

Abstract
The diagnosis of adult-onset Still's disease (AOSD) can be very difficult. There are no specific tests available, and diagnosis is usually based on a symptom complex and the well-described typical evanescent rash seen in the majority of patients. However, in recent years, other atypical cutaneous manifestations of AOSD have been reported. These atypical skin eruptions often present in addition to the typical evanescent rash but may also be the only skin manifestation, resulting in delayed diagnosis because of under-recognition.
In this study, we present 3 new cases of AOSD with atypical cutaneous manifestations diagnosed during a 30-year period in our department and review 78 additional cases previously reported (PubMed 1990–2016). These 81 patients form the basis of the present analysis.
The overall prevalence of atypical cutaneous manifestations in our AOSD population was 14%. These manifestations may appear at any time over the course of the disease, and usually occur in patients who have persistent and severe disease, with a considerable frequency of clinical complications (23%), including serositis, myopericarditis, lung involvement, abdominal pain, neurologic involvement, and reactive hemophagocytic syndrome.
The most representative and frequent lesion among the nonclassical skin rashes is the development of persistent pruritic papules and/or plaques. Interestingly, these lesions show a distinctive histological pattern. Other, less frequently observed lesions include urticaria and urticaria-like eruptions, generalized or widespread non-pruritic persistent erythema, vesiculopustular eruptions, a widespread peau d’orange appearance of the skin, and edema of the eyelids mimicking dermatomyositis without any accompanying skin lesion.
The great majority of these patients required medium or high doses of glucocorticoids (including intravenous methylprednisolone pulse therapy in some cases) and, in nearly 40%, a more potent or maintenance immunotherapy with immunosuppressant drugs and/or biologic agents (mainly anakinra or tocilizumab) to control or manage symptoms because of a polycyclic or chronic course. The development of atypical cutaneous manifestations seems to be associated with a potentially worse prognosis, with a mortality rate reaching 8% primarily because of infectious complications related to immunosuppressive therapy.
In conclusion, the appearance of atypical cutaneous manifestations is not uncommon in AOSD. Recognition of this clinical variant is crucial for the early diagnosis of AOSD, as it might imply persistent disease activity and the need for more aggressive treatment.
Keywords: adult onset Still's disease, atypical cutaneous manifestations, persistent eruptions
1. Introduction
Adult-onset Still's disease (AOSD) is an uncommon, acute, systemic inflammatory disease of unknown etiology that is classically characterized by intermittent high spiking fevers, arthralgias/arthritis, typical evanescent rash, sore throat, generalized lymphadenopathy, liver dysfunction, and splenomegaly.[1] Other features less frequently reported include serositis, myopericarditis, interstitial lung disease, and neurologic involvement.[1] Furthermore, reactive hemophagocytic syndrome is occasionally seen.[1] Therefore, it is important to make an early diagnosis. Several classification criteria have been proposed, being the Yamaguchi's criteria[2] the most commonly used. However, the clinical presentation of AOSD is heterogeneous, and the spectrum of differential diagnoses is wide, including infection, neoplasia, and other autoimmune disorders, which should be ruled out before the diagnosis of AOSD can be made.[3]
Laboratory markers, particularly the presence of leukocytosis with neutrophilia and marked hyperferritinemia, are suggestive of AOSD, but clinicians often rely on the typical evanescent, salmon-pink, maculopapular eruption to make the diagnosis.[3] Though an evanescent eruption is the classic cutaneous finding, a recent literature has highlighted atypical rashes associated with AOSD. These atypical cutaneous manifestations often present in addition to the typical evanescent rash but may also be the only skin manifestation, resulting in delayed diagnosis because of under-recognition.
The objective of this study is to describe 3 new cases of AOSD with atypical skin features and review the clinical spectrum of atypical cutaneous manifestations described in this disease.
2. Patient and methods
2.1. Patient selection
We retrospectively reviewed all patients with AOSD diagnosed between January 1985 and December 2015 by the Department of Rheumatology at the Hospital Universitario de Bellvitge (Barcelona, Spain), a referral tertiary care hospital. Diagnosis of AOSD was based on the criteria set by Yamaguchi et al,[2] with special attention being given to the serum ferritin value.[4] The major criteria are high fever for >1 week, arthalgias for >2 weeks, neutrophilic leukocytosis (>10,000/mm3 with >80% of neutrophils), and the typical evanescent rash. Minor criteria include sore throat, lymphadenopathy and/or splenomegaly, liver dysfunction, and the absence of rheumatoid factor and antinuclear antibody. A minimum of 5 criteria, including at least 2 major criteria, were required for diagnosis, as well as the exclusion of infections, malignancy, and other rheumatologic diseases. The recent criteria proposed by Fautrel et al[5] were not used because most patients were diagnosed before these criteria were published, and owing to lack of data for glycosylated ferritin.
Inpatient and outpatient charts were comprehensively reviewed to obtain clinical, laboratory, and disease course data. From a total of 21 patients with AOSD, we selected those who presented with any cutaneous manifestation different from the typical evanescent rash, excluding dermographism.
In accordance with the guidelines of our institutional ethics committee, formal approval for this study was not required. The local ethics committee agreed that the findings in this report were based on normal clinical practice and were therefore suitable for dissemination. Informed patient consent was obtained and clinical records and patient data were anonymized. This study was conducted in accordance with the principles of the Declaration of Helsinki and the International Conference for Harmonization.
2.2. Literature search strategy and selection criteria
Searches were conducted in the PubMed database (including MEDLINE, National Library of Medicine, and PubMed Central) for the period between January 1990 and November 2016, using strategies recommended by the Cochrane handbook. Search terms included “adult onset Still's disease,” “cutaneous manifestations,” “skin rash,” “persistent,” or “atypical.” Inclusion criteria included case reports or series reporting specific characteristics of an atypical skin eruption in patients fulfilling the diagnostic criteria of Still's disease proposed by Yamaguchi et al.[3] Only English, French, Portuguese, and Spanish reports were considered. The references of the studies obtained were also examined to identify additional reports.
The MEDLINE search resulted in 113 articles. The abstracts or the full text was screened for inclusion and 84 of them were excluded. The remaining 29 articles were evaluated, together with 6 additional reports identified from review of the references.
Therefore, 35 articles were finally selected for review (32 case reports and 3 case series) and 78 well-documented cases of AOSD with atypical skin eruptions were identified.[6–40] Lee et al[6] reported 28 cases in aggregate form only; 1 of them was previously accurately described by Yang et al.[24] Nagai et al[18] also reported 6 patients in aggregate form only.
2.3. Statistical analysis
Qualitative variables are reported as frequencies and percentages and quantitative variables as mean or median ± standard deviation (SD) and range.
3. Results
Among a total of 21 patients with AOSD diagnosed during a 30-year period in our Department, 3 patients (14%) presented with atypical persistent cutaneous manifestations: 1 case with persistent refractory urticaria (Fig. 1), 1 case with persistent pruritic papules and plaques with flagellate erythema-type appearance (Fig. 2), and 1 case with persistent pruritic plaques on the chest and concomitantly evanescent urticarial rash (Fig. 3). Table 1 summarizes the main clinical characteristics and outcomes of these 3 patients.
Figure 1.
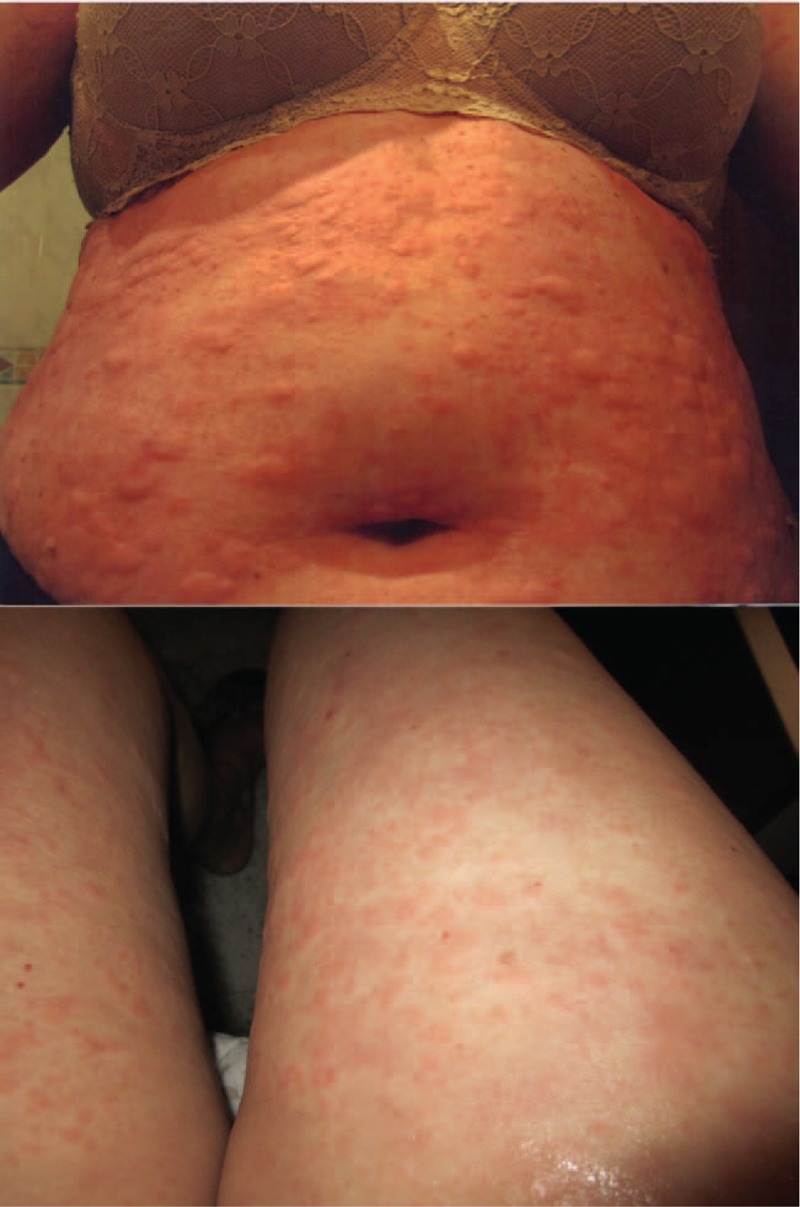
Patient 1: Clinical images of a 54-year-old woman with a persistent urticarial rash on the trunk and limbs as disease onset.
Figure 2.
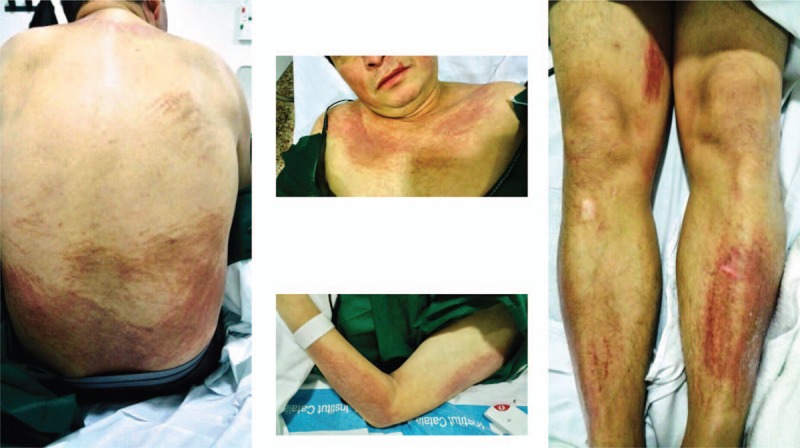
Patient 2: Clinical images of a 52-year-old Hispanic man with persistent pruritic papules and plaques with flagellate erythema-type appearance at disease onset.
Figure 3.
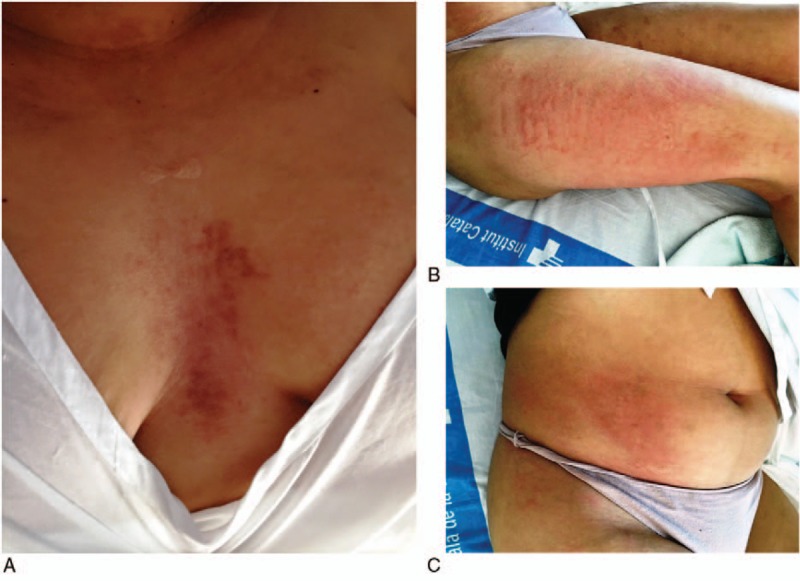
Patient 3: Clinical images of a 29-year-old Hispanic woman who developed persistent pruritic plaques on the chest (A) and concomitant evanescent urticarial rash with intermittent high spiking fevers (B and C) during a disease flare 29 months after diagnosis of the disease.
Table 1.
Main clinical characteristics and outcome of our patients.
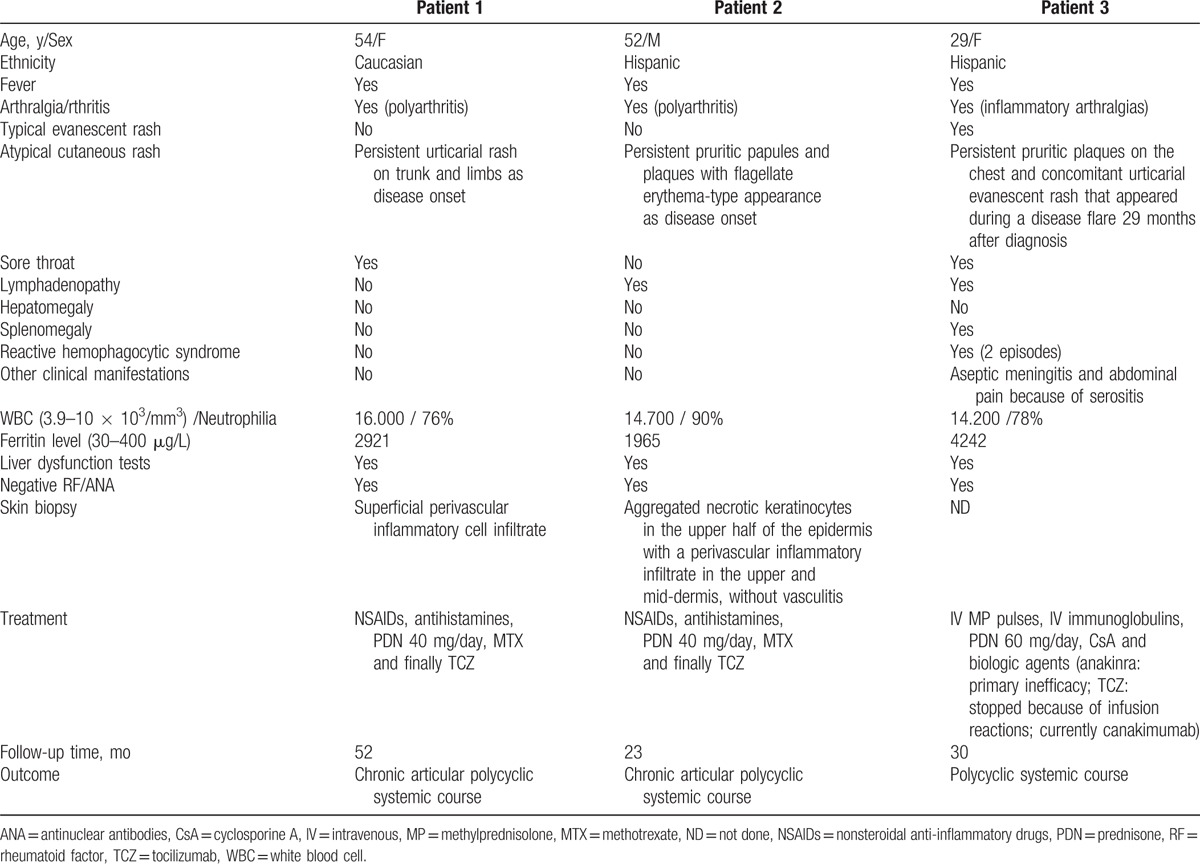
Table 2 summarizes the main clinical characteristics and outcome of the 78 patients obtained from the literature. These 78 cases, together with our 3 patients, form the basis of the present analysis. Because it is a review of the literature and the series of Lee et al[6] and Nagai et al[18] reported their cases in aggregate form only, not all analyzed variables were recorded in all included cases. Thus, the results (percentages) in each variable were calculated considering only the number of patients in which the data were documented.
Table 2.
Clinical characteristics and outcome of the 78 reported cases with adult-onset Still's disease and atypical skin rashes.

The global analysis of the 81 patients available for review provided the following information:
3.1. Demographic data
Of the 81 patients, the great majority were women (48/55; 87%) with ages ranging from 18 to 83 years (median 36 ± 16 years). Most of the cases (52/81; 64%) were reported from East Asia.
3.2. Atypical cutaneous lesions reported in association with AOSD
In the great majority of cases, the atypical skin eruption presented at the time of disease onset concurrently with systemic symptoms, or shortly afterward. Rarely, it was the presenting feature of AOSD preceding the appearance of the classic symptoms of the disease from a few weeks to 3 years, or it appeared during a disease flare several months after the initial diagnosis (as late as 29 months). In 57% (46/81) of patients, the atypical cutaneous lesions presented concurrently with the typical evanescent rash.
The most frequent atypical skin lesions described were the development of persistent papules and/or plaques (61/81; 75%). These lesions were usually, but not always, pruritic (53/61; 87%). Descriptors for the color varied from erythematous or brown in the majority of cases to, less commonly, violaceous. They may present with scales or crusts. The most frequent locations were the trunk (in order of frequency: back, chest, and abdomen) and the extensor surface of the extremities. This type of eruption often had a linear configuration, possibly because of the Koebner phenomenon, thus resembling a flagellate erythema. Other commonly reported morphologic patterns included urticarial papules, lichenoid papules, pigmented plaques, prurigo pigmentosa-like, dermatomyositis-like, and lichen amyloidosis-like rashes. More than 1 morphology or distribution pattern was observed in several patients.
The dermatomyositis-like eruption was pruritic and tended to have extensive cutaneous involvement manifesting as erythematous to violaceous or dusky red maculopapules or patches in a clear photodistribution. In addition, there was erythema and swelling of the upper eyelids resembling a heliotrope sign as well as the presence of erythematous maculopapules on the knuckles of the fingers mimicking Gottron papules. The prurigo pigmentosa-like eruption manifested as pruritic, erythematous urticarial to brownish lichenoid papules, which were arranged in a reticulate pattern on the upper back and chest, mimicking the morphology and distribution of prurigo pigmentosa.
Another frequently reported atypical lesion was urticaria and urticaria-like eruptions (11/81;14%). With the exception of one of our patients, all other cases were persistent and refractory, with poor response to treatment with antihistamines and low doses of oral corticosteroids. Most of these patients had dermographism, suggesting that patients in the active stage are susceptible to urticaria, and some presented with concomitant angioedema of the eyelids, lips, palms, and soles.
Other skin eruptions less frequently described were generalized or widespread non-pruritic persistent erythema (6/81; 7%), which was pigmented in more than half of the cases; more rarely, vesiculopustular eruptions reported on the hands and feet or involving the trunk and limbs (2/81; 2.5%); a widespread peau d’orange appearance of the skin (cutaneous mucinosa) (1/81; 1%); and edema of the eyelids mimicking dermatomyositis without any accompanying skin lesion (1/81; 1%).
3.3. Other clinical manifestations of AOSD
In addition to the skin eruption and classic symptoms of Still's disease (fever, arthralgias/arthritis, and sore throat), 57% (46/81) of these patients presented with lymphadenopathy, 44% (36/81) with hepatomegaly and/or splenomegaly, and 77% (62/81) with liver dysfunction.
Other important clinical manifestations were reported in 20% of the cases (16/81; 2 patients developed 2 complications) including serositis (6 patients), myopericarditis (2), lung involvement (3), abdominal pain (4), and neurologic involvement (3).
Excluding the 33 patients reported in aggregate form in the studies of Lee et al[6] and Nagai et al,[18] in the remaining 48 cases the mean values in the Systemic Score System proposed by Pouchot et al[41] was 5.41 ± 1.5 (range, 3–9). This score system assings 1 point to each of 12 manifestations present at the time of disease onset: fever, typical rash, pleuritis, pneumonitis, pericarditis, hepatomegaly or abnormal function tests, splenomegaly, lymphadenopathy, leukocytosis >15,000/mm3, sore throat, myalgia, and abdominal pain (maximum score: 12 points). Twenty-one percent of cases (10/48) had a score ≥7.
Reactive hemophagocytic syndrome was reported in 6% (5/81) patients. In total, 23% (19/81) of patients developed clinical complications (including reactive hemophagocytic syndrome).
3.4. Histopathology
Skin biopsy data were available for 90% (73/81) of patients. Histopathologically, the most frequently observed pattern (58/73; 79%) was characterized by singly or aggregated dyskeratotic/necrotic keratinocytes in the upper layers of the epidermis in association with a perivascular (and sometimes also interstitial) inflammatory infiltrate in the upper and mid-dermis, without vasculitis. The inflammatory infiltrate was composed mainly of lymphocytes and neutrophils, with the occasional presence of eosinophils. Dermal mucin deposition was sometimes seen.
In the group of patients with persistent papules and/or plaques, cutaneous biopsy was performed in 59 of 61 patients. This distinctive pattern was observed in 95% (56/59) of cases: in 51 (96%) of the 53 patients who presented with persistent papules and plaques, in the 2 patients with persistent plaques in whom the lesions were biopsied (in 2 patients skin biopsy was not performed), and in 3 (75%) of the 4 patients with persistent papules. Of interest, in the 3 patients without this distinctive histopathology, the lesions were non-pruritic.
In the group of patients with urticaria and urticaria-like eruptions, skin biopsy data were available in 8 of the 10 patients. The most frequent finding in these cases was a superficial dermal edema with perivascular, and sometimes interstitial, neutrophilic infiltrate, with the occasional presence of lymphocytes and histiocytes.
In the group of patients with generalized or widespread non-pruritic persistent erythema, a cutaneous biopsy was performed in 3 of the 6 cases. In 1 patient with persistent pigmented eruption, a skin biopsy showed the distinctive pattern observed in patients with persistent papules and/or plaques. In the remaining 2 cases, a biopsy showed a moderate perivascular mononuclear cell infiltration in the edematous upper-mid-dermis, with the occasional presence of neutrophils. Finally, in the 2 cases with vesiculopustular eruption, a skin biopsy showed epidermal spongiosis and significant edema and mononuclear infiltration in the upper and mid-dermis.
3.5. Treatment
Information about treatment was available in 76 of the 81 patients. The great majority (71/76; 93%) of patients were treated with glucocorticoids, usually (69/76; 91%) of medium or high doses (initial doses ≥30 mg/daily of prednisone or equivalent). Seventeen percent of patients (13/76) received intravenous methylprednisolone pulse therapy (1 g daily for 3 days), followed by oral corticosteroids.
In 38% (29/76) of patients, immunosuppressive agents were used in conjunction with steroids from the initiation of treatment, or when the patient failed to improve. The most frequently used were methotrexate (22 cases), azathioprine (5), and cyclosporine A (4).
Concomitant biologic therapy was employed as a rescue treatment in refractory cases (7/76; 9%), with good efficacy and safety profile. The biologic agents used were anakinra (in 4 cases, effective only in 3), tocilizumab (3 cases), canakimumab (1 case), and etanercept (in 1 case, with primary inefficacy). Intravenous immunoglobulin was administered in 2 patients (3%) with reactive hemophagocytic syndrome.
Nonsteroidal anti-inflammatory drugs (NSAIDs), alone or in combination with glucocorticoids, were employed in 54% (41/76) of patients. Information about the duration of applied treatment, particularly with respect to glucocorticoid regimen and tapering, was not included in the considered articles.
3.6. Outcome
Information about follow-up was available for 75 of the 81 patients. Of the 75 patients, 12 (16%) had a self-limiting or monocyclic systemic disease[42]. Six patients (8%) died: 3 within the first 3 months after diagnosis of the disease, 1 within 4 months, and 2 within a year of diagnosis. Four of these deaths were because of sepsis following immunosuppressive therapy. One patient died because of disease (she presented with high fever and pleural effusion at the disease onset and died shortly thereafter of respiratory failure despite immunosuppressive therapy), and 1 died shortly after the diagnosis of AOSD from unrelated causes.
The remaining patients (57/75; 76%%) had a polycyclic systemic or chronic articular polycyclic systemic disease[42] with persistent but temporarily resolved or controlled symptoms.
4. Discussion
The diagnosis of AOSD can be very difficult. There are no specific tests and reliance is usually placed on a symptom complex and the well-described typical rash seen in most patients. However, in recent years, other atypical cutaneous manifestations of AOSD have been reported; these rare presentations are not routinely recognized[6–38] Atypical cutaneous features often present in addition to the typical evanescent rash, but in 43% of the cases they are the only skin manifestation.[6,8,11,14,15,17,25,28–33,37,38]. A delayed diagnosis in these cases is still common, as the cutaneous lesions are often misdiagnosed as an allergic reaction to drugs, usually NSAIDs prescribed for joint symptoms or fever.
The appearance of atypical skin features in AOSD is not uncommon, with a prevalence of 14% according to our experience during a 30-year period. They may appear at any time over the course of the disease. In the great majority of cases, the atypical skin eruption presented at the time of disease onset concurrently with systemic symptoms, or shortly afterward. Rarely, cutaneous lesions were the presenting feature of AOSD preceding the appearance of the classic symptoms of the disease[9,13,23,29,30] or appeared during a flare several months after the diagnosis.[17,26,37]
The most representative and frequent lesion among the nonclassical skin rash is the development of persistent pruritic papules and/or plaques.[6–27] Descriptors for the color of these lesions varied from erythematous or brown in the majority of cases to, less commonly, violaceous. They may present with scales or crusts, and usually involve the back, upper chest, abdomen, and extensor surface of the extremities. This type of eruption often has a linear configuration, possibly because of the Koebner phenomenon, thus resembling a flagellate erythema.[6–19] Other commonly reported morphologic patterns include urticarial papules, lichenoid papules, pigmented plaques, prurigo pigmentosa-like, dermatomyositis-like, and lichen amyloidosis-like rashes.[6–18,20–27] More than 1 morphology or distribution pattern was observed in several patients.[6–27] Interestingly, persistent pruritic papules and plaques show a distinctive histological pattern characterized by singly or aggregated dyskeratotic/necrotic keratinocytes in the upper layers of the epidermis in association with a perivascular inflammatory infiltrate in the upper and mid-dermis.[6–13,15–20,23–27,43] Dermal mucin deposition is sometimes seen.
Other nonclassical skin lesions less frequently observed include urticaria and urticaria-like eruptions,[28–34] generalized or widespread non-pruritic persistent erythema,[18,35–37] vesiculopustular eruptions,[38,39] a widespread peau d’orange appearance of the skin (cutaneous mucinosa),[40] and edema of the eyelids mimicking dermatomyositis without any accompanying skin lesion.[18] In cases of urticaria/urticaria-like eruptions and generalized (widespread) nonpruritic persistent erythema, biopsy specimens reveal a nonspecific slight to moderate inflammatory infiltrate in the upper dermis, similar to that observed in biopsies of the typical evanescent skin rash.[1–3,43] Thus, a skin biopsy of atypical eruptions is recommended (specially in patients who do not yet fulfill the Yamaguchi criteria) because the histologic features and the previously discussed distinctive pattern can be considered to be highly suggestive of AOSD.
Additionally, association or co-occurrence of alopecia, acne-like lesions, erythema chronicum migrans, cutaneous polyarteritis nodosa, cutaneous vasculitis associated with mixed cryoglobulinemia, and Sweet's syndrome has been reported,[44–47] although it is not clear in these cases if the association is casual or a coincidence.
The great majority of patients with AOSD and atypical cutaneous lesions had persistent and severe disease, with a considerable frequency (23%) of clinical complications, including serositis, myopericarditis, lung involvement, abdominal pain, neurologic involvement, and reactive hemophagocytic syndrome[7,10,16–19,24,29,30,31,37]. Thus, most patients required medium or high doses of glucocorticoids (including intravenous methylprednisolone pulse therapy in some cases) and, in nearly 40%, a more potent or maintenance immunotherapy consisting of immunosuppressant drugs (including methotrexate, azathioprine, cyclosporine A, and hydroxychloroquine)[6,8–10,12,15,16,18–20,24,25,28,29,31,34,39,40] and/or biologic agents (mainly anakinra or tocilizumab)[8,10,15,21] to control or manage symptoms because they had an intermittent/polycyclic or chronic systemic course. The development of atypical cutaneous manifestations seems to be associated with a potentially worse prognosis (especially those with persistent pruritic papules and plaques with dermatomyositis-type appearance), with a mortality rate that reached 8% primarily because of infectious complications related to the immunosuppressive therapy[6,10,24]. In this sense, 21% of these patients had a score ≥7 at the time of disease onset in the Systemic Score System proposed by Pouchot et al.[41] This score system has a proven predictive value and a score ≥7.0 has demonstrated a strong prognostic impact in identifying patients at risk of AOSD-related death.[48]
In addition, 96% of patients with atypical cutaneous manifestations presented hyperferritinemia. During AOSD, persistent high serum levels of ferritin may be observed; it is an iron storage protein composed of 24 subunits, heavy (H) subunits and light (L) subunits. The ferritin enriched in L subunits (L-ferritin) and the ferritin enriched in H subunits (H-ferritin) may be observed in different tissues.[49] Recently, Ruscitti et al[49] have demonstrated an increased skin expression of H-ferritin in the biopsies obtained from persistent cutaneous lesions of AOSD patients, associated with a strong infiltrate of CD68+/H-ferritin+ cells. This finding seems to have clinical implications as according to their findings there is a correlation between both, the tissue H-ferritin levels and the CD68+/H-ferritin+ cells, with the severity of the clinical picture and the multi-visceral involvement of the disease.
In conclusion, the appearance of atypical cutaneous manifestations is not uncommon in AOSD. Recognition of this clinical variant is crucial for the early diagnosis of AOSD, as it might imply persistent disease activity and the need for more aggressive treatment. However, in interpreting the results of our study, we cannot ignore the pitfalls inherent in any systematic review including the relatively small number of identified patients, the retrospective design, and incomplete follow-up data in some cases. More studies are needed to confirm these results.
Footnotes
Abbreviations: AOSD = adult-onset Still's disease, NSAIDs = nonsteroidal anti-inflammatory drugs, SD = standard deviation.
All authors have made substantial contributions to all of the following: JN the conception and design of the study, or acquisition of data, or analysis and interpretation of data, (2) drafting the article or revising it critically for important intellectual content, and (3) final approval of the version to be submitted.
Ethics approval: In accordance with the guidelines of our institutional ethics committee, formal approval for this study was not required. The local ethics committee agreed that the findings in this report were based on normal clinical practice and were therefore suitable for dissemination. Informed consent of the patients was obtained, and their clinical records and information were anonymized. This study was conducted in accordance with the principles of the Declaration of Helsinki and the International Conference for Harmonization.
Submission declaration: The manuscript has not been accepted for publication elsewhere, is not being considered for publication elsewhere, and does not duplicate material already published. All authors have read the final version of the manuscript and agree to be coauthors.
The authors have no conflicts of interest to disclose.
References
- [1].Gerfaud-Valentin M, Jamilloux Y, Iwaz J, et al. Adult-onset Still's disease. Autoimmun Rev 2014;13:708–22. [DOI] [PubMed] [Google Scholar]
- [2].Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol 1992;19:424–30. [PubMed] [Google Scholar]
- [3].Efthimiou P, Laik PK, Bielory L. Diagnosis and management of adult onset Still's disease. Ann Rheum Dis 2006;65:564–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fautrel B, Le Moël G, Saint-Marcoux B, et al. Diagnostic value of ferritin and glycosylated ferritin in adult onset Still's disease. J Rheumatol 2001;28:322–9. [PubMed] [Google Scholar]
- [5].Fautrel B, Zing E, Golmard JL, et al. Proposal for a new set of classification criteria for adult-onset Still disease. Medicine (Baltimore) 2002;81:194–200. [DOI] [PubMed] [Google Scholar]
- [6].Lee JY, Hsu CK, Liu MF, et al. Evanescent and persistent pruritic eruptions of adult-onset Still disease: a clinical and pathologic study of 36 patients. Semin Arthritis Rheum 2012;42:317–26. [DOI] [PubMed] [Google Scholar]
- [7].Lübbe J, Hofer M, Chavaz P, et al. Adult-onset Still's disease with persistent plaques. Br J Dermatol 1999;141:710–33. [DOI] [PubMed] [Google Scholar]
- [8].Sun NZ, Brezinski EA, Berliner J, et al. Updates in adult-onset Still disease: atypical cutaneous manifestations and associations with delayed malignancy. Am Acad Dermatol 2015;73:294–303. [DOI] [PubMed] [Google Scholar]
- [9].Cho YT, Liao YH. Prurigo pigmentosa-like persistent papules and plaques in a patient with adult-onset Still's disease. Acta Derm Venereol 2014;94:102–3. [DOI] [PubMed] [Google Scholar]
- [10].Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still's disease. J Cutan Pathol 2010;37:932–7. [DOI] [PubMed] [Google Scholar]
- [11].Woods MT, Gavino AC, Burford HN, et al. The evolution of histopathologic findings in adult Still disease. Am J Dermatopathol 2011;33:736–9. [DOI] [PubMed] [Google Scholar]
- [12].Suzuki K, Kimura Y, Aoki M, et al. Persistent plaques and linear pigmentation in adult-onset Still's disease. Dermatology 2001;202:333–5. [DOI] [PubMed] [Google Scholar]
- [13].Kaur S, Bambery P, Dhar S. Persistent dermal plaque lesions in adult onset Still's disease. Dermatology 1994;188:241–2. [DOI] [PubMed] [Google Scholar]
- [14].Perez C, Montes M, Gallego M, et al. Atypical presentation of adult Still's disease with generalized rash and hyperferritinaemia. Br J Dermatol 2001;145:187–8. [DOI] [PubMed] [Google Scholar]
- [15].Wolgamot G, Yoo J, Hurst S, et al. Unique histopathologic findings in a patient with adult-onset Still disease. Am J Dermatopathol 2007;29:194–6. [DOI] [PubMed] [Google Scholar]
- [16].Said NH, Wong SN, Tan WH. A case of adult-onset Still's disease presenting with urticated plaques and acute myopericarditis. Indian J Dermatol 2013;58:405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kikuchi N, Satoh M, Ohtsuka M, et al. Persistent pruritic papules and plaques associated with adult-onset Still's disease: report of six cases. J Dermatol 2014;41:407–10. [DOI] [PubMed] [Google Scholar]
- [18].Nagai Y, Hasegawa M, Okada E, et al. Clinical follow-up study of adult-onset Still's disease. J Dermatol 2012;39:898–901. [DOI] [PubMed] [Google Scholar]
- [19].Ciliberto H, Kumar MG, Musiek A. Flagellate erythema in a patient with fever. JAMA Dermatol 2013;149:1425–6. [DOI] [PubMed] [Google Scholar]
- [20].Kavusi S, Paravar T, Hasteh F, et al. Atypical eruption but Still's: case report and review of the literature. Int J Dermatol 2015;54:3154–9. [DOI] [PubMed] [Google Scholar]
- [21].Michailidou D, Shin J, Forde I, et al. Typical evanescent and atypical persistent polymorphic cutaneous rash in an adult Brazilian with Still's disease: a case report and review of the literature. Auto Immun Highlights 2015;6:39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fernández-Guarino M, González C, Bardal Ruiz A, et al. Adult Still's disease with atypical skin manifestations. Actas Dermosifiliogr 2006;97:591–3. [DOI] [PubMed] [Google Scholar]
- [23].Tomaru K, Nagai Y, Ohyama N, et al. Adult-onset Still's disease with prurigo pigmentosa-like skin eruption. J Dermatol 2006;33:55–8. [DOI] [PubMed] [Google Scholar]
- [24].Yang CC, Lee JY, Liu MF, et al. Adult-onset Still's disease with persistent skin eruption and fatal respiratory failure in a Taiwanese woman. Eur J Dermatol 2006;16:593–4. [PubMed] [Google Scholar]
- [25].Yoshifuku A, Kawai K, Kanekura T. Adult-onset Still disease with peculiar persistent plaques and papules. Clin Exp Dermatol 2014;39:503–5. [DOI] [PubMed] [Google Scholar]
- [26].Affleck AG, Littlewood SM. Adult-onset Still's disease with atypical cutaneous features. J Eur Acad Dermatol Venereol 2005;19:360–3. [DOI] [PubMed] [Google Scholar]
- [27].Thien Huong NT, Pitche P, Minh Hoa T, et al. Persistent pigmented plaques in adult-onset Still's disease. Ann Dermatol Venereol 2005;132:693–6. [DOI] [PubMed] [Google Scholar]
- [28].Salaffi F, Filosa G, Bugatti L, et al. Urticaria as a presenting manifestation of adult-onset Still's disease. Clin Rheumatol 2010;19:389–91. [DOI] [PubMed] [Google Scholar]
- [29].Criado RF, Criado PR, Vasconcellos C, et al. Urticaria as a cutaneous sign of adult-onset Still's disease. J Cutan Med Surg 2006;10:99–103. [DOI] [PubMed] [Google Scholar]
- [30].Criado PR, de Carvalho JF, Ayabe LA, et al. Urticaria and dermographism in patients with adult-onset Still's disease. Rheumatol Int 2012;32:2551–5. [DOI] [PubMed] [Google Scholar]
- [31].Cozzi A, Papagrigoraki A, Biasi D, et al. Cutaneous manifestations of adult-onset Still's disease: a case report and review of literature. Clin Rheumatol 2016;35:1377–82. [DOI] [PubMed] [Google Scholar]
- [32].Setterfield JF, Hughes GR, Kobza Black A. Urticaria as a presentation of adult Still's disease. Br J Dermatol 1998;138:906–8. [DOI] [PubMed] [Google Scholar]
- [33].Soy M. A case of adult-onset Still's disease presenting as angioedema. Clin Rheumatol 2004;23:92. [DOI] [PubMed] [Google Scholar]
- [34].Tseng HC, Lee CH. Refractory urticaria in adult-onset Still's disease. Rheumatol Int 2014;34:1029–30. [DOI] [PubMed] [Google Scholar]
- [35].Fujii K, Konishi K, Kanno Y, et al. Persistent generalized erythema in adult-onset Still's disease. Int J Dermatol 2003;42:824–5. [DOI] [PubMed] [Google Scholar]
- [36].Maza A, Renard L, Monestier S, et al. Fever with skin rash and polyarthralgia in a genetically black-skinned woman. Med Trop (Mars) 2008;68:297–9. [PubMed] [Google Scholar]
- [37].Sarkar RN, Bhattacharya R, Bhattacharyya K, et al. Adult onset Still's disease with persistent skin lesions complicated by secondary hemophagocytic lymphohistiocytosis. Int J Rheum Dis 2014;17:118–21. [DOI] [PubMed] [Google Scholar]
- [38].Lee JB, Kim JW, Lee SS, et al. Adult onset Still's disease with vesiculopustules on the hands and feet. J Korean Med Sci 2002;17:852–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bachmeyer C, Blum L, Petitjean B, et al. Vesiculopustules in adult-onset Still's disease. J Am Acad Dermatol 2006;54(5 suppl):S247–8. [DOI] [PubMed] [Google Scholar]
- [40].Phillips WG, Weller R, Handfield-Jones SE, et al. Adult Still's disease. Br J Dermatol 1994;130:511–3. [DOI] [PubMed] [Google Scholar]
- [41].Pouchot J, Sampalis JS, Beaudet F, et al. Adult Still's disease: manifestations, disease course, and outcome in 62 patients. Medicine (Baltimore) 1991;70:118–36. [PubMed] [Google Scholar]
- [42].Cush JJ, Medsger TA, Jr, Wallace CC, et al. Adult-onset Still's disease. Clinical course and outcome. Arthritis Rheum 1987;30:186–94. [DOI] [PubMed] [Google Scholar]
- [43].Lee JY, Yang C, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still's disease. J Am Acad Dermatol 2005;52:1003–8. [DOI] [PubMed] [Google Scholar]
- [44].Ohta A, Yamaguchi M, Kaneoka H, et al. Adult Still's disease: review of 228 cases from the literature. J Rheumatol 1987;14:1139–46. [PubMed] [Google Scholar]
- [45].Mylona E, Vadala C, Papadakos V, et al. Cutaneous polyarteritis nodosa in adult onset Still's disease. Eur J Dermatol 2009;19:621–2. [DOI] [PubMed] [Google Scholar]
- [46].Elezoglou AV, Giamarelos-Bourboulis E, Katsilambros N, et al. Cutaneous vasculitis associated with mixed cryoglobulinemia in adult Still's disease. Clin Exp Rheumatol 2003;21:405–6. [PubMed] [Google Scholar]
- [47].Elinav H, Maly A, Ilan Y, et al. The coexistence of Sweet's syndrome and Still's disease: is it merely a coincidence? J Am Acad Dermatol 2004;50:S90–2. [DOI] [PubMed] [Google Scholar]
- [48].Ruscitti P, Cipriani P, Masedu F, et al. Adult-onset Still's disease: evaluation of prognostic tools and validation of the systemic score by analysis of 100 cases from three centers. BMC Med 2016;14:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ruscitti P, Cipriani P, Ciccia F, et al. H-ferritin and CD68(+)/H-ferritin(+) monocytes/macrophages are increased in the skin of adult-onset Still's disease patients and correlate with the multi-visceral involvement of the disease. Clin Exp Immunol 2016;186:30–8. [DOI] [PMC free article] [PubMed] [Google Scholar]