

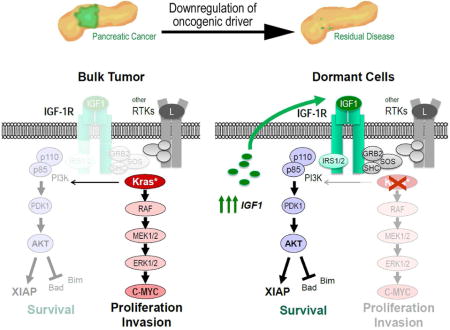
Abstract
Mutant KRAS and c-MYC are oncogenic drivers and rational therapeutic targets for the treatment of pancreatic cancer. While tumor growth and homeostasis is largely dependent on these oncogenes, a few residual cancer cells are able to survive the ablation of mutant KRAS and c-MYC. By performing a genome-wide gene expression analysis of in vivo-derived bulk tumor cells and residual cancer cells lacking the expression of mutant KRAS or c-MYC, we have identified an increase in autocrine IGF1/AKT signaling as a common survival mechanism in dormant cancer cells. The pharmacological inhibition of IGF-1R reduces residual disease burden and cancer recurrence, suggesting this molecular pathway is crucial for the survival of cancer cells in the absence of the primary oncogenic drivers.
Graphical abstract
Introduction
Although effective therapies are still lacking to treat pancreatic ductal adenocarcinoma (PDAC), significant advances have been made over the past two decades in the identification of molecular pathways that play key roles in the initiation of this malignancy. Oncogenic driver mutations in the KRAS gene have been validated to play a pivotal role in the genesis of premalignant lesions as well as PDAC progression (Bardeesy and DePinho, 2002; Hruban et al., 2000). More recent studies in genetic models have demonstrated that expression of mutant KRAS is equally required for the maintenance of primary and metastatic lesions (Collins et al., 2012a; Collins et al., 2012b; Ying et al., 2012). A critical downstream effector of mutant KRAS is the c-MYC proto-oncogene, which is suggested to play a pivotal role in the metabolism of pancreatic cancer cells (Ying et al., 2012). We found that c-MYC is upregulated in all human pancreatic cancer cell lines as well as in many primary human PDAC cases and in KRAS-induced pancreatic tumors in mice (Lin et al., 2013). Amplifications of the c-MYC locus are more often associated with adenosquamous carcinomas and seem to be linked to a very dismal prognosis (Witkiewicz et al., 2015). In line with this observation, we demonstrated in genetically engineered mice that upregulation of c-MYC in pancreatic progenitors was entirely sufficient to induce metastatic pancreatic cancer after a short latency (Lin et al., 2013). Moreover, expression of c-MYC was required for cancer cell survival at primary and metastatic sites regardless of the expression of wildtype p53 or a loss-of-heterozygosity of Cdkn2a. Collectively, the genetic studies in pancreatic cancer models expressing mutant KRAS and c-MYC provide a sound rationale for the development of targeted therapies against oncogenic drivers to treat early and advanced stages of pancreatic cancer.
Despite what appeared to be a complete remission of primary and metastatic pancreatic neoplasms in response to the downregulation of c-MYC, we were able to detect residual cancer cells within the unremodeled tumor-associated fibrous stroma (Lin et al., 2013). We used a genetic cell-fate mapping technique in combination with orthotopic transplantation into wildtype recipients to validate that the residual disease was not caused by a de novo transformation of normal cells. These genetically labeled dormant cancer cells lacked expression of endogenous and exogenous c-MYC, and they were not proliferating or undergoing cell death. In comparison to the parental bulk tumor cells, a significantly larger subset of dormant cancer cells expressed cancer stem cell markers, and they exhibited a higher rate of engraftment into secondary recipients (Lin et al., 2013; Lin et al., 2014). The swift emergence of invasive cancer following re-expression of c-MYC provided experimental evidence that dormant cancer cells were the cellular basis for disease recurrence. Residual disease was also observed in a KRAS-dependent PDAC model (Collins et al., 2012b), and it is therefore evident that cancer stem cell dormancy will likely present a lingering challenge in the development of targeted therapies to effectively treat PDAC (Lin et al., 2014). This view was substantiated in a more recent study by Viale et al. (2014) that shows that explanted pancreatic cancer cells that remained viable following the ablation of oncogenic KRAS depend on oxidative phosphorylation for their survival. In conclusion, all studies that have been performed in reversible pancreatic cancer models highlighted the importance for the development of adjuvant therapeutic strategies in addition to targeting oncogenic drivers to effectively eradicate residual cancer cells and to prevent disease recurrence.
In an effort to identify common, cancer cell-intrinsic molecular pathways that mediate residual disease following the ablation of oncogenic drivers, we performed a genome-wide gene expression analysis of in vivo-derived bulk tumor cells and dormant cancer cells that survived the ablation of oncogenic KRAS or a downregulation of exogenous c-MYC. The results presented here illuminate the biological significance of the activation of an IGF1/IGF-1R autocrine signaling loop and the downstream activation of AKT as a common mechanism that promotes cancer cell survival and dormancy following the specific ablation of oncogenic KRAS or c-MYC. Co-targeting IGF-1R signaling along with the downregulation of the oncogenic drivers resulted in a reduction in minimal residual disease, which might be a crucial step in developing more effective therapeutic strategies for the treatment of PDAC.
Results
Quiescent pancreatic cancer cells that survive the downregulation of oncogenic KRAS expression are the cellular basis for cancer recurrence upon reactivation of the oncogene
We used a Cre recombinase-induced and ligand-controlled transgene expression system (Lin et al., 2013) to generate genetically engineered mice that express mutant KRAS (KRASG12D) in a doxycycline (Dox)-controlled manner in the pancreas (Fig. 1A). In contrast to previous studies (Collins et al., 2012b; Ying et al., 2012), the upregulation of this oncogene in our model occurs in the absence of Dox (i.e., Tet-OFF system), and the downstream activation of MAP kinases can be swiftly repressed through administration of the ligand (Fig. 1B, 1C). Another unique feature of this genetic system is the predominant expression of the transactivator (tTA)-driven responder genes in the ductal epithelium of the pancreas (Lin et al., 2013). GFP-labeled pancreatic intraepithelial neoplastic (PanIN) lesions were readily detectable in mice that co-expressed KRASG12D and nuclear GFP under control of the tTA (Fig. 1D). Although the initiation and maintenance of hyper-proliferative PanIN lesions were dependent on the expression of mutant KRAS, only 3 out of 11 mice that were maintained for up to 18 month developed primary PDAC. A progression of these lesions into metastatic PDAC was, however, not observed in these animals. The introduction of only one Cdkn2a knockout allele initiated the development of PDAC in all KRASG12D-expressing animals after more than one year, and complete deficiency in Cdkn2a accelerated significantly the carcinogenic process (Fig. 1E). As expected, the liver and lung were the main sites for metastatic growth in diseased mice (Fig. 1F). Interestingly, induction of acute or chronic pancreatitis seemed to have no discernable effect on the genesis of primary and metastatic tumors in this model (not shown). The molecular analysis of primary pancreatic cancers from aging Cdkn2a heterozygous knockout mice revealed that tumorigenesis was associated with the loss of the Cdkn2a wildtype allele and an upregulation of MDM2 (Fig. 1G, 1H). This confirms that the extended tumor-free survival in Cdkn2a heterozygous knockouts mice is a consequence of the tumor suppressive functions of p16Ink4a and p19Arf encoded by the remaining Cdkn2a wildtype allele. Similar to previous reports (Collins et al., 2012b; Ying et al., 2012), the Dox-controlled suppression of mutant KRAS expression in our model led to an induction of cell death and a swift regression of pancreatic ductal lesions and invasive adenocarcinomas (Suppl. Fig. S1). Hence, the survival of the vast majority of primary and metastatic cancer cells was still dependent on the sustained expression of mutant KRAS in the absence of the tumor suppressive functions of p16Ink4a and p19Arf.
Figure 1. Expression of oncogenic KRASG12D in required for the onset and maintenance of primary and metastatic pancreatic ductal adenocarcinoma (PDAC).
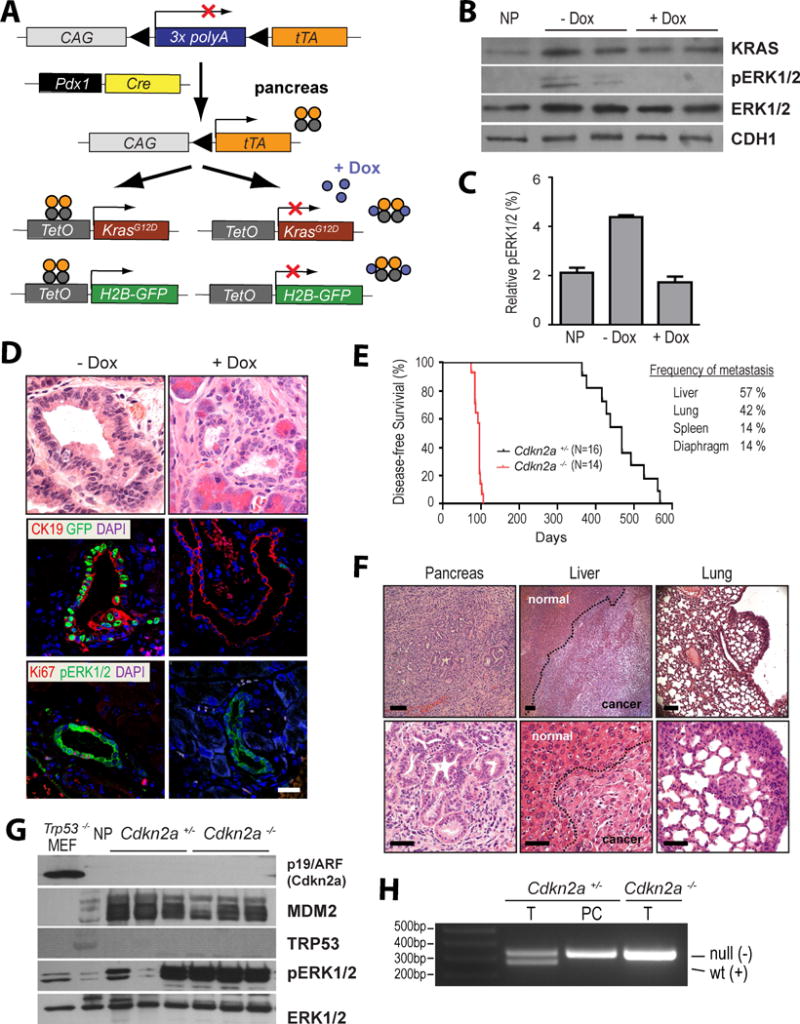
A. Generation of a genetically engineered mouse model that permits a temporally and spatially controlled expression of oncogenic KRAS and a H2B-GFP reporter in the pancreas in a doxycycline (Dox)-repressible manner (TET-OFF). B. Total KRAS protein expression as well as downstream activation of ERK1/2 in triple transgenic mice before and after administration of Dox; NP, normal pancreas of a littermate control that lacks the TetO-KRASG12D transgene. C. Quantitative analysis of relative ERK1/2 activation as determined by capillary electrophoresis of triplicate sets of tissues shown in panel B on a ProteinSimple NanoPro 1000 machine. D. H&E stained histological sections and immunofluorescent labeling of CK19, GFP, Ki67, and pERK1/2 in pancreatic specimens of 3-month-old Pdx1-Cre, CAG-LSL-tTA, TetO-KRASG12D, TetO-H2B-GFP quadruple transgenic mice prior to (-Dox) and after 7 days of Dox treatment; bar represents 50 μm. E. Kaplan–Meier survival plot of mice that conditionally express mutant KRAS in a Cdkn2a heterozygous (Cdkn2a+/−) or homozygous knockout background (Cdkn2a−/−). The table illustrates the relative incidence of metastatic lesions in these mice. F. Representative images of H&E stained sections of primary PDAC as well as metastatic lesions in liver and lung. G. Expression of p19/Arf, MDM2, and p53 as well as activation of ERK1/2 in primary pancreatic tumors of mice that express oncogenic KRAS in the Cdkn2a heterozygous and homozygous knockout background; NP, normal pancreas of a wildtype mouse. Trp53-deficient mouse embryonic fibroblasts (MEF) served as additional control. H. PCR assay to validate the loss of exon 2 of Cdkn2a in the DNA of purified pancreatic cancer cells (PC) of a mouse that was genotyped as a heterozygous Cdkn2a knockout (Cdkn2a+/−) using DNA from the tail (T). Tail DNA from a homozygous Cdkn2a knockout (Cdkn2a−/−) was used as a control.
The widespread expression of KRAS in the pancreata of transgenic mice caused numerous PanINs and multifocal disease onset which made it virtually impossible to discriminate dormant cancer cells from preneoplastic cells following the downregulation of oncogenic KRAS. We therefore employed a Cre/loxP–based cell-fate-labeling method in combination with cancer cell transplantation (Fig. 2A) to study the entire process of engraftment and growth of GFP-labeled tumor cells as well as regression and residual disease in response to the downregulation of exogenous KRAS. Small GFP-labeled pancreatic tumor fragments of approximately 1 mm3 in size were isolated from the central regions of primary cancers of transgenic donors and transplanted orthotopically into the pancreata of 8 to 12-week-old wildtype recipients. All recipient mice subsequently developed unifocal, GFP-positive pancreatic tumors (Fig. 2B) that were histologically indistinguishable from the donor tissues (Suppl. Fig. S2A). Despite macroscopic remission of primary pancreatic cancers following the ablation of mutant KRAS and expression of pERK (Suppl. Fig. S2B), we were able to detect GFP-positive residual cancer cells under the fluorescent stereoscope (Fig. 2B) or in histological sections (Fig. 2C). In contrast to bulk tumor cells prior to the administration of Dox, residual cancer cells did not proliferate and did not undergo programmed cell death (Fig. 2C). These cells remained in a dormant state for a prolonged period, and re-expression of oncogenic KRAS following the withdrawal of Dox led to a swift recurrence of GFP-positive cancers (Fig. 2D). This suggested that, identical to our previous findings in a c-MYC-associated pancreatic cancer model, few KRASG12D-induced cancer cells were capable of surviving the downregulation of the oncogene and remained dormant. These cells can serve as a reservoir for cancer recurrence upon reactivation of the transforming oncogene.
Figure 2. Pancreatic cancer cell dormancy following tumor regression in response to the ablation of oncogenic KRAS is a mediator for disease recurrence.
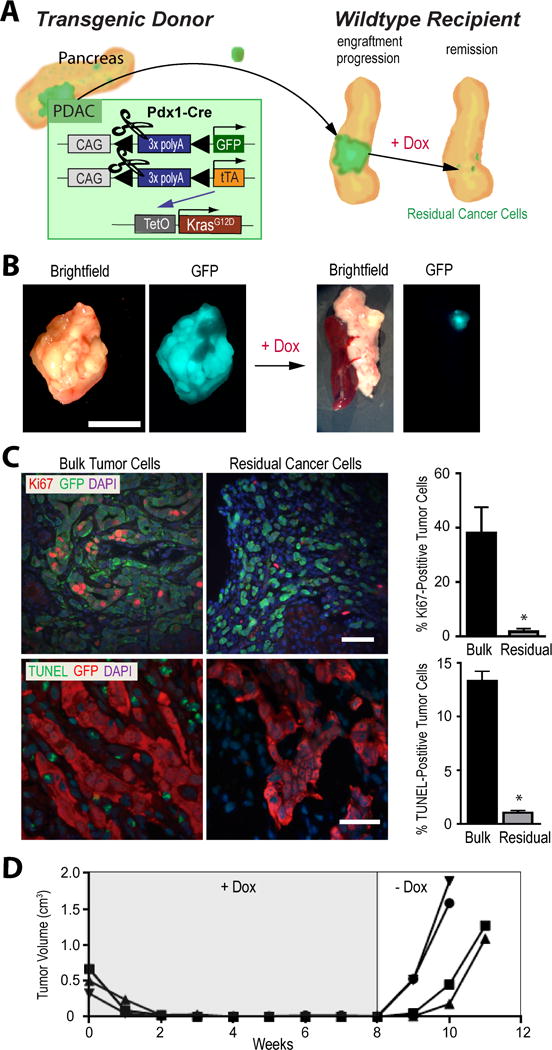
A. Schematic outline of the cell-fate-labeling method in combination with cancer cell transplantation to study tumor regression and cancer cell dormancy in the absence of mutant KRAS expression. B. Stereoscopic bright-field and GFP fluorescent images of pancreatic bulk tumors and residual cancer tissues before and four weeks after doxycycline (Dox)-mediated downregulation of mutant KRAS; bar represents 1 cm. C. Immunofluorescence staining of Ki67 and GFP as well as TUNEL labeling of GFP-positive pancreatic cancer cells on histological sections of bulk tumors and residual cancer cells (four weeks after downregulation of mutant KRAS); bar represents 50 μm. Diagrams show the relative number of proliferating and apoptotic cells within both tissue types D. Growth suppression of individual pancreatic tumors in wildtype recipient mice that lack oncogenic KRAS expression. Re-expression of mutant KRAS following withdrawal of Dox led to swift disease recurrence.
Ablation of oncogenic KRAS leads to a compensatory increase in IGF-1R signaling and activation of AKT in dormant cancer cells
To gain insight into the underlying mechanism(s) by which residual cancer cells evade cell death in the absence of KRASG12D as the oncogenic driver, we performed a comprehensive, genome-wide RNA-Seq analysis on GFP-expressing bulk tumor cells and isolated GFP-positive residual cancer cells. The remarkable fidelity of the RNA-Seq method also allowed us to confirm the sustained downregulation of exogenous KRAS on the transcriptional level (Suppl. Fig. S3A, S3B). The Gene Set Enrichment Analyses (GSEA) of the RNA-Seq data of three bulk tumor tissues and three residual cancer samples shown in Suppl. Fig. S3C exemplifies the deregulated expression of genes that are associated with MAP kinase signaling and pancreatic cancer. Residual cancer cells also exhibited the anticipated switch in the expression of cell cycle regulatory genes including c-MYC (Suppl. Fig. S3D), and we identified differentially expressed gene sets related to certain metabolic pathways such as those that control the biosynthesis of N- and O-glycan (Suppl. Fig. S3C). We did not observe a significantly upregulated expression of gene sets associated with the electron transport chain and peroxisomal β-oxidation that were previously identified in a gene array-based transcriptome analysis of cultured tumor spheres that were conditionally deficient in mutant KRAS (Viale et al., 2014). However, one of the most consistently deregulated sets of genes in the in vivo-derived residual cancer cells with a P value of less than 0.001 (FDR<0.001; NES=2.34) were those that cluster with the IGF1 pathway (Fig. 3A). Despite a reduced expression of Igf1r transcripts, the significantly elevated mRNA expression of its ligand led to a sustained higher activation of this signaling pathway. Using immunohistochemistry and immunoblot analysis, we confirmed that, compared to bulk tumor cells, residual cancer cells showed a significantly elevated phosphorylation of the IGF-1 receptor (IGF-1R) as well as downstream activation of AKT (Fig. 3B and 3C), which is known to exert its pro-survival functions at the pre- and post-mitochondrial level. A higher expression of the pro-apoptotic proteins BIM and BAD in residual cancer cells was compensated by an AKT-mediated increase in their functionally inactive (i.e., phosphorylated) isoforms. Moreover, we observed a significantly elevated expression of the X-linked inhibitor of apoptosis protein (XIAP) and its more stable phosphorylated form in residual cancer cells in vivo. As anticipated, residual cancer cells exhibited a lower expression of cleaved Caspases, which corresponded to the observed reduction in apoptosis and confirmed their quiescent characteristics (Fig. 2C).
Figure 3. Dormant cancer cells lacking oncogenic KRAS exhibit a compensatory increase in IGF-1R/AKT signaling.
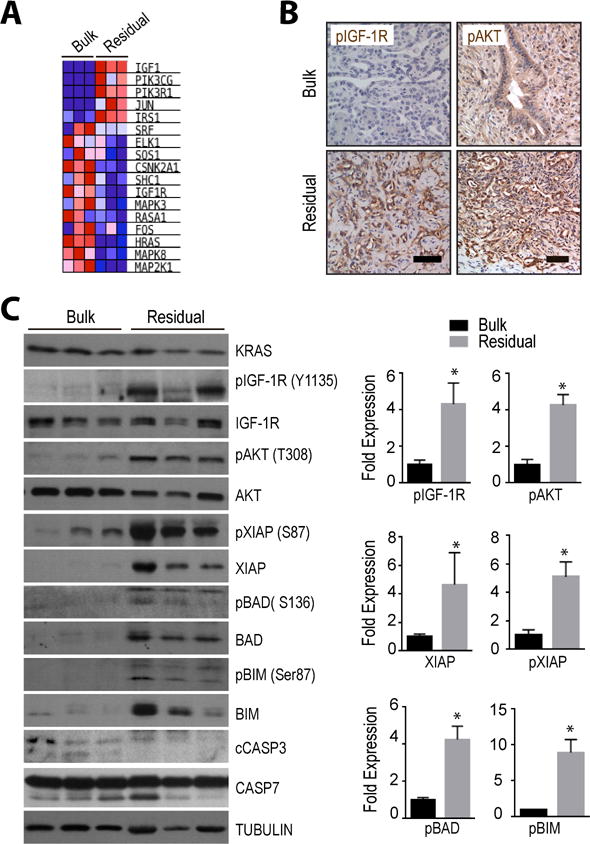
A. Heat map of genes that cluster with IGF1 signaling and that exhibit a deregulated expression in residual cancer cells following compared to pancreatic bulk tumors. B. Immunohistochemical staining of the activated IGF1 receptor (pIGF-1R) as well as pAKT in oncogenic KRAS-expressing bulk tumor cells and in residual cancer cells two weeks after the ablation of the oncogene; bar represents 50 μm. C. Immunoblot analysis comparing the expression and activation of IGF-1R and AKT as well as levels of downstream regulators of cell survival and cell death between bulk tumors and residual cancer tissues; cCASP3, cleaved Caspase 3. Bar graphs show Image-J quantification of selected protein bands. All data are represented as mean ± SEM; * represents P<0.05.
Next, we established two GFP-positive cell lines from bulk tumors to assess whether the compensatory increase in IGF-1R/AKT signaling following the downregulation of mutant KRAS is a cancer cell intrinsic phenomenon. Although the total levels of KRAS were only marginally elevated prior to Dox treatment, the phosphorylation of ERK1/2 was significantly reduced in response to the specific inhibition of the oncogene (Suppl. Fig. S4A and S4B). The downregulation of mutant KRAS led to the anticipated decelerated growth, but more importantly cells acquired a more differentiated epithelial morphology (Suppl. Fig. S4C). Unlike bulk tumor cells in vivo, however, the downregulation of mutant KRAS in 2D cultures did not induce widespread apoptosis, which is validated by the lack of expression of cleaved Caspase 3 and PARP. These cells were quite similar to residual cancer cells in vivo where the prolonged inhibition of oncogenic KRAS over a period of 7 days resulted in a compensatory increase in IGF-1R activation as well as a sustained phosphorylation of AKT and its downstream effectors BIM and the ribosomal protein S6 kinase (Suppl. Fig. S4D). In summary, the collective findings on in vivo-derived bulk and residual tumor cells as well as cultured pancreatic cancer cells revealed that ablation of oncogenic KRAS expression resulted in a synchronous increase in the activation of the IGF-1/AKT signaling axis.
Inhibition of IGF-1R signaling reduces the survival of dormant cancer cells and delays cancer recurrence upon re-expression of oncogenic KRAS
To address whether increased signaling through the IGF1 receptor tyrosine kinase and the AKT pathway plays a role in cancer cell dormancy, we used a pharmacological approach to selectively inhibit IGF-1R activation in residual cancer cells (Fig. 4A). The treatment of diseased animals following the downregulation of KRAS and tumor remission with linsitinib (OSI-906) for 5 days effectively blocked IGF-1R and AKT activation in dormant cancer cells (Fig. 4B), and in turn, this resulted in an increase in the relative number of apoptotic cells within the GFP-positive residual cancer cell population (Fig. 4C and Suppl. Fig. S5A). As a biological readout to assess the effectiveness of the linsitinib-mediated eradication of dormant cancer cells, we examined the recurrence of tumors for a period of 4 weeks following the withdrawal of doxycycline and re-expression of mutant KRAS (Fig. 4D). We observed a significant delay in the reemergence of cancers within the pancreata of recipient mice, suggesting that the survival of a substantial subset of dormant cancer cells in the absence of oncogenic KRAS was dependent on IGF-1R signaling. In a control experiment, we treated bulk tumors that express oncogenic KRAS (i.e., without Dox administration) with linsitinib or vehicle control for 5 days, which corresponded to the same treatment period of animals with residual disease that received Dox. As anticipated, the growth and survival of bulk tumor cells that lack a high activation of the IGF-1R was not affected by the treatment with linsitinib (Suppl. Fig. S5B). The collective results suggest that an inhibition of IGF-1R results in a selective ablation of dormant cancer cells that also lack mutant KRAS expression.
Figure 4. Co-inhibition of IGF-1R signaling reduces minimal residual disease following the targeted downregulation of oncogenic KRAS.
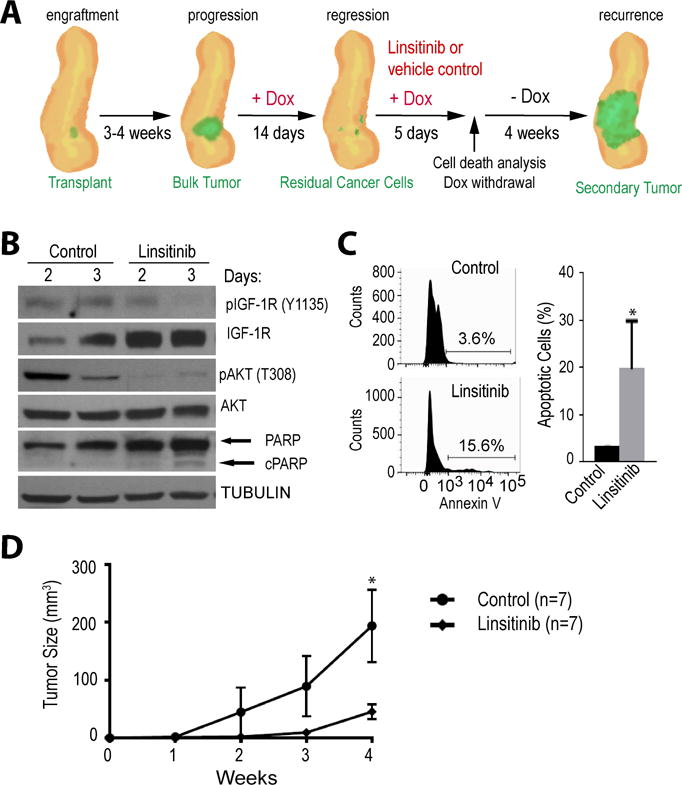
A. Schematic outline of the experimental design to study tumor regression in response to mutant KRAS ablation and the pharmacological inhibition of IGF-1R with linsitinib to selectively eliminate residual cancer cells. Tumor recurrence following oncogenic KRAS re-expression was being used as readout for the presence of remaining dormant cancer cells. B. Immunoblot analysis to validate the pharmacological inhibition of IGF-1R and AKT signaling as well as activation of cleaved PARP (cPARP) in residual cancer cells in vivo. C. Flow cytometric analysis of GFP-labeled residual cancer cells and quantitative assessment of dying cells in vivo in response to IGF-1R inhibition. D. Tumor growth curves comparing cancer recurrence following reactivation of mutant KRAS between controls and animals that were treated with the IGF1-R inhibitor.
Elevated levels of active IGF-1R and AKT mediate cancer cell dormancy in a c-MYC-induced reversible pancreatic tumor model
The vast majority but not all human pancreatic ductal adenocarcinomas carry mutations in KRAS. We have demonstrated previously that the c-MYC protein is overexpressed in an array of human pancreatic cancer cell lines including BXPC-3 and Hs766T cells that are wildtype for KRAS (Lin et al., 2013). To identify common molecular pathways that mediate pancreatic cancer cell dormancy following the ablation of oncogenic drivers other than KRAS, we carried out a genome-wide RNA-Seq analysis on pancreatic tumors that originated in response to the overexpression of c-MYC. The comparison of enriched gene sets between c-MYC overexpressing bulk tumor cells and their descendant residual cancer cells after the downregulation of c-MYC showed the expected decrease in the expression of genes associated with basal transcription factors, DNA replication, and the cell cycle (Suppl. Fig. S6A). A closer examination of individual genes revealed that residual cancer cells from the c-MYC model show some striking similarities to gene expression profiles of dormant cancer cells from the KRAS-induced tumor model (Suppl. Fig. S6B). Specifically, we observed identical changes in expression of genes related to IGF-1R signaling (e.g. Igf1, Jun, Pik3cg, Pik3ca). Ablation of c-MYC also led to the same compensatory upregulation of IGF1 receptor phosphorylation and activation of AKT as well as an increase in the expression of total and phosphorylated XIAP (Fig. 5A and Suppl. Fig. S7A). While elevated levels in XIAP seemed to be a common feature of dormant cancer cells in both pancreatic tumor models, the expression of the pro-apoptotic Bcl2 family protein BAD was reduced in residual cancer cells that lack exogenous c-MYC. Similarly, we observed a downregulation of pro-apoptotic gene Bax on the transcriptional level and an upregulation of the pro-survival genes Bcl2 and Bcl2l1. We reported previously that a significant subset of c-MYC-induced adenocarcinomas were poorly differentiated (Lin et al., 2013), but it was interesting to note that residual cancer cells showed a significantly higher expression of the ductal marker CK19 (Fig. 5A).
Figure 5. IGF-1R/AKT signaling is critical for cancer cell dormancy in a reversible pancreatic tumor model that is based on the conditional expression of c-MYC as the primary oncogenic driver.
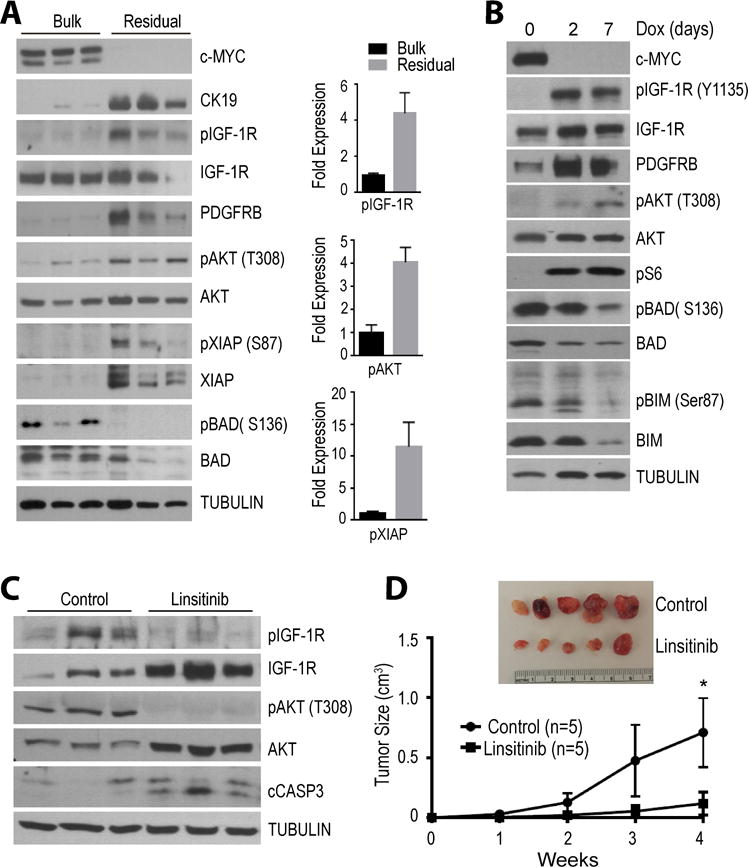
A. Immunoblot analysis comparing the expression and activation of IGF-1R, AKT, and the levels of downstream cell survival/death effectors between bulk tumors and residual cancer tissues in vivo. Bar graphs show Image-J quantification of selected protein bands. All data are represented as mean ± SEM; * represents P<0.05. B. Activation of the IGF-1R/AKT signaling in cultured pancreatic cancer cells following the doxycycline (Dox)-mediated downregulation of the oncogenic driver c-MYC. C. Validation of the pharmacological inhibition of IGF-1R and AKT signaling as well as activation of cleaved caspase 3 (cCASP3) in residual cancer cells in vivo. D. Tumor growth curves comparing cancer recurrence following re-expression of exogenous c-MYC between controls and animals that were treated with the IGF1-R inhibitor linsitinib.
The compensatory upregulation of IGF-1R/AKT signaling was confirmed in cultured pancreatic cancer cell from the c-MYC model (Fig. 5B). This suggested that, identical to the KRAS-associated cancer model, the activation of IGF-1R signaling in the absence of c-MYC as the oncogenic driver is a cell intrinsic phenomenon. Similarly, the inhibition of IGF-1R phosphorylation with linsitinib was sufficient to block the activation of AKT (Fig. 5C), and residual cancer cells that were treated with the IGF-1R inhibitor showed an increased expression of active Caspase 3. Withdrawal of Dox and therefore re-expression of c-MYC resulted in significantly delayed cancer recurrence and reduced tumor burden in linsitinib-treated animals compared to vehicle-treated controls with residual disease (Fig. 5D). Collectively, these findings suggest that, similar to the KRAS-induced PDAC model, inhibition of IGF-1R/AKT signaling leads to a selective eradication of dormant cancer cells in the pancreatic tumor model that is driven by c-MYC.
Human pancreatic cancer cells engage in higher IGF-1R/AKT signaling upon targeted downregulation of KRAS in vitro and in vivo
We generated two human pancreatic cancer cell lines (AsPC-1 and MIA PaCa-2) that express a previously validated shRNA against KRAS in a Dox-inducible manner (Tet-ON-shKRAS) (Shao et al., 2014) to address whether reduced levels of this oncogenic driver led to the same compensatory increase in IGF-1R signaling that was observed in the two diverse murine cancer models. A main difference of targeting KRAS at the posttranscriptional state is that the small hairpin RNA leads to a downregulation of the total protein level regardless of the mutational status of the endogenous alleles. As anticipated, reduced expression of total KRAS over a course of 7 days of treatment with Dox led to a significant increase in cell death (Fig. 6A), and the surviving cells in culture exhibited a high activation of the IGF1 receptor (Fig. 6B). Similar to the two mouse cancer models, both human cell lines also showed elevated expression levels of active AKT as well as total and phosphorylated XIAP. To study the effects of KRAS ablation in vivo, AsPC-1 cells were transplanted into immunocompromised recipient mice that were treated with Dox two weeks following successful engraftment (Fig. 6C). Inhibition of KRAS expression led to an arrest in cancer cell growth and a slight reduction in tumor size in the majority of recipients. Compared to tumors from untreated animals, the suppression of KRAS expression in tumors from Doxtreated mice expressing shKRAS showed the expected upregulation in IGF-1R/AKT signaling and elevated levels of total and phosphorylated XIAP (Fig. 6D). The absence of a more substantial regression of tumors associated with true cancer cell dormancy in the xenograft model was likely due to selective mechanisms by which cancer cells were able to bypass the RNA interference to restore expression of KRAS as shown in Fig. 6D. This was also the case following the xenotransplantation of single cancer cell clones that exhibited the most effective downregulation of KRAS in vitro (data not shown).
Figure 6. Compensatory increase in IGF-1R/AKT signaling in human pancreatic cancer cells in response to KRAS downregulation in vitro and in vivo.
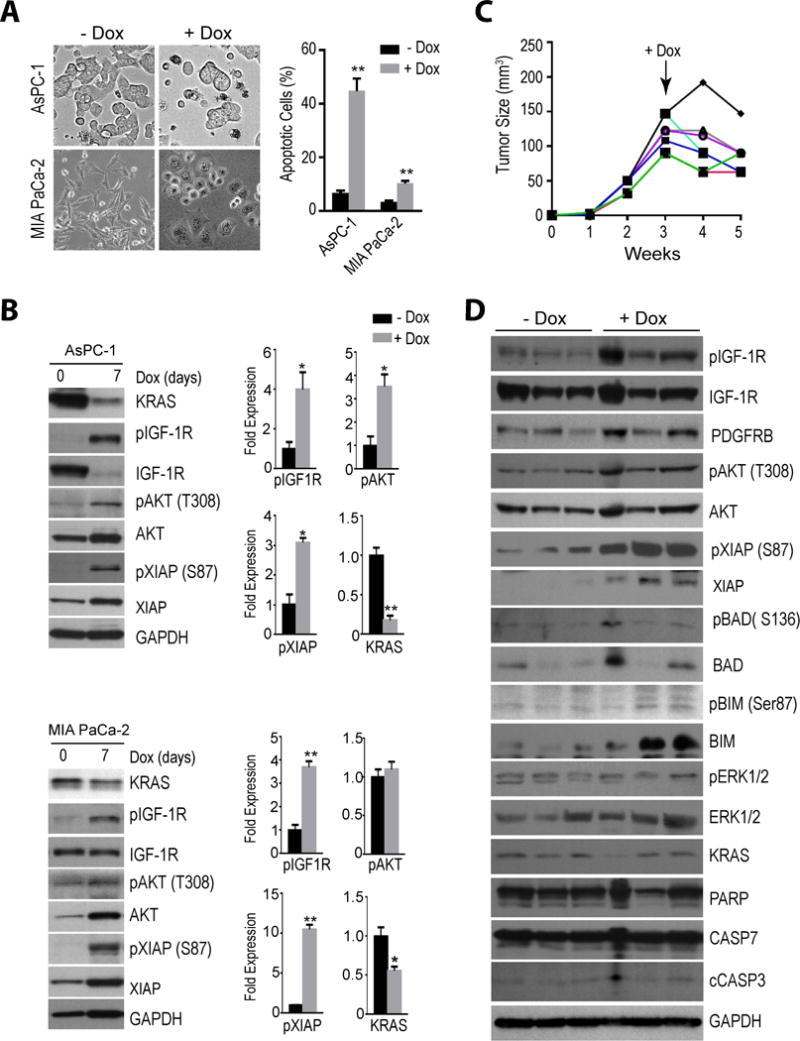
A. Bright-field images of cultured AsPC-1 and MIA PaCa-2 cells before (−Dox) and after seven days of a doxycycline-induced expression (+Dox) of a KRAS-specific shRNA. The graphs illustrate the significant increase in apoptotic cells in both cell lines following KRAS downregulation. The data is represented as mean ± SEM; ** P<0.01. B. Immunoblot analysis comparing the activation of the IGF-1R/AKT signaling axis in both cell lines before and after 7 days of a Dox-controlled knockdown of KRAS. Bar graphs show Image-J quantification of selected protein bands. All data are represented as mean ± SEM; * P<0.05; ** P<0.01. C. Partial growth inhibition of tumors with xenografted AsPC-1 cells following a Dox-inducible knockdown of KRAS. D. Comparison of the expression and activation of the IGF-1R/AKT pathway before (−Dox) and two weeks after the Dox-inducible knockdown of KRAS (+Dox) in xenografted AsPC-1 cells.
Activation of IGF-1R is a consequence of IGF1 autocrine signaling in both mouse and human pancreatic cancer cells that conditionally lack oncogenic KRAS or c-MYC overexpression
A detailed analysis of RNA-Seq data sets of both the KRAS and c-MYC-induced mouse models for pancreatic cancer revealed that Igf1 mRNA transcripts were virtually absent in bulk tumors and significantly upregulated in dormant cancer cells (Suppl. Fig. S7B). Using immunohistochemistry, we observed a stronger IGF1 staining specifically in residual cancer cells compared to bulk tumors in both reversible cancer models (Fig. 7A). More importantly, we were able to validate by quantitative RT-PCR on cultured pancreatic cancer cells that the significant increase in the transcriptional activation of Igf1 was a direct consequence of the conditional downregulation of oncogenic KRAS or exogenous c-MYC (Fig. 7B). This observation was also confirmed in the two human pancreatic cancer cell lines following a ligand-induced knockdown of KRAS (Fig. 7C). Interestingly, the more substantial transcriptional increase in Igf1 expression in AsPC-1 cells compared to MIA PaCa-2 cells was directly proportional to the degree in IGF-1R phosphorylation and activation of AKT (Fig 6B). The collective results from the RNA-Seq analyses and immunostaining of dormant tumor cells in vivo as well as quantitative RT-PCR experiments of isolated cancer cells in culture demonstrated very consistently that the increase in IGF-1R signaling in cells that conditionally lack the primary oncogenic drivers (i.e., KRASG12D or c-MYC) is a result of a cell-intrinsic upregulation of its ligand and the establishment of an autocrine signaling loop.
Figure 7. Upregulation of IGF1 is a cancer cell-intrinsic mechanism that promotes minimal residual disease in the absence of mutant KRAS or c-MYC as oncogenic drivers.
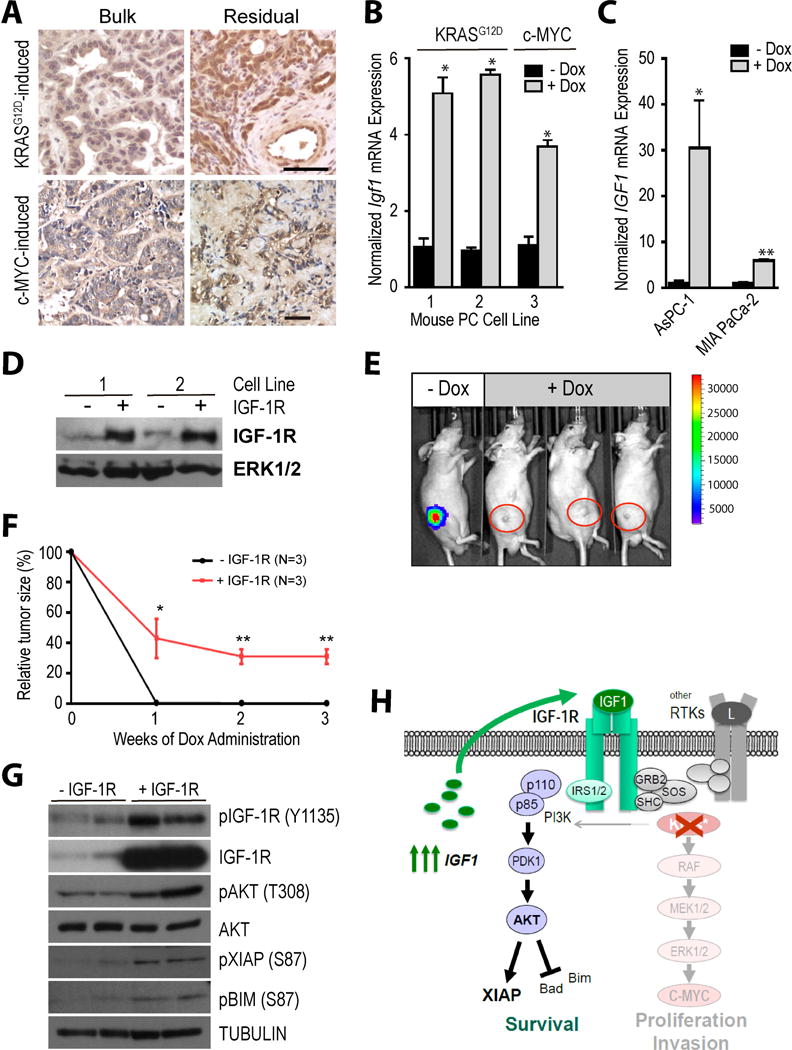
A. Immunohistochemical staining of IGF1 in mutant KRAS or c-MYC-driven bulk tumors as well as the corresponding residual cancer cells after the targeted downregulation of these oncogenic drivers in both models. B. Bar graphs illustrating the results of a quantitative real-time RT-PCR to assess the levels of Igf1 mRNA expression in cultured cancer cells from KRAS and c-MYC-induced mouse pancreatic tumors prior to (−Dox) or after the Dox-mediated downregulation of mutant KRAS or c-MYC (+Dox). C. Quantitative real-time RT-PCR results of IGF1 transcripts in human pancreatic cancer cell lines before and after Dox-mediated knockdown of KRAS. D. Immunoblot to validate the elevated levels of exogenous IGF-1R in two mouse pancreatic cancer cell lines that conditionally express mutant KRAS. E. Bioluminescence imaging to validate the co-repression of luciferase in mice with minimal residual disease following the Dox-mediated ablation of mutant KRAS. F. Average reduction in tumor sizes comparing cancers with and without expression of exogenous IGF-1R during 3 weeks of mutant KRAS ablation. G. Immunoblot to validate the sustained expression and activation of exogenous IGF-1R as well as downstream expression of active AKT and its effectors XIAP and BIM in dormant cancer cells following downregulation of mutant KRAS. H. Schematic outline of the establishment of an IGF1 autocrine loop that promotes cancer cell dormancy in the absence of mutant KRAS expression.
Since dormant cancer cells exhibit a compensatory upregulation of the Igf1 gene, elevating the levels of the receiving unliganded IGF-1R might be sufficient to sustain cancer cell survival and promote dormancy. To experimentally address this idea, we generated two cell lines from the KRAS-associated pancreatic cancer model that overexpress the wildtype IGF1 receptor (Fig. 7D). IGF-1R-expressing and isogenic control cells were transplanted subcutaneously into wildtype recipients. Following engraftment and establishment of bulk tumors, these mice were treated with Dox for three weeks to assess the presence of residual disease. Since both isogenic cancer cell lines co-expressed the TetO-driven luciferase reporter, we were able to monitor the sustained Dox-mediated co-repression of oncogenic KRAS and luciferase in vivo (Fig. 7E). As anticipated, upregulation of exogenous IGF-1R was sufficient to partially rescue the loss of oncogenic KRAS and to significantly increase the extent of minimal residual disease (Fig. 7F). The elevated levels of total and phosphorylated IGF-1R in residual cancer cells that lack expression of mutant KRAS for three weeks was confirmed by immunoblot (Fig. 7G). These cells also exhibited higher levels of active AKT and downstream phosphorylation of XIAP and BIM.
Our collective studies provide evidence that IGF-1R signaling plays a key role in pancreatic cancer cell dormancy following the downregulation of mutant KRAS or c-MYC as oncogenic drivers. While a pharmacological inhibition of IGF-1R led to a reduced number of dormant cancer cells, the overexpression the wildtype receptor was sufficient to counteract the therapeutic effects of the downregulation of the oncogenic driver and to significantly increase residual disease burden. Finally, we provided several lines of evidence that show that the activation of the IGF-1R is mediated by the increased expression of its ligand in a cancer cell-intrinsic manner (Fig. 7H).
Discussion
The dismal prognosis of pancreatic cancer emphasizes the urgent need for the development of targeted therapies to more effectively treat this malignancy. Due to the very frequent occurrence of gain-of-function mutations within the KRAS gene in PDAC, its encoded, constitutively active GTPase is a rational therapeutic target. Mutant KRAS is not only a key player during the initiation of PDAC, but as more recent studies (Collins et al., 2012a; Collins et al., 2012b; Ying et al., 2012) as well as the inaugural findings of this work show, this oncogene is equally important for the maintenance of PDAC at primary and metastatic sites. However, the swift recurrence of the disease following reactivation of the oncogenic driver might indicate that some cancer cells or advanced preneoplastic lesions were still present following the downregulation of mutant KRAS (Collins et al., 2012b). Using a cell-fate labeling method in an alternative, reversible PDAC model that conditionally overexpresses c-MYC, our team demonstrated previously that a few pancreatic cancer cells that possess stem cell features were able to survive in a quiescent state in the absence of the primary oncogenic driver (Lin et al., 2013; Lin et al., 2014). Similar to our c-MYC-associated cancer model, we showed here that residual tumor cells that conditionally lack oncogenic KRAS exhibit the characteristics of true cancer dormancy. Unlike the phenomenon previously defined as tumor mass dormancy where cancer cells exist in an equilibrium of cell division and cell death (Aguirre-Ghiso, 2007), these residual pancreatic cancer cells do not proliferate and they do not undergo cell death. In line with this notion, Viale and colleagues (2014) have shown recently that pancreatic cancer cells that survive the ablation of mutant KRAS ex vivo are enriched for cancer-initiating cells. Their observations and our earlier findings on the expression of stem cell markers in residual pancreatic cancer cells of mice that conditionally express c-MYC collectively support the paradigm that tumor-initiating cells seem to be the cellular basis for cancer dormancy.
Dormant cancer cells can remain in a quiescent state for an extended period and serve as the cellular reservoir for a speedy recurrence of a clinically overt disease upon receiving favorable cell intrinsic cues such as the re-expression of the oncogenic driver. While maintaining diseased mice on Dox for an extended period, we were unable to detect any large recurring tumors that reemerged in the complete absence of the transforming oncogenes (i.e., c-MYC or mutant KRAS). The effectiveness of the Dox-mediated suppression of the transgenes in these mice was repeatedly monitored by the activity of a TetO-driven luciferase transgene. Based on these observations, our two PDAC models did not provide any clear evidence for a compensatory activation of any alternative molecular pathways that effectively mediate cancer recurrence in the complete absence of the primary oncogenic drivers such as transcriptional upregulation or amplification of Yap1 as reported recently (Kapoor et al., 2014; Shao et al., 2014). Additionally, the analysis of our RNA-Seq data did not reveal any differences in the expression of Yap1 mRNA between bulk tumors and residual cancer cells in both animal models that conditionally express mutant KRAS or c-MYC. This suggests that this transcription factor does not seem to play a key role in cancer cell dormancy in our two models in contrast to its proposed function in disease recurrence. Whether deficiency in wildtype p53 in the KRAS-associated PDAC model by Kapoor et al. (2014), as opposed to the loss of Cdkn2a in our models, is a defining factor for the reported gain-of-function of YAP1 remains to be investigated.
Regardless of subsequent molecular events that may promote disease recurrence, it is equally, if not more, important to understand the mechanisms that mediate cancer cell dormancy in an effort to eliminate residual disease following first-line therapy. Using genome-wide transcriptome analyses in two genetically engineered pancreatic cancer models, we have identified the activation of autocrine IGF1 signaling and the downstream activation of AKT as a common mechanism that promotes cancer cell survival and dormancy following the specific ablation of KRASG12D and c-MYC as oncogenic drivers. Moreover, we have validated that targeting KRAS in human pancreatic cancer cells leads to a compensatory upregulation of IGF-1R/AKT signaling. We also demonstrate that co-targeting mutant KRAS or c-MYC and IGF-1R reduces the number of residual cancer cells and delays tumor recurrence. Conversely, overexpression of wildtype IGF-1R was sufficient to counteract the therapeutic effects of targeting KRAS leading to more extensive residual disease. IGF-1R signaling has been previously reported to be crucial for neoplastic transformation and survival of epithelial cells in the mammary gland and pancreas in response to the expression of oncogenic mutants of KRAS and BRAF (Appleman et al., 2012; Klinakis et al., 2009). Hirakawa et al. (2013) reported that high levels of IGF-1R expression correlate with aggressiveness and a poorer survival of patients with resectable PDAC, which is indicative for the presence of residual disease. A more direct association of enhanced IGF-1R and AKT signaling and the development of drug resistance following the targeted inhibition of BRAF has been demonstrated recently in human melanoma and a corresponding mutant BRAF expressing mouse model (Perna et al., 2015; Villanueva et al., 2010).
Our findings in this work on the MYC-associated PDAC model suggest that the activation of the IGF-1R/AKT pathway in dormant pancreatic cancer cells seems not to be restricted to mutations in the RAS/RAF cascade, and they might not even be specific for targeted therapies per se. A role for IGF1 autocrine signaling has been recognized earlier as a potential mechanism for prostate cancer cells to evade androgen deprivation therapy (Nickerson et al., 2001), and a more recent report highlighted the significance of IGF1 in cancer stem cells that are resistant to radiation (Osuka et al., 2013). The pharmacological inhibition of IGF-1R signaling was however insufficient to eradicate all residual cancer cells in our KRAS and c-MYC-associated, reversible PDAC models. Flow cytometric results from our previous studies have shown that dormant pancreatic cancer cell may represent a heterogeneous population based on differences in the expression levels of various stem cell markers (Lin et al., 2013; Lin et al., 2014). It is likely that a subset of these residual tumor cells also engages in a compensatory increase in the activation of other receptor tyrosine kinases such as PDGFRB, which seems to play a role in the recurrence of melanoma following treatment with vemurafenib (Nazarian et al., 2010). Since we observed a co-upregulation of PDGFRB on the transcriptional and protein level in dormant cancer cells of both murine tumor models as well as human AsPC-1 cells in response to the shRNA-mediated downregulation of KRAS, in might be valid to assess in future studies the combinatorial effects of the inhibition of PDGFRB and IGF-1R for an even more effective eradication of minimal residual disease following the ablation of oncogenic KRAS and c-MYC.
Experimental Procedures
Mouse models, orthotopic transplantation of pancreatic tumors, and in vivo bioluminescence imaging
The generation and genotyping of the TetO-KRASG12D, TetO-MYC strains as well as the CAG-LSL-tTA and TetO-ccnd1-Luc transgenic lines were described previously (Felsher and Bishop, 1999; Fisher et al., 2001; Zhang et al., 2011; Zhang et al., 2010). The CAG-LSL-GFP reporter strain was generated by Kawamoto et al. (2000). Pdx1-Cre transgenic mice (Hingorani et al., 2003) and the Cdkn2a knockout strain (Serrano et al., 1996) were obtained from the NCI repository. TetO-H2B/GFP transgenic mice (Tumbar et al., 2004) were purchased from the Jackson Laboratory. Experimental mice were maintained in a mixed genetic background comprised of the above mentioned strains (50% C57/Bl6, and 25% 129Sv, and 25% FVB). The orthotopic transplantation of pancreatic cancer tissues from transgenic mice (i.e., Pdx1-Cre CAG-LSL-tTA CAG-LSL-GFP TetO-KRASG12D Cdkn2a−/− and Pdx1-Cre CAG-LSL-tTA CAG-LSL-GFP TetO-MYC Cdkn2a+/−) into pancreata of 8 to 12 weeks-old wildtype recipients (Athymic nude mice, NCr strain) was performed as described previously (Lin et al., 2013). The human PDAC cells were transplanted subcutaneously into wildtype recipients. All animals used in this study were treated humanely and in accordance with institutional guidelines and federal regulations.
In vivo bioluminescence imaging and treatment with doxycycline and linsitinib
The administration of doxycycline (Dox) in the drinking water and the use of the IVIS200 (Caliper Life Sciences, Alameda, CA) for in vivo bioluminescence imaging were described in our previous publications (Lin et al., 2013; Zhang et al., 2011; Zhang et al., 2010). Wildtype recipient with residual pancreatic cancer cells following downregulation of oncogenic KRAS or c-MYC were treated with the IGF-1R inhibitor linsitinib (OSI-906; ChemieTek LLC, Indianapolis) or vehicle control. The inhibitor was administered by oral gavage (50 mg/kg) once daily for five consecutive days.
Histologic analysis and immunostaining
A description of the preparation of histological sections from pancreatic cancer tissues for immunostaining can be found elsewhere (Lin et al., 2013). A list of commercially available primary and secondary antibodies and staining conditions will be provided upon request. TUNEL staining was carried out using the in situ cell death detection kit (Roche Applied Sciences, Indianapolis, IN). Stained slides were examined with an Axio Imager microscope (Carl Zeiss) or a LSM5 PASCAL confocal microscope.
Isolation of GFP-labeled cancer cells and flow cytometric analysis
The isolation of bulk tumor cells as well as residual cancer cells was performed prior to or two to four weeks after treatment with Dox and downregulation of the oncogenic driver. This extended period, which exceeded the time required for a macroscopic regression of the tumors by more than 7 to 21 days (Lin et al., 2013), was chosen to ensure that the cancers had completely regressed and only dormant cancer cells were analyzed. GFP-positive bulk tumors or areas with small residual tumor masses that contained GFP-positive cells were isolated from the pancreata under a fluorescent stereoscope. To determine the number of GFP-positive apoptotic cells using flow cytometry, excised tissues were processed for enzymatic dissociation and biotinylated anti-CD31 and anti-CD45 antibodies (BD Pharmingen) were used for elimination of endothelial and hematopoietic lineages using AutoMACS Pro (Miltenyi Biotec) as described previously (Lin et al., 2013). A PE-conjugated Annexin V antibody (BD Pharmingen) was used for staining of apoptotic cells. The flow cytometric data was acquired on a BD FACSCalibur at the UNMC Cell Analysis Core Facility. Data was analyzed using the FlowJo V9.8 software (Tree Star Inc.).
Cell culture
GFP-positive pancreatic cancer cells were derived from primary tumors of mice that conditionally overexpress mutant KRAS and c-MYC. Cells were maintained in DMEM medium supplemented with 10% FBS, L-glutamine, nonessential amino acids as well as 10 μg/ml penicillin/streptomycin and 50 μg/ml gentamicin. The human pancreatic cancer cell lines AsPC-1 and MIA PaCa-2 that carry different KRAS mutations (G12D and G12C) were maintained in RPMI-1640 medium using the same supplements listed above. The inducible lentiviral shRNA vector targeting human KRAS [PGK-rtTA3/Tet-ON-shKRAS] (Shao et al., 2014) was a generous gift from Dr. W.C. Hahn (Dana-Farber Cancer Institute). Cells infected with the lentiviral constructs were selected with 2 μg/ml puromycin. Mouse and human pancreatic cancer cells were treated with 2 μg/ml doxycycline to repress the tTA-mediated expression of TetO-driven transgenes (i.e., KRASG12D, c-MYC, and luciferase) or to upregulate the rtTA-inducible shRNA to downregulate endogenous KRAS. The retroviral vector to overexpress IGF-1R in mutant KRAS-expressing cells was obtained from Addgene (#11212).
Immunoblot analysis
Detailed experimental procedures for western blot analysis were described elsewhere (Sakamoto et al., 2007). A list of commercially available primary and secondary antibodies and experimental conditions will be provided upon request.
RNA-Seq analysis and quantitative RT-PCR (qRT-PCR)
Total RNA was extracted from flash-frozen bulk tumor specimen or residual cancer tissues following the downregulation of oncogenic KRAS or c-MYC using the RNeasy Mini Kit (Qiagen). The methodologies for the generation of expression libraries, next generation sequencing, and bioinformatics analyses can be found in the Supplemental Experimental Procedures section. Quantitative detection of IGF1 mRNA transcripts was performed using iQ SYBR green Supermix (Bio-Rad), and primer sequences are available from the authors upon request. The quantitative PCRs (qPCRs) were carried out in triplicate in a CFX96 real-time PCR detection system (Bio-Rad). IGF1 gene expression data were normalized against either Actin (mouse) or GAPDH (human) as internal control using the 2−ΔΔCt method and expressed as arbitrary units.
Statistical analysis
All graphic illustrations and statistics were performed with Prism 6 software (GraphPad Software, Inc., La Jolla, CA). Data are expressed as mean ±SEM unless otherwise indicated and were compared using an unpaired Student t test. A P value of less than .05 was considered significant.
Supplementary Material
Acknowledgments
The authors thank the UNMC Genomics Core Facility for his assistance in next-generation sequencing (NGS) service and the UNMC Cell Analysis Core facility for flow cytometry. We are grateful to Karen K. Dulany for the preparation of histological sections.
Funding Information
Financial support provided to K.-U.W. by the Nebraska Cancer and Smoking Disease Research Program (NE DHHS LB506 2011-36 and LB506 2016-54) was imperative to finance the maintenance of mutant mice and the collection of tumor tissues for transcriptome analysis. Additional funding was provided by the Public Health Service grant CA202917 (K.-U.W.). The work on the c-MYC-associated pancreatic cancer model was supported by the Public Health Service grant R21 CA155175 (K.-U.W.). N.R and W.-C.L. were supported through a research assistantship from the UNMC Graduate Studies Office. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement: The authors have nothing to disclose.
Accession Numbers
The accession number for the sequencing data reported in this paper is GEO: GSE93946.
Author Contributions
K.-U.W. formulated the overarching research goals and aims and supervised the research. N.R. and W.-C. L. developed the KRAS and c-MYC-associated pancreatic cancer models and conducted the molecular and biological analyses. B.L.W. assisted in specific experiments. N.R. and A.A.T. generated the gene expression libraries and performed the computational analyses of the RNA-Seq data sets. K.-U.W. wrote the manuscript.
References
- Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7:834–846. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleman VA, Ahronian LG, Cai J, Klimstra DS, Lewis BC. KRAS(G12D)- and BRAF(V600E)-induced transformation of murine pancreatic epithelial cells requires MEK/ERK-stimulated IGF1R signaling. Mol Cancer Res. 2012;10:1228–1239. doi: 10.1158/1541-7786.MCR-12-0340-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, Rakshit S, Flannagan KS, Adsay NV, Pasca di MM. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012a;122:639. doi: 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MA, Brisset JC, Zhang Y, Bednar F, Pierre J, Heist KA, Galban CJ, Galban S, di Magliano MP. Metastatic pancreatic cancer is dependent on oncogenic Kras in mice. PLoS One. 2012b;7:e49707. doi: 10.1371/journal.pone.0049707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4:199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- Fisher GH, Wellen SL, Klimstra D, Lenczowski JM, Tichelaar JW, Lizak MJ, Whitsett JA, Koretsky A, Varmus HE. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15:3249–3262. doi: 10.1101/gad.947701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- Hirakawa T, Yashiro M, Murata A, Hirata K, Kimura K, Amano R, Yamada N, Nakata B, Hirakawa K. IGF-1 receptor and IGF binding protein-3 might predict prognosis of patients with resectable pancreatic cancer. BMC Cancer. 2013;13:392. doi: 10.1186/1471-2407-13-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruban RH, Wilentz RE, Kern SE. Genetic progression in the pancreatic ducts. Am J Pathol. 2000;156:1821–1825. doi: 10.1016/S0002-9440(10)65054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, Zhong Y, Wu CJ, Sadanandam A, Hu B, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell. 2014;158:185–197. doi: 10.1016/j.cell.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto S, Niwa H, Tashiro F, Sano S, Kondoh G, Takeda J, Tabayashi K, Miyazaki J. A novel reporter mouse strain that expresses enhanced green fluorescent protein upon Cre-mediated recombination. FEBS Lett. 2000;470:263–268. doi: 10.1016/s0014-5793(00)01338-7. [DOI] [PubMed] [Google Scholar]
- Klinakis A, Szabolcs M, Chen G, Xuan S, Hibshoosh H, Efstratiadis A. Igf1r as a therapeutic target in a mouse model of basal-like breast cancer. Proc Natl Acad Sci U S A. 2009;106:2359–2364. doi: 10.1073/pnas.0810221106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WC, Rajbhandari N, Liu C, Sakamoto K, Zhang Q, Triplett AA, Batra SK, Opavsky R, Felsher DW, DiMaio DJ, et al. Dormant cancer cells contribute to residual disease in a model of reversible pancreatic cancer. Cancer Res. 2013;73:1821–1830. doi: 10.1158/0008-5472.CAN-12-2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WC, Rajbhandari N, Wagner KU. Cancer cell dormancy in novel mouse models for reversible pancreatic cancer: a lingering challenge in the development of targeted therapies. Cancer Res. 2014;74:2138–2143. doi: 10.1158/0008-5472.CAN-13-3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson T, Chang F, Lorimer D, Smeekens SP, Sawyers CL, Pollak M. In vivo progression of LAPC-9 and LNCaP prostate cancer models to androgen independence is associated with increased expression of insulin-like growth factor I (IGF-I) and IGF-I receptor (IGF-IR) Cancer Res. 2001;61:6276–6280. [PubMed] [Google Scholar]
- Osuka S, Sampetrean O, Shimizu T, Saga I, Onishi N, Sugihara E, Okubo J, Fujita S, Takano S, Matsumura A, Saya H. IGF1 receptor signaling regulates adaptive radioprotection in glioma stem cells. Stem Cells. 2013;31:627–640. doi: 10.1002/stem.1328. [DOI] [PubMed] [Google Scholar]
- Perna D, Karreth FA, Rust AG, Perez-Mancera PA, Rashid M, Iorio F, Alifrangis C, Arends MJ, Bosenberg MW, Bollag G, et al. BRAF inhibitor resistance mediated by the AKT pathway in an oncogenic BRAF mouse melanoma model. Proc Natl Acad Sci U S A. 2015;112:E536–545. doi: 10.1073/pnas.1418163112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, Creamer BA, Triplett AA, Wagner KU. The Janus kinase 2 is required for expression and nuclear accumulation of cyclin D1 in proliferating mammary epithelial cells. Mol Endocrinol. 2007;21:1877–1892. doi: 10.1210/me.2006-0316. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, Schinzel AC, Sood S, Rosenbluh J, Kim JW, et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell. 2014;158:171–184. doi: 10.1016/j.cell.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbar T, Guasch G, Greco V, Blanpain C, Lowry WE, Rendl M, Fuchs E. Defining the epithelial stem cell niche in skin. Science. 2004;303:359–363. doi: 10.1126/science.1092436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–632. doi: 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744. doi: 10.1038/ncomms7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Sakamoto K, Liu C, Triplett AA, Lin WC, Rui H, Wagner KU. Cyclin D3 compensates for the loss of cyclin D1 during ErbB2-induced mammary tumor initiation and progression. Cancer Res. 2011;71:7513–7524. doi: 10.1158/0008-5472.CAN-11-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Triplett AA, Harms DW, Lin WC, Creamer BA, Rizzino A, Wagner KU. Temporally and spatially controlled expression of transgenes in embryonic and adult tissues. Transgenic Res. 2010;19:499–509. doi: 10.1007/s11248-009-9329-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.