macromolecules (3, 10, 20, and 40 kDa FITC-dextrans; FITC-labeled ovalbumin; and Alexa Fluor 647 IgG) was controlled by changing the chemical identity of the polyol esters and by using multiple polyol esters within a single hydrogel. For example, FITC-labeled ovalbumin was released from the hydrogel over the course of ∼ 4 days to ∼ 25 days, depending on which of the six thiol-containing polyol esters was used to form the hydrogel (Fig. 5D). In a follow-up work, trehalose, a disaccharide known for its protein-stabilizing properties, was incorporated within these hydrogels.139 Trehalose diacrylate was mixed with PEG-diacrylate at various ratios and reacted with thiol-functionalized polyol esters for a total of 25 wt% polymer in the final hydrogel. When 100% of the acrylate groups came from trehalose (i.e., 0% from PEG-diacrylate), nearly 100% of active horseradish peroxidase encapsulated in the gel was recovered within ∼ 4 days. In contrast, when 6.25% of the acrylate groups came from trehalose (i.e., 93.75% from PEG-diacrylate), less than 3% of the horseradish peroxidase was recovered in active form within ∼ 12 days. Covalent incorporation of trehalose also maintained the activity of two other model proteins, glucose oxidase and α-chymotrypsin. Although the mechanism of trehalose-mediated protein stabilization is not fully understood, trehalose is produced by many plants and animals in response to osmotic, high temperature, and other stresses, and it is thought to stabilize proteins by making protein unfolding more thermodynamically unfavorable.38 This concept may motivate additional approaches for stabilization of proteins in hydrogels.
Natural polymers and their derivatives also have been used to make hydrogels for protein release. For instance, Peng et al. reacted dextran-maleimide with thiol-containing azobenzene to form a photo-responsive dextran hydrogel.140 The cis/trans isomerization of azobenzene was used to release green fluorescent protein in a light-responsive manner (100 W at 365 nm). Beyond model proteins, hyaluronic acid-based hydrogels have been used to deliver two growth factors, stromal cell-derived factor-1 (SDF-1) and BMP-2, to promote hMSC infiltration and differentiation for bone regeneration in an animal model.141 These hydrogels were formed in situ by a Michael-type reaction between hyaluronic acid-maleimide, a dithiol MMP-degradable peptide (GCRDVPMS↓MRGGDRCG), and a thiol-functionalized RGD peptide (GCGYGRGDSPG). Limited release of these growth factors occurred by diffusion, but in the presence of the enzyme collagenase, the gel crosslinks were degraded and growth factors were released from the hydrogel. Complete degradation was observed within 9 days with 1 U ml-1 collagenase and within 4 days with 2 U ml-2 collagenase. Toward modulating myofibroblast activities and tissue regeneration after myocardial infarction, Burdick and coworkers also created injectable, MMP-degradable, hyaluronic acid-based hydrogel for delivery a recombinant tissue inhibitor of MMPs (rTIMP-3).142 The hydrogel was formed by conjugating a thiol- and hydrazide-functionalized MMP-degradable peptide (GGRMSMPV) to maleimide-functionalized hyaluronic acid. The hydrazide-functionalized MMP-degradable hyaluronic acid macromer was then reacted with aldehyde-functionalized hyaluronic acid to form a hydrogel in situ upon injection. Additionally, aldehyde-functionalized dextran sulphate was incorporated into the hydrogel to sequester encapsulated rTIMP-3 through electrostatic interactions. The hydrogel/TIMP-3 construct was delivered to a region of MMP overexpression in a porcine myocardial infarction model, resulting in significantly reduced MMP activity and adverse left ventricular remodeling. The study demonstrated the ability to locally release an MMP-inhibitor as needed in response to MMP-overexpressing pathologies.
4.3 Cell delivery
The delivery of mammalian cells to the site of injury or disease tissues is an emerging therapeutic approach, where the cell is able to exert positive effects on its surroundings by a variety of mechanisms including the deposition of ECM or the secretion of effector molecules (e.g., cytokines, chemokines) that can influence surrounding cell behavior.143 In this regard, hydrogels have been used to i) improve the viability or decrease the immunogenicity of transplanted cells, ii) provide mechanical integrity to the construct of transplanted cells, and iii) control the transport of cells and biomolecules. In these applications, the material can prevent the uncontrolled release of cells throughout the body, limit contact between the transplanted cells and the immune system, and support (or prevent) the diffusion of nutrients and waste to and from the transplanted cells.144 In particular, hydrogels formed by thiol–ene chemistry are advantageous for cell delivery owing to their often cytocompatible nature, their ability to form in neutral pH, aqueous, oxygen-rich solutions, their often rapid reaction kinetics, and reaction specificity and high yield.145 Additional information on cell delivery from hydrogels, including those formed by thiol–ene reactions, can be found in depth in several recent reviews.146-148
5. Conclusions and future perspectives
Significant progress has been made in the synthesis and characterization of thiol–ene hydrogels for the controlled delivery of therapeutics. Many of the systems overviewed here have shown promise with model cargo molecules, such as BSA, IgG, and lysozyme, and these studies provide well-defined formulations that now can be adapted for the delivery of specific therapeutics in vivo. However, several challenges need to be addressed toward broad clinical translation of thiol–ene hydrogels. The ability to retain the bioactivity of cargo molecules after exposure to the hydrogel environment, whether during formation or degradation, is one key area that further needs to be studied and addressed. For example, incorporation of natural polymers, such as silk, trehalose, or heparin, can be used to influence the stability of bioactive cargoes within otherwise synthetic hydrogels to provide protection during the encapsulation process.149 Maintaining precise control over cargo release is another persistent challenge that needs to be further explored. The ability to control release of cargo from thiol–ene hydrogels post-administration for patient-specific treatment regimes (e.g., triggered changes in material properties and release with light or enzymes) will be beneficial for efficient disease management. Furthermore, the light-directed control afforded by radical-mediated thiol–ene reactions may provide opportunities to create drug-releasing constructs of arbitrary shapes and sizes with rapidly evolving three-dimensional printing systems and other photolithographic processes, such as those recently demonstrated by DeSimone and coworkers.150 Taken together, thiol–ene hydrogels are promising therapeutic delivery vehicles owing to the range of facile, often biocompatible, chemistries available for their formation and modification providing tunability over material mechanical properties and cargo release profiles.
Acknowledgments
The authors gratefully acknowledge support, for related work in their laboratories, from the National Institutes of Health (NIH) Chemistry-Biology Interface program at the University of Delaware (NIH T32GM008550), the Delaware COBRE program with a grant from the National Institute of General Medical Sciences (NIGMS P20GM104316) from the NIH, the Burroughs Wellcome Fund, a grant from the California Institute for Regenerative Medicine (RN3-06460), and the University of Delaware Research Foundation. The authors thank Prof. Christopher Kloxin for helpful discussions on thiol–ene chemistry.
References
- 1.Hou JJC, Codamo J, Pilbrough W, Hughes B, Gray PP, Munro TP. New frontiers in cell line development: challenges for biosimilars. J Chem Technol Biotechnol. 2011;86(7):895–904. doi: 10.1002/jctb.2574. [DOI] [Google Scholar]
- 2.Audie J, Boyd C. The Synergistic Use of Computation, Chemistry and Biology to Discover Novel Peptide-Based Drugs: The Time is Right. Curr Pharm Design. 2011;16(5):567–582. doi: 10.2174/138161210790361425. [DOI] [PubMed] [Google Scholar]
- 3.Pirogova E, Istivan T, Gan E, Cosic I. Advances in methods for therapeutic peptide discovery, design and development. Curr Pharm Biotechnol. 2011;12(8):1117–27. doi: 10.2174/138920111796117436. [DOI] [PubMed] [Google Scholar]
- 4.Li N, Zhuang CY, Wang M, Sui CG, Pan WS. Low molecular weight chitosan-coated liposomes for ocular drug delivery: in vitro and in vivo studies. Drug Deliv. 2011;19(1):28–35. doi: 10.3109/10717544.2011.621994. [DOI] [PubMed] [Google Scholar]
- 5.Yang B, Geng SY, Liu XM, Wang JT, Chen YK, Wang YL, Wang JY. Positively charged cholesterol derivative combined with liposomes as an efficient drug delivery system, in vitro and in vivo study. Soft Matter. 2011;8(2):518–525. doi: 10.1039/c1sm06087b. [DOI] [Google Scholar]
- 6.Chen F, Hong H, Zhang Y, Valdovinos HF, Shi S, Kwon GS, Theuer CP, Barnhart TE, Cai W. In vivo tumor targeting and image-guided drug delivery with antibody-conjugated, radiolabeled mesoporous silica nanoparticles. ACS Nano. 2011;7(10):9027–39. doi: 10.1021/nn403617j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y, Ding X, Li J, Luo Z, Hu Y, Liu J, Dai L, Zhou J, Hou C, Cai K. Enzyme responsive drug delivery system based on mesoporous silica nanoparticles for tumor therapy in vivo. Nanotechnology. 2011;26(14):145102. doi: 10.1088/0957-4484/26/14/145102. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Q, Wang XL, Li PZ, Nguyen KT, Wang XJ, Luo Z, Zhang HC, Tan NS, Zhao YL. Biocompatible, Uniform, and Redispersible Mesoporous Silica 27 Nanoparticles for Cancer- Targeted Drug Delivery In Vivo. Adv Funct Mater. 2011;24(17):2450–2461. doi: 10.1002/adfm.201302988. [DOI] [Google Scholar]
- 9.Anajafi T, Mallik S. Polymersome-based drug-delivery strategies for cancer therapeutics. Ther Deliv. 2011;6(4):521–34. doi: 10.4155/tde.14.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Egli S, Nussbaumer MG, Balasubramanian V, Chami M, Bruns N, Palivan C, Meier W. Biocompatible functionalization of polymersome surfaces: a new approach to surface immobilization and cell targeting using polymersomes. Journal of the American Chemical Society. 2011;133(12):4476–83. doi: 10.1021/ja110275f. [DOI] [PubMed] [Google Scholar]
- 11.Ge X, Zhang Q, Cai Y, Duan S, Chen S, Lv N, Jin T, Chen Y, Yuan W. PEG-PCL-DEX polymersome-protamine vector as an efficient gene delivery system via PEG-guided self-assembly. Nanomedicine. 2011;9(8):1193–207. doi: 10.2217/nnm.13.83. [DOI] [PubMed] [Google Scholar]
- 12.Souza JG, Dias K, Silva SA, de Rezende LC, Rocha EM, Emery FS, Lopez RF. Transcorneal iontophoresis of dendrimers: PAMAM corneal penetration and dexamethasone delivery. J Controlled Release. 2015;200:115–24. doi: 10.1016/j.jconrel.2014.12.037. [DOI] [PubMed] [Google Scholar]
- 13.Tang Y, Li YB, Wang B, Lin RY, van Dongen M, Zurcher DM, Gu XY, Banaszak Holl MM, Liu G, Qi R. Efficient in vitro siRNA delivery and intramuscular gene silencing using PEG-modified PAMAM dendrimers. Mol Pharm. 2011;9(6):1812–21. doi: 10.1021/mp3001364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raphael AP, Primiero CA, Ansaldo AB, Keates HL, Soyer HP, Prow TW. Elongate microparticles for enhanced drug delivery to ex vivo and in vivo pig skin. J Controlled Release. 2011;172(1):96–104. doi: 10.1016/j.jconrel.2013.07.025. [DOI] [PubMed] [Google Scholar]
- 15.Boudou T, Kharkar P, Jing J, Guillot R, Pignot-Paintrand I, Auzely-Velty R, Picart C. Polyelectrolyte multilayer nanoshells with hydrophobic nanodomains for delivery of Paclitaxel. J Controlled Release. 2011;159(3):403–12. doi: 10.1016/j.jconrel.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seib FP, Pritchard EM, Kaplan DL. Self-assembling doxorubicin silk hydrogels for the focal treatment of primary breast cancer. Adv Funct Mater. 2011;23(1):58–65. doi: 10.1002/adfm.201201238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu J, Strandman S, Zhu JX, Barralet J, Cerruti M. Genipin-crosslinked catechol-chitosan mucoadhesive hydrogels for buccal drug delivery. Biomaterials. 2015;37:395–404. doi: 10.1016/j.biomaterials.2014.10.024. [DOI] [PubMed] [Google Scholar]
- 18.Lin CC, Anseth KS. PEG hydrogels for the controlled release of biomolecules in regenerative medicine. Pharm Res. 2011;26(3):631–43. doi: 10.1007/s11095-008-9801-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oh EJ, Park K, Kim KS, Kim J, Yang JA, Kong JH, Lee MY, Hoffman AS, Hahn SK. Target specific and long-acting delivery of protein, peptide, and nucleotide therapeutics using hyaluronic acid derivatives. J Controlled Release. 2011;141(1):2–12. doi: 10.1016/j.jconrel.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 20.Bhattarai N, Gunn J, Zhang M. Chitosan-based hydrogels for controlled, localized drug delivery. Adv Drug Deliv Rev. 2011;62(1):83–99. doi: 10.1016/j.addr.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 21.Liang Y, Kiick KL. Heparin-functionalized polymeric biomaterials in tissue engineering and drug delivery applications. Acta Biomater. 2011;10(4):1588–600. doi: 10.1016/j.actbio.2013.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yucel T, Lovett ML, Kaplan DL. Silk-based biomaterials for sustained drug delivery. J Controlled Release. 2014;190:381–97. doi: 10.1016/j.jconrel.2014.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gombotz WR, Wee SF. Protein release from alginate matrices. Adv Drug Deliv Rev. 2011;64(SUPPL):194–205. doi: 10.1016/j.addr.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 24.Elia R, Newhide DR, Pedevillano PD, Reiss GR, Firpo MA, Hsu EW, Kaplan DL, Prestwich GD, Peattie RA. Silk-hyaluronan-based composite hydrogels: a novel, securable vehicle for drug delivery. J Biomater Appl. 2011;27(6):749–62. doi: 10.1177/0885328211424516. [DOI] [PubMed] [Google Scholar]
- 25.Anumolu SS, DeSantis AS, Menjoge AR, Hahn RA, Beloni JA, Gordon MK, Sinko PJ. Doxycycline loaded poly(ethylene glycol) hydrogels for healing vesicant-induced ocular wounds. Biomaterials. 2011;31(5):964–74. doi: 10.1016/j.biomaterials.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee S, Tong X, Yang F. The effects of varying poly(ethylene glycol) hydrogel crosslinking density and the crosslinking mechanism on protein accumulation in three-dimensional hydrogels. Acta Biomater. 2011;10(10):4167–74. doi: 10.1016/j.actbio.2014.05.023. [DOI] [PubMed] [Google Scholar]
- 27.Zustiak SP, Leach JB. Hydrolytically degradable poly(ethylene glycol) hydrogel scaffolds with tunable degradation and mechanical properties. Biomacromolecules. 2011;11(5):1348–57. doi: 10.1021/bm100137q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kharkar PM, Kiick KL, Kloxin AM. Designing degradable hydrogels for orthogonal control of cell microenvironments. Chem Soc Rev. 2011;42(17):7335–72. doi: 10.1039/c3cs60040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Appel EA, Forster RA, Rowland MJ, Scherman OA. The control of cargo release from physically crosslinked hydrogels by crosslink dynamics. Biomaterials. 2011;35(37):9897–9903. doi: 10.1016/j.biomaterials.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 30.Lin CC. Recent advances in crosslinking chemistry of biomimetic poly(ethylene glycol) hydrogels. RSC Adv. 2011;5(50):39844–398583. doi: 10.1039/C5RA05734E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kolb HC, Finn MG, Sharpless KB. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew Chem Int Ed Engl. 2011;40(11):2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 32.Nimmo CM, Owen SC, Shoichet MS. Diels-Alder Click cross-linked hyaluronic acid hydrogels for tissue engineering. Biomacromolecules. 2011;12(3):824–30. doi: 10.1021/bm101446k. [DOI] [PubMed] [Google Scholar]
- 33.Hoyle CE, Bowman CN. Thiol-ene click chemistry. Angew Chem Int Ed Engl. 2011;49(9):1540–73. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 34.Hoyle CE, Lowe AB, Bowman CN. Thiol-click chemistry: a multifaceted toolbox for small molecule and polymer synthesis. Chem Soc Rev. 2011;39(4):1355–87. doi: 10.1039/b901979k. [DOI] [PubMed] [Google Scholar]
- 35.Xi WX, Scott TF, Kloxin CJ, Bowman CN. Click Chemistry in Materials Science. Adv Funct Mater. 2011;24(18):2572–2590. doi: 10.1002/adfm.201302847. [DOI] [Google Scholar]
- 36.Impellitteri NA, Toepke MW, Levengood SKL, Murphy WL. Specific VEGF sequestering and release using peptide-functionalized hydrogel microspheres. Biomaterials. 2011;33(12):3475–3484. doi: 10.1016/j.biomaterials.2012.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nelson DL, Lehninger AL, Cox MM. Lehninger principles of biochemistry. Macmillan; 2008. [Google Scholar]
- 38.Jain NK, Roy I. Effect of trehalose on protein structure. Protein Sci. 2011;18(1):24–36. doi: 10.1002/pro.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zustiak SP, Leach JB. Characterization of protein release from hydrolytically degradable poly (ethylene glycol) hydrogels. Biotechnol Bioeng. 2011;108(1):197–206. doi: 10.1002/bit.22911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shih H, Lin CC. Photo-click hydrogels prepared from functionalized cyclodextrin and poly (ethylene glycol) for drug delivery and in situ cell encapsulation. Biomacromolecules. 2011;16(7):1915–1923. doi: 10.1021/acs.biomac.5b00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang Y, Chen J, Deng C, Suuronen EJ, Zhong Z. Click hydrogels, microgels and nanogels: emerging platforms for drug delivery and tissue engineering. Biomaterials. 2011;35(18):4969–85. doi: 10.1016/j.biomaterials.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 42.O'Shea TM, Aimetti AA, Kim E, Yesilyurt V, Langer R. Synthesis and characterization of a library of in-situ curing, nonswelling ethoxylated polyol thiol-ene hydrogels for tailorable macromolecule delivery. Adv Mater. 2011;27(1):65–72. doi: 10.1002/adma.201403724. [DOI] [PubMed] [Google Scholar]
- 43.He C, Zhuang X, Tang Z, Tian H, Chen X. Stimuli-sensitive synthetic polypeptide-based materials for drug and gene delivery. Adv Healthc Mater. 2011;1(1):48–78. doi: 10.1002/adhm.201100008. [DOI] [PubMed] [Google Scholar]
- 44.Patterson J, Hubbell JA. Enhanced proteolytic degradation of molecularly engineered PEG hydrogels in response to MMP-1 and MMP-2. Biomaterials. 2011;31(30):7836–45. doi: 10.1016/j.biomaterials.2010.06.061. [DOI] [PubMed] [Google Scholar]
- 45.Van Hove AH, Beltejar MJ, Benoit DS. Development and in vitro assessment of enzymatically-responsive poly(ethylene glycol) hydrogels for the delivery of therapeutic peptides. Biomaterials. 2011;35(36):9719–30. doi: 10.1016/j.biomaterials.2014.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seliktar D. Designing cell-compatible hydrogels for biomedical applications. Science. 2011;336(6085):1124–8. doi: 10.1126/science.1214804. [DOI] [PubMed] [Google Scholar]
- 47.Nguyen MK, Alsberg E. Bioactive factor delivery strategies from engineered polymer hydrogels for therapeutic medicine. Prog Polym Sci. 2011;39(7):1236–1265. doi: 10.1016/j.progpolymsci.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arslan M, Gevrek TN, Sanyal R, Sanyal A. Fabrication of poly(ethylene glycol)-based cyclodextrin containing hydrogels via thiol-ene click reaction. Eur Polym J. 2015;62:426–434. doi: 10.1016/j.eurpolymj.2014.08.018. [DOI] [Google Scholar]
- 49.Kade MJ, Burke DJ, Hawker CJ. The power of thiol-ene chemistry. J Polym Sci A Polym Chem. 2011;48(4):743–750. [Google Scholar]
- 50.Alge DL, Azagarsamy MA, Donohue DF, Anseth KS. Synthetically tractable click hydrogels for three-dimensional cell culture formed using tetrazine-norbornene chemistry. Biomacromolecules. 2011;14(4):949–53. doi: 10.1021/bm4000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lowe AB. Thiol-ene “click” reactions and recent applications in polymer and materials synthesis. Polym Chem. 2011;1(1):17–36. [Google Scholar]
- 52.Nguyen KT, West JL. Photopolymerizable hydrogels for tissue engineering applications. Biomaterials. 2011;23(22):4307–14. doi: 10.1016/s0142-9612(02)00175-8. [DOI] [PubMed] [Google Scholar]
- 53.Toepke MW, Impellitteri NA, Theisen JM, Murphy WL. Characterization of Thiol-Ene Crosslinked PEG Hydrogels. Macromol Mater Eng. 2011;298(6):699–703. doi: 10.1002/mame.201200119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fairbanks BD, Schwartz MP, Bowman CN, Anseth KS. Photoinitiated polymerization of PEG-diacrylate with lithium phenyl-2,4,6-trimethylbenzoylphosphinate: polymerization rate and cytocompatibility. Biomaterials. 2011;30(35):6702–7. doi: 10.1016/j.biomaterials.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shih H, Lin CC. Visible-Light-Mediated Thiol-Ene Hydrogelation Using Eosin-Y as the Only Photoinitiator. Macromol Rapid Commun. 2011;34(3):269–273. doi: 10.1002/marc.201200605. [DOI] [PubMed] [Google Scholar]
- 56.Cramer NB, Scott JP, Bowman CN. Photopolymerizations of thiol-ene polymers without photoinitiators. Macromolecules. 2011;35(14):5361–5365. doi: 10.1021/ma0200672. [DOI] [Google Scholar]
- 57.Terella A, Mariner P, Brown N, Anseth K, Streubel SO. Repair of a calvarial defect with biofactor and stem cell–embedded polyethylene glycol scaffold. JAMA Facial Plast Surg. 2011;12(3):166–171. doi: 10.1001/archfacial.2010.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mustafa F, Jaafar M. Comparison of wavelength-dependent penetration depths of lasers in different types of skin in photodynamic therapy. Indian J Phys. 2011;87(3):203–209. [Google Scholar]
- 59.Elisseeff J, Anseth K, Sims D, McIntosh W, Randolph M, Langer R. Transdermal photopolymerization for minimally invasive implantation. Proc Natl Acad Sci U S A. 2011;96(6):3104–3107. doi: 10.1073/pnas.96.6.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maverakis E, Miyamura Y, Bowen MP, Correa G, Ono Y, Goodarzi H. Light, including ultraviolet. J Autoimmun. 2011;34(3):J247–J257. doi: 10.1016/j.jaut.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Müller G, Zalibera M, Gescheidt G, Rosenthal A, Santiso-Quinones G, Dietliker K, Grützmacher H. Simple One-Pot Syntheses of Water-Soluble Bis (acyl) phosphane Oxide Photoinitiators and Their Application in Surfactant-Free Emulsion Polymerization. Macromol Rapid Commun. 2011;36(6):553–557. doi: 10.1002/marc.201400743. [DOI] [PubMed] [Google Scholar]
- 62.Benedikt S, Wang J, Markovic M, Moszner N, Dietliker K, Ovsianikov A, Grützmacher H, Liska R. Highly efficient water-soluble visible light photoinitiators. J Polym Sci A Polym Chem. 2015 [Google Scholar]
- 63.Li Z, Torgersen J, Ajami A, Mühleder S, Qin X, Husinsky W, Holnthoner W, Ovsianikov A, Stampfl J, Liska R. Initiation efficiency and cytotoxicity of novel water-soluble two-photon photoinitiators for direct 3D microfabrication of hydrogels. RSC Advances. 2011;3(36):15939–15946. [Google Scholar]
- 64.Hermanson GT. Bioconjugate techniques. Academic press; 2013. [Google Scholar]
- 65.Place ES, Nair R, Chia HN, Szulgit G, Lim EH, Stevens MM. Latent TGF-beta hydrogels for cartilage tissue engineering. Adv Healthc Mater. 2011;1(4):480–4. doi: 10.1002/adhm.201200038. [DOI] [PubMed] [Google Scholar]
- 66.Shah SS, Kim M, Cahill-Thompson K, Tae G, Revzin A. Micropatterning of bioactive heparin-based hydrogels. Soft Matter. 2011;7(7):3133–3140. doi: 10.1039/c0sm00771d. [DOI] [Google Scholar]
- 67.Chan JW, Yu B, Hoyle CE, Lowe AB. Convergent synthesis of 3-arm star polymers from RAFT-prepared poly (N, N-diethylacrylamide) via a thiol–ene click reaction. Chem Commun. 2008(40):4959–4961. doi: 10.1039/b813438c. [DOI] [PubMed] [Google Scholar]
- 68.Sletten EM, Bertozzi CR. Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew Chem Int Ed Engl. 2011;48(38):6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Absar S, Nahar K, Choi S, Ahsan F, Yang VC, Kwon YM. Serum albumin-protamine conjugate for biocompatible platform for targeted delivery of therapeutic macromolecules. J Biomed Mater Res A. 2011;102(8):2481–90. doi: 10.1002/jbm.a.34916. [DOI] [PubMed] [Google Scholar]
- 70.Zhuang Y, Su Y, Peng Y, Wang D, Deng H, Xi X, Zhu X, Lu Y. Facile fabrication of redox-responsive thiol-containing drug delivery system via RAFT polymerization. Biomacromolecules. 2011;15(4):1408–18. doi: 10.1021/bm500018s. [DOI] [PubMed] [Google Scholar]
- 71.Lutolf MP, Hubbell JA. Synthesis and physicochemical characterization of end-linked poly(ethylene glycol)-co-peptide hydrogels formed by Michael-type addition. Biomacromolecules. 2011;4(3):713–22. doi: 10.1021/bm025744e. [DOI] [PubMed] [Google Scholar]
- 72.McGann CL, Levenson EA, Kiick KL. Resilin-Based Hybrid Hydrogels for Cardiovascular Tissue Engineering. Macromolecules. 2011;214(2):203–213. doi: 10.1002/macp.201200412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peng G, Wang J, Yang F, Zhang S, Hou J, Xing W, Lu X, Liu C. In situ formation of biodegradable dextran-based hydrogel via Michael addition. J Appl Polym Sci. 2011;127(1):577–584. [Google Scholar]
- 74.Lutolf MR, Weber FE, Schmoekel HG, Schense JC, Kohler T, Muller R, Hubbell JA. Repair of bone defects using synthetic mimetics of collagenous extracellular matrices. Nat Biotechnol. 2011;21(5):513–518. doi: 10.1038/nbt818. [DOI] [PubMed] [Google Scholar]
- 75.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: engineering cell-invasion characteristics. Proc Natl Acad Sci U S A. 2011;100(9):5413–8. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nair DP, Podgórski M, Chatani S, Gong T, Xi W, Fenoli CR, Bowman CN. The thiol-Michael addition click reaction: a powerful and widely used tool in materials chemistry. Chem Mater. 2011;26(1):724–744. [Google Scholar]
- 77.Baldwin AD, Kiick KL. Tunable degradation of maleimide-thiol adducts in reducing environments. Bioconjug Chem. 2011;22(10):1946–53. doi: 10.1021/bc200148v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Knight P. Hydrolysis of p-NN'-phenylenebismaleimide and its adducts with cysteine. Implications for cross-linking of proteins. Biochem J. 2011;179(1):191–7. doi: 10.1042/bj1790191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jensen LH, Renodon-Corniere A, Wessel I, Langer SW, Sokilde B, Carstensen EV, Sehested M, Jensen PB. Maleimide is a potent inhibitor of topoisomerase II in vitro and in vivo: A new mode of catalytic inhibition. Mol Pharm. 2011;615(5):1235–1243. 1235. doi: 10.1124/Mol.61. [DOI] [PubMed] [Google Scholar]
- 80.Phelps EA, Enemchukwu NO, Fiore VF, Sy JC, Murthy N, Sulchek TA, Barker TH, Garcia AJ. Maleimide cross-linked bioactive PEG hydrogel exhibits improved reaction kinetics and cross-linking for cell encapsulation and in situ delivery. Adv Mater. 2011;24(1):64–70. 2. doi: 10.1002/adma.201103574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fu Y, Kao WJ. In situ forming poly(ethylene glycol)-based hydrogels via thiol-maleimide Michael-type addition. J Biomed Mater Res A. 2011;98(2):201–11. doi: 10.1002/jbm.a.33106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Baldwin AD, Kiick KL. Reversible maleimide-thiol adducts yield glutathione-sensitive poly(ethylene glycol)-heparin hydrogels. Polym Chem. 2011;4(1):133–143. doi: 10.1039/C2PY20576A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kharkar PM, Kloxin AM, Kiick KL. Dually degradable click hydrogels for controlled degradation and protein release. J Mater Chem B. 2011;2(34):5511–5521. doi: 10.1039/c4tb00496e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS. A VersatileSynthetic Extracellular Matrix Mimic via Thiol-Norbornene Photopolymerization. Adv Mater. 2011;21(48):5005–10. doi: 10.1002/adma.200901808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hao Y, Shih H, Munoz Z, Kemp A, Lin CC. Visible light cured thiol-vinyl hydrogels with tunable degradation for 3D cell culture. Acta Biomater. 2011;10(1):104–14. doi: 10.1016/j.actbio.2013.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cramer NB, Reddy SK, O'Brien AK, Bowman CN. Thiol-ene photopolymerization mechanism and rate limiting step changes for various vinyl functional group chemistries. Macromolecules. 2011;36(21):7964–7969. doi: 10.1021/ma034667s. [DOI] [Google Scholar]
- 87.Gramlich WM, Kim IL, Burdick JA. Synthesis and orthogonal photopatterning of hyaluronic acid hydrogels with thiol-norbornene chemistry. Biomaterials. 2011;34(38):9803–11. doi: 10.1016/j.biomaterials.2013.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lin CC, Ki CS, Shih H. Thiol-Norbornene Photoclick Hydrogels for Tissue Engineering Applications. J Appl Polym Sci. 2011;132(8) doi: 10.1002/app.41563. doi:Artn 41563 10.1002/App.41563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Alge DL, Anseth KS. T hiol-X Reactions in Tissue. Thiol-X Chemistries in Polymer and Materials Science. 2013;6:165. [Google Scholar]
- 90.Zhang Y, Chan HF, Leong KW. Advanced materials and processing for drug delivery: the past and the future. Adv Drug Deliv Rev. 2011;65(1):104–20. doi: 10.1016/j.addr.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Salinas CN, Anseth KS. Mixed mode thiol-acrylate photopolymerizations for the synthesis of PEG-peptide hydrogels. Macromolecules. 2011;41(16):6019–6026. doi: 10.1021/ma800621h. [DOI] [Google Scholar]
- 92.Rydholm AE, Bowman CN, Anseth KS. Degradable thiol-acrylate photopolymers: polymerization and degradation behavior of an in situ forming biomaterial. Biomaterials. 2011;26(22):4495–506. doi: 10.1016/j.biomaterials.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 93.Dan K, Pan R, Ghosh S. Aggregation and pH responsive disassembly of a new acid-labile surfactant synthesized by thiol-acrylate Michael addition reaction. Langmuir. 2011;27(2):612–7. doi: 10.1021/la104445h. [DOI] [PubMed] [Google Scholar]
- 94.Hiemstra C, van der Aa LJ, Zhong ZY, Dijkstra PJ, Feijen J. Rapidly in situ-forming degradable hydrogels from dextran thiols through michael addition. Biomacromolecules. 2011;8(5):1548–1556. doi: 10.1021/bm061191m. [DOI] [PubMed] [Google Scholar]
- 95.Censi R, Fieten PJ, di Martino P, Hennink WE, Vermonden T. In Situ Forming Hydrogels by Tandem Thermal Gelling and Michael Addition Reaction between Thermosensitive Triblock Copolymers and Thiolated Hyaluronan. Macromolecules. 2011;43(13):5771–5778. doi: 10.1021/ma100606a. [DOI] [PubMed] [Google Scholar]
- 96.Sawicki LA, Kloxin AM. Design of thiol-ene photoclick hydrogels using facile techniques for cell culture applications. Biomater Sci. 2011;2(11):1612–1626. doi: 10.1039/c4bm00187g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yang T, Malkoch M, Hult A. Sequential interpenetrating poly (ethylene glycol) hydrogels prepared by UV-initiated thiol–ene coupling chemistry. J Polym Sci A Polym Chem. 2011;51(2):363–371. [Google Scholar]
- 98.Yang T, Long H, Malkoch M, Kristofer Gamstedt E, Berglund L, Hult A. Characterization of well-defined poly (ethylene glycol) hydrogels prepared by thiol-ene chemistry. J Polym Sci A Polym Chem. 2011;49(18):4044–4054. [Google Scholar]
- 99.Zhu W, Xiong L, Wang H, Zha G, Du H, Li X, Shen Z. Sustained drug release from an ultrathin hydrogel film. Polym Chem. 2011;6(40):7097–7099. [Google Scholar]
- 100.Zhu W, Gao L, Luo Q, Gao C, Zha G, Shen Z, Li X. Metal and light free “click” hydrogels for prevention of post-operative peritoneal adhesions. Polym Chem. 2011;5(6):2018–2026. [Google Scholar]
- 101.Xu K, Cantu DA, Fu Y, Kim J, Zheng X, Hematti P, Kao WJ. Thiol-ene Michael-type formation of gelatin/poly (ethylene glycol) biomatrices for three-dimensional mesenchymal stromal/stem cell administration to cutaneous wounds. Acta biomaterialia. 2011;9(11):8802–8814. doi: 10.1016/j.actbio.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Baldwin AD, Robinson KG, Militar JL, Derby CD, Kiick KL, Akins RE. In situ crosslinkable heparin-containing poly (ethylene glycol) hydrogels for sustained anticoagulant release. J Biomed Mater Res A. 2011;100(8):2106–2118. doi: 10.1002/jbm.a.34050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Smeets NM, Bakaic E, Patenaude M, Hoare T. Injectable and tunable poly (ethylene glycol) analogue hydrogels based on poly (oligoethylene glycol methacrylate) Chem Commun. 2011;50(25):3306–3309. doi: 10.1039/c3cc48514e. [DOI] [PubMed] [Google Scholar]
- 104.Jo S, Kim S, Cho TH, Shin E, Hwang SJ, Noh I. Effects of recombinant human bone morphogenic protein-2 and human bone marrow-derived stromal cells on in vivo bone regeneration of chitosan–poly (ethylene oxide) hydrogel. J Biomed Mater Res A. 2011;101(3):892–901. doi: 10.1002/jbm.a.34354. [DOI] [PubMed] [Google Scholar]
- 105.Bakaic E, Smeets NM, Hoare T. Injectable hydrogels based on poly (ethylene glycol) and derivatives as functional biomaterials. RSC Advances. 2011;5(45):35469–35486. [Google Scholar]
- 106.Rehmann MS, Garibian AC, Kloxin AM. Hydrolytically degradable thiol–ene hydrogels for protein release. Macromol Symp. 2011;329(1):58–65. doi: 10.1002/masy.201200133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shih H, Lin CC. Cross-linking and degradation of step-growth hydrogels formed by thiol-ene photoclick chemistry. Biomacromolecules. 2011;13(7):2003–12. doi: 10.1021/bm300752j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang C, Mariner PD, Nahreini JN, Anseth KS. Cell-mediated delivery of glucocorticoids from thiol-ene hydrogels. J Controlled Release. 2011;162(3):612–8. doi: 10.1016/j.jconrel.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kharkar PM, Kiick KL, Kloxin AM. Design of thiol-and light-sensitive degradable hydrogels using Michael-type addition reactions. Polym Chem. 2011;6(31):5565–5574. doi: 10.1039/C5PY00750J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rydholm AE, Anseth KS, Bowman CN. Effects of neighboring sulfides and pH on ester hydrolysis in thiol-acrylate photopolymers. Acta Biomater. 2011;3(4):449–55. doi: 10.1016/j.actbio.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Khetan S, Guvendiren M, Legant WR, Cohen DM, Chen CS, Burdick JA. Degradation-mediated cellular traction directs stem cell fate in covalently crosslinked three-dimensional hydrogels. Nat Mater. 2011;12(5):458–465. doi: 10.1038/nmat3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Balendiran GK, Dabur R, Fraser D. The role of glutathione in cancer. Cell Biochem Funct. 2011;22(6):343–52. doi: 10.1002/cbf.1149. [DOI] [PubMed] [Google Scholar]
- 113.Pritchard CD, O'Shea TM, Siegwart DJ, Calo E, Anderson DG, Reynolds FM, Thomas JA, Slotkin JR, Woodard EJ, Langer R. An injectable thiol-acrylate poly(ethylene glycol) hydrogel for sustained release of methylprednisolone sodium succinate. Biomaterials. 2011;32(2):587–97. doi: 10.1016/j.biomaterials.2010.08.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Stevens DM, Rahalkar A, Spears B, Gilmore K, Douglas E, Muthukumar M, Harth E. Semibranched polyglycidols as “fillers” in polycarbonate hydrogels to tune hydrophobic drug release. Polym Chem. 2015;6:1096–1102. [Google Scholar]
- 115.Chun C, Lee SM, Kim CW, Hong KY, Kim SY, Yang HK, Song SC. Doxorubicin-polyphosphazene conjugate hydrogels for locally controlled delivery of cancer therapeutics. Biomaterials. 2011;30(27):4752–62. doi: 10.1016/j.biomaterials.2009.05.031. [DOI] [PubMed] [Google Scholar]
- 116.Cuggino JC, Contreras CB, Jimenez-Kairuz A, Maletto BA, Alvarez Igarzabal CI. Novel poly(NIPA-co-AAc) functional hydrogels with potential application in drug controlled release. Mol Pharm. 2011;11(7):2239–49. doi: 10.1021/mp400615n. [DOI] [PubMed] [Google Scholar]
- 117.Atmani H, Chappard D, Basle MF. Proliferation and differentiation of osteoblasts and adipocytes in rat bone marrow stromal cell cultures: effects of dexamethasone and calcitriol. J Cell Biochem. 2011;89(2):364–72. doi: 10.1002/jcb.10507. [DOI] [PubMed] [Google Scholar]
- 118.Testa B. Prodrugs: bridging pharmacodynamic/pharmacokinetic gaps. Curr Opin Chem Biol. 2011;13(3):338–44. doi: 10.1016/j.cbpa.2009.04.620. [DOI] [PubMed] [Google Scholar]
- 119.Lu Y, Yang J, Sega E. Issues related to targeted delivery of proteins and peptides. The AAPS journal. 2011;8(3):E466–78. doi: 10.1208/aapsj080355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bruno BJ, Miller GD, Lim CS. Basics and recent advances in peptide and protein drug delivery. Ther Deliv. 2011;4(11):1443–67. doi: 10.4155/tde.13.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Synthetic therapeutic peptides: science and market. Drug Discov Today. 2011;15(1-2):40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 122.Wang H, Han A, Cai Y, Xie Y, Zhou H, Long J, Yang Z. Multifunctional biohybrid hydrogels for cell culture and controlled drug release. Chem Commun. 2011;49(67):7448–50. doi: 10.1039/c3cc43711f. [DOI] [PubMed] [Google Scholar]
- 123.Koehler KC, Anseth KS, Bowman CN. Diels-Alder mediated controlled release from a poly(ethylene glycol) based hydrogel. Biomacromolecules. 2011;14(2):538–47. doi: 10.1021/bm301789d. [DOI] [PubMed] [Google Scholar]
- 124.Van Hove AH, Antonienko E, Burke K, Brown E, 3rd ; Benoit, D.S. Temporally Tunable, Enzymatically Responsive Delivery of Proangiogenic Peptides from Poly(ethylene glycol) Hydrogels. Adv Healthc Mater. 2015 doi: 10.1002/adhm.201500304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kontos S, Hubbell JA. Drug development: longer-lived proteins. Chem Soc Rev. 2011;41(7):2686–95. doi: 10.1039/c2cs15289d. [DOI] [PubMed] [Google Scholar]
- 126.Vermonden T, Censi R, Hennink WE. Hydrogels for protein delivery. Chem Rev. 2011;112(5):2853–88. doi: 10.1021/cr200157d. [DOI] [PubMed] [Google Scholar]
- 127.Smith MR. Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene. 2011;22(47):7359–68. doi: 10.1038/sj.onc.1206939. [DOI] [PubMed] [Google Scholar]
- 128.Siegel RA, Rathbone MJ. Fundamentals and Applications of Controlled Release Drug Delivery. Springer; 2012. Overview of controlled release mechanisms; pp. 19–43. [Google Scholar]
- 129.Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Hydrogels in biology and medicine: from molecular principles to bionanotechnology. Adv Mater. 2011;18(11):1345. [Google Scholar]
- 130.Hennink W, Van Nostrum CF. Novel crosslinking methods to design hydrogels. Adv Drug Deliv Rev. 2012;64:223–236. doi: 10.1016/s0169-409x(01)00240-x. [DOI] [PubMed] [Google Scholar]
- 131.Buwalda SJ, Dijkstra PJ, Feijen J. In Situ Forming Poly(ethylene glycol)- Poly(L-lactide) Hydrogels via Michael Addition: Mechanical Properties, Degradation, and Protein Release. Macromol Chem Phys. 2011;213(7):766–775. doi: 10.1002/macp.201100640. [DOI] [Google Scholar]
- 132.Chung BG, Lee KH, Khademhosseini A, Lee SH. Microfluidic fabrication of microengineered hydrogels and their application in tissue engineering. Lab Chip. 2011;12(1):45–59. doi: 10.1039/c1lc20859d. [DOI] [PubMed] [Google Scholar]
- 133.Impellitteri NA, Toepke MW, Lan Levengood SK, Murphy WL. Specific VEGF sequestering and release using peptide-functionalized hydrogel microspheres. Biomaterials. 2011;33(12):3475–84. doi: 10.1016/j.biomaterials.2012.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Piossek C, Schneider-Mergener J, Schirner M, Vakalopoulou E, Germeroth L, Thierauch KH. Vascular endothelial growth factor (VEGF) receptor II-derived peptides inhibit VEGF. J Biol Chem. 2011;274(9):5612–9. doi: 10.1074/jbc.274.9.5612. [DOI] [PubMed] [Google Scholar]
- 135.McCall JD, Anseth KS. Thiol-Ene Photopolymerizations Provide a Facile Method To Encapsulate Proteins and Maintain Their Bioactivity. Biomacromolecules. 2011;13(8):2410–2417. doi: 10.1021/bm300671s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Aimetti AA, Machen AJ, Anseth KS. Poly(ethylene glycol) hydrogels formed by thiol-ene photopolymerization for enzyme-responsive protein delivery. Biomaterials. 2011;30(30):6048–54. doi: 10.1016/j.biomaterials.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Aimetti AA, Tibbitt MW, Anseth KS. Human neutrophil elastase responsive delivery from poly(ethylene glycol) hydrogels. Biomacromolecules. 2011;10(6):1484–9. doi: 10.1021/bm9000926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Alconcel SN, Baas AS, Maynard HD. FDA-approved poly (ethylene glycol)–protein conjugate drugs. Polym Chem. 2011;2(7):1442–1448. [Google Scholar]
- 139.O'Shea TM, Webber MJ, Aimetti AA, Langer R. Covalent Incorporation of Trehalose within Hydrogels for Enhanced Long-Term Functional Stability and Controlled Release of Biomacromolecules. Adv Healthc Mater. 2015;4:1802–1812. doi: 10.1002/adhm.201500334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Peng K, Tomatsu I, Kros A. Light controlled protein release from a supramolecular hydrogel. Chem Commun. 2011;46(23):4094–6. doi: 10.1039/c002565h. [DOI] [PubMed] [Google Scholar]
- 141.Holloway JL, Ma H, Rai R, Hankenson KD, Burdick JA. Synergistic Effects of SDF-1alpha and BMP-2 Delivery from Proteolytically Degradable Hyaluronic Acid Hydrogels for Bone Repair. Macromol Biosci. 2015 doi: 10.1002/mabi.201500178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Purcell BP, Lobb D, Charati MB, Dorsey SM, Wade RJ, Zellars KN, Doviak H, Pettaway S, Logdon CB, Shuman JA, Freels PD, Gorman JH, 3rd, Gorman RC, Spinale FG, Burdick JA. Injectable and bioresponsive hydrogels for on-demand matrix metalloproteinase inhibition. Nat Mater. 2011;13(6):653–61. doi: 10.1038/nmat3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang C, Varshney RR, Wang DA. Therapeutic cell delivery and fate control in hydrogels and hydrogel hybrids. Adv Drug Deliv Rev. 2011;62(7-8):699–710. doi: 10.1016/j.addr.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 144.Schmidt JJ, Rowley J, Kong HJ. Hydrogels used for cell-based drug delivery. J Biomed Mater Res A. 2011;87(4):1113–1122. doi: 10.1002/jbm.a.32287. [DOI] [PubMed] [Google Scholar]
- 145.Alge DL, Anseth KS. Thiol-X Reactions in Tissue Engineering. Lowe AB, Bowman CN, editors. Thiol-X Chemistries in Polymer and Materials Science. 2013:165–194. [Google Scholar]
- 146.Schmidt JJ, Rowley J, Kong HJ. Hydrogels used for cell-based drug delivery. J Biomed Mater Res A. 2011;87(4):1113–22. doi: 10.1002/jbm.a.32287. [DOI] [PubMed] [Google Scholar]
- 147.Wan AC, Ying JY. Nanomaterials for in situ cell delivery and tissue regeneration. Adv Drug Deliv Rev. 2011;62(7-8):731–40. doi: 10.1016/j.addr.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 148.Noth U, Rackwitz L, Steinert AF, Tuan RS. Cell delivery therapeutics for musculoskeletal regeneration. Adv Drug Deliv Rev. 2011;62(7-8):765–83. doi: 10.1016/j.addr.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 149.Zhang J, Pritchard E, Hu X, Valentin T, Panilaitis B, Omenetto FG, Kaplan DL. Stabilization of vaccines and antibiotics in silk and eliminating the cold chain. Proc Natl Acad Sci U S A. 2011;109(30):11981–6. doi: 10.1073/pnas.1206210109. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 150.Tumbleston JR, Shirvanyants D, Ermoshkin N, Janusziewicz R, Johnson AR, Kelly D, Chen K, Pinschmidt R, Rolland JP, Ermoshkin A, Samulski ET, DeSimone JM. Additive manufacturing. Continuous liquid interface production of 3D objects. Science. 2011;347(6228):1349–52. doi: 10.1126/science.aaa2397. [DOI] [PubMed] [Google Scholar]