

Summary
Atherosclerosis is characterized by a proliferation of vascular smooth muscle cells (VSMCs) and their migration to the intima, which induces thickening of the intima itself, but the mechanism remains poorly understood. Low molecular weight heparin (LMWH) inhibits the proliferation of VSMCs. Previous studies have shown that a LMWH, parnaparin (PNP), acts on the processes of atherogenesis and atheroprogression in experimental animal models. The aim of this study was to investigate the involvement of oxidative stress, inflammation and VSMCs in the regulation of vascular wall homeostasis. We also considered the possibility of restoring vascular pathological changes using PNP treatment. In order to evaluate vascular remodelling in this study we have analysed the morphological changes in aortas of an animal model of atherosclerosis, apolipoprotein E‐deficient mice (ApoE−/−) fed with a normal or a western diet without treatment or treated with PNP. We also analysed, by immunohistochemistry, the expression of proteins linked to atherogenesis and atheroprogression – an enzyme involved in oxidative stress, iNOS, examples of inflammatory mediators, such as tumour necrosis factor alpha (TNF‐α), interleukins 1 and 6 (IL‐1 and IL‐6), and markers of VSMC changes, in particular plasminogen activator inhibitor‐1 and thrombospondin‐1 (PAI‐1 and TSP‐1). Our results could suggest that PNP downregulates VSMC proliferation and migration, mediated by PAI‐1 and TSP‐1, and reduces inflammation and oxidative stress in vessels. These data suggested that LMWH, in particular PNP, could be a theoretically practical tool in the prevention of atherosclerotic vascular modification.
Keywords: atherosclerosis, intimal hyperplasia, low molecular weight heparin, parnaparin
In recent years, atherosclerosis has become a serious health challenge. This is a progressive disease that is still considered a major cause of mortality in the industrialized world. The genesis and the progression of atherosclerotic plaque have been well described in morphological studies on arterial wall of genetically modified mice, although the aetiology and pathophysiological mechanism remain the object for continuing discussions (Musumeci et al. 2014a). This disease is a complicated process and not a simple passive accumulation of lipids within the vascular wall. Endothelial dysfunction, characterized by an increase in adhesion molecules, is a key early event in plaque genesis that leads to the infiltration of leucocytes and macrophages into the subendothelial space. These cells, originating from monocytes, differentiate into phagocytes and ingest the accumulated lipoproteins, forming ‘foam cells’ that promote disease progression (Tian et al. 2005; Musumeci et al. 2014b). These steps are characterized by inflammation, metabolic alterations and oxidative stress. Oxidative stress, which is characterized by high levels of reactive oxygen species (ROS), damages the cellular components and leads to a response to damage that involves inflammation.
Inflammation, if it is not able to neutralize harmful agents, induces proliferation and migration of vascular smooth muscle cells (VSMCs) from the media layer into the intima layer, producing extracellular matrix that acts as a scaffold of the plaque (Bonomini et al. 2008; Musumeci et al. 2014b).
Reactive oxygen species are overproduced in the atherosclerotic process correlated with alterations of endogenous endothelial nitric oxide synthase (eNOS), which induces a decrease in available endothelial nitric oxide (NO). The decrease in NO has several significant effects on arteries inducing proinflammatory, prothrombotic and proconstrictive vascular activities (Bonomini et al. 2008; Breen et al. 2012). On the other hand, ROS increase inducible nitric oxide synthase (iNOS) expression in macrophages and smooth muscle cells in different stages of atherosclerotic lesions. iNOS plays a role in inflammation through the production of prostanoids and NO and is thus also implicated in the development of atherosclerotic lesions with proatherosclerotic effects (Napoli et al. 2006).
Recently, it was shown that atherosclerosis is characterized by both an increase in the number of smooth muscle cells (SMCs) and migration of these cells to the intimal layer, leading to intimal thickening (hyperplasia) (Karki et al. 2013). The vascular homeostasis alterations, leading to arterial dysfunction, are still intriguing and poorly understood (Rodella et al. 2007).
The mechanisms of intimal hyperplasia and consequent restenosis can be described as the complex involvement of several factors and cytokines including tumour necrosis factor‐α (TNF‐α), interleukin 1 (IL‐1) and 6 (IL‐6) that induce proliferation and migration of VSMCs (Takeda et al. 2005).
Our research group demonstrated that during vascular disease, VSMCs exhibited a phenomenon of dedifferentiation involving the expression of thrombospondin‐1 (TSP‐1), a member of a family of related glycoproteins. In particular, TSP‐1 is secreted by numerous cell types, including platelets, endothelial cells, macrophages, fibroblasts and VSMCs. TSP‐1 expression is elevated with hypercholesterolaemia in vivo. In the vessel wall of atherosclerotic mice there is an increase in plasminogen activator inhibitor‐1 (PAI‐1) that affects on infiltration of cells into plaque, proliferation, migration and apoptosis of VSMCs and accumulation and composition of extracellular matrix in plaque modulating atherogenesis (Rodella et al. 2012).
Over the past decade apolipoprotein E (ApoE)‐deficient mice (ApoE−/− mice) have been commonly used as a model for experimental atherosclerosis research and to examine nutritional and pharmacological interventions. The outstanding success of this animal model, which exhibits delayed clearance of lipoproteins, is due to its ready availability and the rapid development of the full morphological spectrum of atherosclerotic lesions as detected in humans (Coleman et al. 2006; Desai et al. 2008).
To date, there are some papers on the potential effects of low molecular weight heparin (LMWH) as anti‐coagulant medications for the treatment of vascular diseases (Wong & Giugliano 2003; Hirsh & Raschke 2004; Clark 2008). In particular, Artico et al. (2011) demonstrated the possible influence of LMWH‐Parnaparin® (PNP) on atherosclerosis in laboratory animals (ApoE‐deficient mice). These experiments have demonstrated a possible role of the PNP in the control of atherogenic disease. In fact, in PNP‐treated animals, there is a reduction in number and size of atherosclerotic lesions in the aortic wall.
On the basis of this previous study, our goal was to evaluate the mechanism by which PNP reduces atherosclerotic alterations in vessels of ApoE−/− mice. Therefore we evaluated the expression of proteins linked to atherogenesis and atheroprogression, in particular an enzyme involved in oxidative stress, iNOS, some inflammatory mediators (TNF‐α, IL‐1, IL‐6) and markers of VSMC alterations (PAI‐1 and TSP‐1). These data support further studies on PNP as a therapeutic strategy.
Methods
Parnaparin
Parnaparin® was kindly provided by the manufacturer, Alfa Wassermann S.p.A. (Macchi & Maggiore 1987; Dettori & Babbini 1992; Marchi 1992; Frampton & Faulds 1994; Dettori 1995). This substance is the sodium salt of a LMWH derived from heparin of porcine intestinal mucosa by radical‐catalysed depolymerization with hydrogen peroxide and cupric salt (Artico et al. 2011).
Ethical approval statement
All the experiments were conducted according to the guidelines formulated by the European Community for experimental animal use (L358‐86/609EEC) and were approved by the Italian Ministry of Health.
Animals
The deletion of the gene for apolipoprotein E (ApoE‐deficient mice) induces severe hypercholesterolaemia and spontaneous atherosclerosis development. Male ApoE‐deficient mice (18–20 g) were fed with a standard diet (A1, A2 and A3 groups: 20 animals) or western diet (B1, B2 and B3 groups: 20 animals) for a period of 18 weeks. The western‐type diet, characterized by 0.2% of cholesterol and 42% of fat, was purchased from Harlan Teklad, Inc. (Td 88137, Indianapolis, IN, USA). These two groups of mice were further divided into six subgroups (three per group): control mice without treatment (groups A1 and B1, five animals for group) or mice subcutaneously injected with PBS (groups A2 and B2, five animals for group) and mice treated for six days/week with subcutaneous injection of PNP in PBS at the dose of 5 mg/kg (groups A3 and B3, 10 animals for group). Animals were weighed weekly before injecting the optimal PNP concentration. The drug concentration used to treat ApoE−/− mice was chosen according to preliminary experiments on ApoE−/− mice (Artico et al. 2011). In the light of the results of these preliminary experiments, 5 mg/kg PNP was chosen as the optimal dose used for animal treatment.
Morphological evaluation
After dissection, aortas were washed in phosphate‐buffered saline (PBS, 0.1 M, pH 7.4), fixed with 10% buffered formalin, embedded in paraffin, using a standard procedure, and cut in serial sections, with thickness of 5 μm, using a microtome. For morphological evaluation, the sections were stained with haematoxylin and eosin, Verhoeff's elastin stain (Verhoeff 1908) and Sirius Red.
Immunohistochemical analysis
For immunohistochemical analysis, sections of aortas were processed according to an avidin complexed with biotinylated peroxidase technique (ABC/HRP). The sections, after dehydration, were immersed in citrate buffer (pH 6) and subjected to microwave irradiation twice for 5 mins. Subsequently, for quenching endogenous peroxidase activity, all sections were immersed for 30 mins in 0.3% hydrogen peroxide in methanol. To block non‐specific binding, the slides were incubated for 1 h at room temperature with mouse Ig‐blocking reagent (M.O.M.) (Vector Laboratories, Burlingame, CA, USA). The sections were then incubated overnight at 4°C with primary antibodies at concentrations assessed in preliminary experiments, in particular rabbit anti‐IL‐1β polyclonal antibody (1:50, Santa Cruz Biotechnology, Santa Cruz, CA, USA); rabbit anti‐IL‐6 polyclonal antibody (1:100, Santa Cruz Biotechnology); mouse anti‐TNF‐α monoclonal antibody (1:100, Santa Cruz Biotechnology); iNOS (1:50, Santa Cruz Biotechnology); PAI‐1 (1:50, Santa Cruz Biotechnology); TSP‐1 (1:50, Santa Cruz Biotechnology). Samples, after incubation with primary antibodies, were rinsed twice in PBS, exposed for 1 h at room temperature to the appropriate secondary biotinylated goat anti‐mouse or anti‐rabbit IgG (Vector Laboratories, BA9200 and BA1000) and then treated with peroxidase‐conjugated avidin (Vectastain Elite ABC Kit Standard* PK 6‐100) for 35 mins. After this step, the slides were treated with 0.05% 3,3‐diaminobenzidine (DAB) with 0.1% H2O2 (DAB substrate kit for peroxidase, Vector Laboratories SK‐4100). The sections were counterstained with Mayer's haematoxylin (TSP‐1, PAI‐1 and iNOS) or with haematoxylin–eosin (IL‐1β, IL‐6 and TNF‐α) and observed using a light microscope. Negative control experiments were carried out in different ways: (i) without the primary antibody incubation; (ii) by substituting the primary antibody with an equivalent amount of immunoglobulins; (iii) by preincubating the primary antibody with the specific blocking peptide (according to the supplier's instructions). The staining was evaluated by two competent observers, and the immunopositivity was assessed microdensitometrically using an IAS 2000 image analyzer (Delta Sistemi, Rome, Italy) connected to the microscope by a camera. The software has a function that allows quantifying the integrated optical density (IOD) measurements. Briefly, after the grey‐scale conversion of the images, we set the threshold values of black and white and created the optical density‐scale calibration using known algorithms. This calibration was then applied for each capture imported in the software for processing. The background obtained in sections exposed to non‐immune serum was considered as zero in system calibration. In each section, ten 100‐μm2 areas were considered. The quantitative results regarding the immunopositivity were analysed statistically using analysis of variance (anova) followed by Duncan's multiple range test.
Results
Morphology
The morphological and biochemical results were considered without distinction between the ApoE−/− groups without treatment and PBS‐treated animals because they were similar.
Morphological evaluation of samples from ApoE−/− mice groups without treatment revealed marked damage, with the presence of atherosclerotic lesions in the vessel wall, in particular in the aortic root, with elastic fibre disorganization, increased connective tissue deposition and fibrosis. The intimal layer was commonly characterized by lipid droplets; in particular, there were lipid‐rich macrophages called ‘foam cells’. The atherosclerotic plaque region was acellular in the central necrotic lipid core, sometimes protruding into the lumen and was characterized by a fibrous cap. PNP treatment in ApoE−/− mice reduced plaque formation and injury‐induced remodelling, especially at the level of the aortic root (Figures 1, 2).
Figure 1.
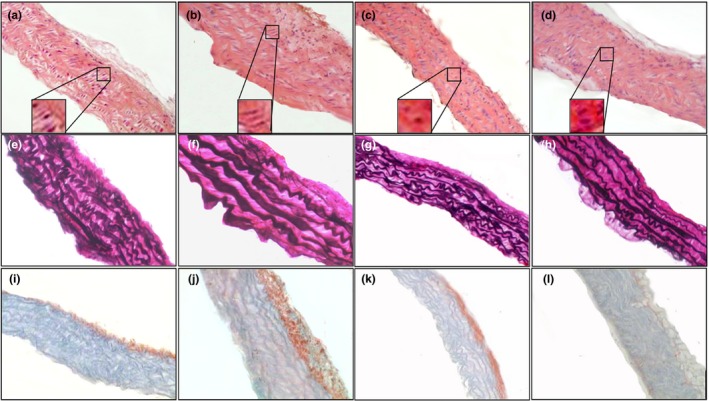
Histology of aorta of ApoE−/− mice fed with normal diet (a, e, i), western diet (b, f, j), normal diet plus parnaparin (PNP) treatment (c, g, k) and western diet plus PNP treatment (d, h, l), stained with haematoxylin and eosin (a–d), Verhoeff's elastin staining (e–h) and Sirius Red staining (i–l) (200×). Insets show the morphology of a typical vascular smooth muscle cell in aorta of ApoE−/− mice fed with normal diet (a), western diet (b), normal diet plus PNP treatment (c) and western diet plus PNP treatment (d) (1000×).
Figure 2.
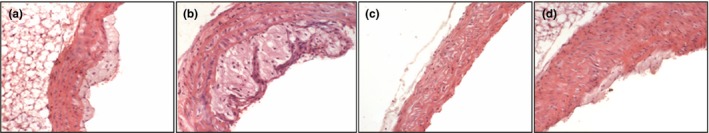
Histology of aortic root of ApoE−/− mice fed with normal diet (a), western diet (b), normal diet plus parnaparin (PNP) treatment (c) and western diet plus PNP treatment (d), stained with haematoxylin and eosin (200×).
Haematoxylin and eosin staining indicated that neo‐intima formation was present in ApoE−/− mice without treatment (Figure 1a,b). VSMCs had a different morphology in ApoE−/− mice vessels compared to PNP‐treated aorta. In the tunica media of ApoE−/− mice, VSMCs were slightly elongated and had cobblestone morphology (Figure 1a,b insets). Most VSMCs also underwent mitosis, which is aligned in a circumferential direction. On the contrary in vessels of mice with PNP administration, VSMCs had a central nucleus and were spindle‐shaped (Figure 1c,d insets). Sometimes, VSMCs underwent mitosis.
Verhoeff‐Van Gieson staining showed that in ApoE−/− mice, there is disorganization of elastic fibres, which sometimes appeared discontinuous, with a consequent reduction in vascular elasticity (Figure 1e,f). The aorta of animals treated with PNP, instead, had a regular pattern of elastic fibres (Figure 1g,h).
Sirius Red staining, a method for the determination of collagen content in the aorta, showed that in ApoE−/− mice, there are high total collagen levels in aorta (Figure 1i,j), whereas the administration of PNP reduced the collagen fibre deposition (Figure 1k,l).
Immunohistochemistry
In the aorta of ApoE−/− mice on a western diet, there was an appreciable immunoreactivity for inflammatory markers. In particular, IL‐1β was present in VSMCs and in the fibrous plaque (Figure 3b). In ApoE−/− mice fed with normal diet or animals treated with PNP, IL‐1 expression was moderate, in particular in the endothelial layer (Figure 3a,c,d).
Figure 3.
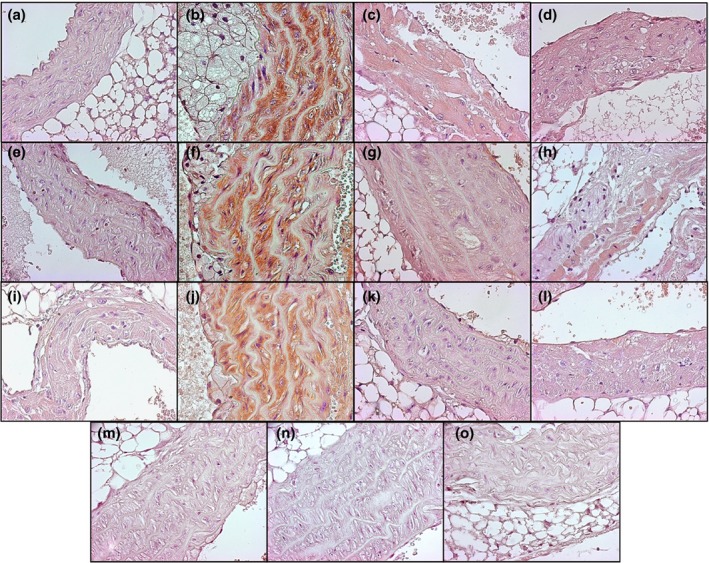
Immunolocalization of inflammatory markers IL‐1β (a–d), IL‐6 (e–h) and TNF‐α (i–l) in aorta of ApoE−/− mice fed with normal diet (a, e, i), western diet (b, f, j), normal diet plus parnaparin (PNP) treatment (c, g, k) and western diet plus PNP treatment (d, h, l). Immunohistochemical negative controls for IL‐1β (m), IL‐6 (n) and TNF‐α (o) (400×).
IL‐6 immunostaining was observed sporadically in the endothelial cells of ApoE−/− animals fed with normal diet (Figure 3e). Strongly positive immunostaining for IL‐6 was observed in endothelial and smooth muscle layers in atherosclerotic mice fed with a western diet (Figure 3f). In particular, the immunopositivity was evident in areas characterized by the infiltration of macrophages. The immunopositivity for IL‐6 was significantly lower in the PNP‐treated mice in comparison with non‐PNP‐treated mice in particular those on a western diet (Figure 3g,h).
Immunohistochemical analysis showed that TNF‐α level was very low in ApoE−/− mice fed with normal diet (Figure 3i). In contrast, immunoreactivity showed that TNF‐α was strongly expressed in mice fed with a western diet (Figure 3j), while in PNP‐treated animals, the protein expression was reduced (Figure 3k,l).
Immunohistochemical analyses of TSP‐1 (Figure 4a–d) and PAI‐1 (Figure 4e–h) proteins showed expression in tunica media of ApoE−/− mice fed with normal diet (Figure 4a,e); however, its staining was diffuse and it was markedly increased in vessels of ApoE−/− mice fed with western diet (Figure 4b,f). The PNP treatment reduced expression of both these proteins in vessels of ApoE−/− mice fed with both diets (Figure 4c,d,g,h).
Figure 4.
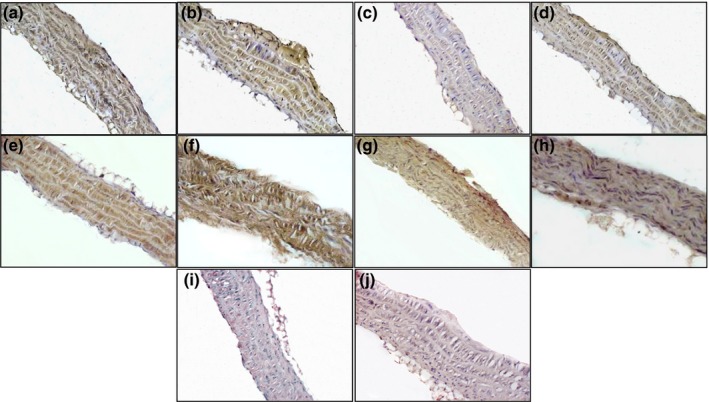
Immunolocalization of TSP‐1 (a–d) and PAI‐1 (e–h) in aorta of ApoE−/− mice fed with normal diet (a, e), western diet (b, f), normal diet plus parnaparin (PNP) treatment (c, g) and western diet plus PNP treatment (d, h). Immunohistochemical negative controls for TSP‐1 (i) and PAI‐1 (j) (200×).
Augmented expression of iNOS protein was evident in ApoE−/− mice without PNP treatment, in particular in animals maintained on western diet. The staining was diffusely and intracellular and was distributed both in tunica intima and in tunica media (Figure 5a,b). The protein expression was reduced in ApoE−/− mice after treatment with PNP either on normal or on western diet (Figure 5c,d).
Figure 5.
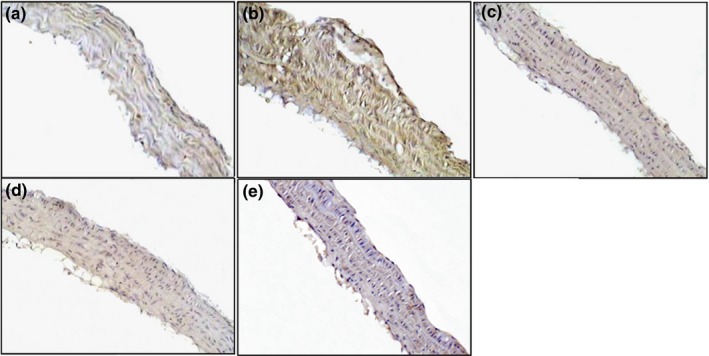
Immunolocalization of iNOS in aorta of ApoE−/− mice fed with normal diet (a), western diet (b), normal diet plus parnaparin (PNP) treatment (c) and western diet plus PNP treatment (d). Immunohistochemical negative control (e) (200×).
The results of immunopositivity were confirmed by statistical analysis (Figure 6).
Figure 6.
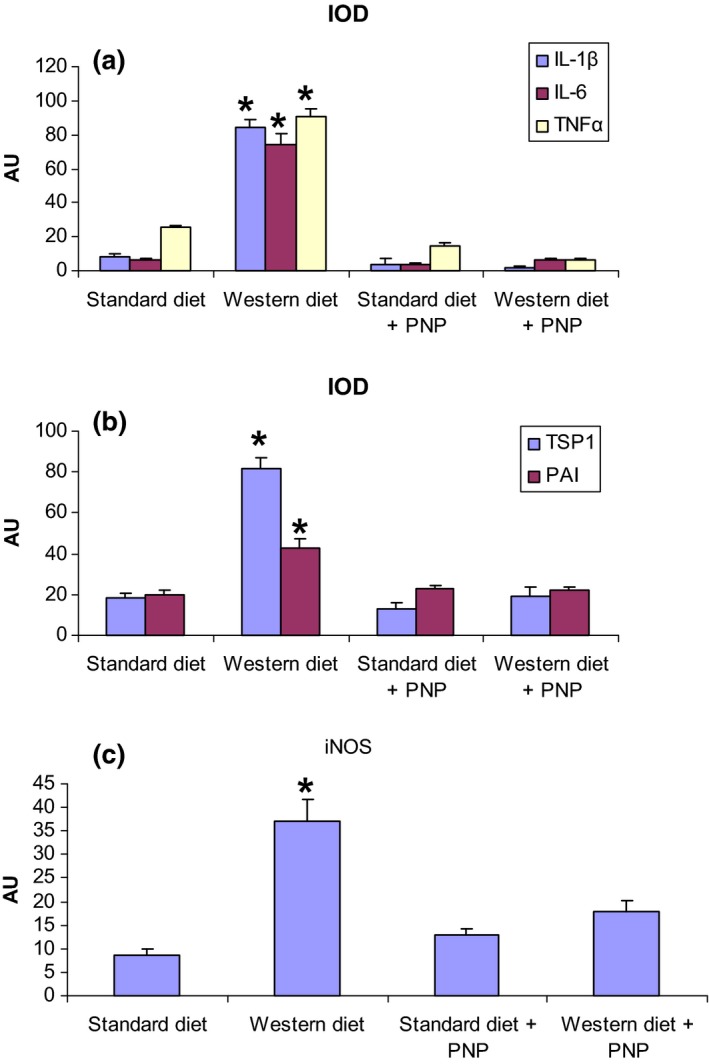
Quantitative analyses of inflammatory mediators (a), of markers of vascular smooth muscle cells alterations (b) and of an enzyme involved in oxidative stress, iNOS (c) presented in bar graphs; representative graphs with integrated optical density (IOD) readings are expressed as arbitrary units (AU) ± standard deviation (SD) (c). *P < 0.05 vs. ApoE−/− with standard diet.
Discussion
From the literature, it is known that in the development of atherosclerotic plaque there are histopathological alterations in both tunica intima and tunica media. In particular during the early stages of lesion formation, there is endothelial dysfunction, appearance and accumulation of lipid‐filled macrophages in the subendothelium leading to ‘fatty streaks’. In the tunica media the elastic membranes become disrupted and discontinuous and the muscle cells appear to proliferate (Tian et al. 2005; Coleman et al. 2006; Rodella et al. 2007; Musumeci et al. 2014a,b). In early atherosclerosis, there are also modifications in localization and expression of some proteins, such as iNOS, VEGF and MMP2 (Rodella et al. 2007).
Previous data from the literature demonstrated that ApoE is an important protein in the pathology of atherosclerosis and the generation of ApoE−/− mice has been one of the most critical advances in the elucidation of factors affecting atherogenesis (Rodella et al. 2007; Bonomini et al. 2010).
Our results showed that PNP treatment in ApoE−/− mice inhibits injury‐induced remodelling of vessels and could be therapeutic in control of the neo‐intimal hyperplasia.
The underlying mechanism responsible for restenosis during atherosclerotic disease is not completely understood. Previous studies have suggested that LMWH, such as PNP, could inhibit VSMC proliferation, which is the major pathologic change in occlusive disease (Zhao et al. 2011).
Low molecular weight heparins, such as PNP, bind to anti‐thrombin (AT) III with a unique pentasaccharide sequence for performing their main anti‐coagulant effect, like heparin, but have a major bioavailability (Artico et al. 2004).
Recent studies have shown that LMWHs inhibited bovine VSMC growth and pulmonary vascular remodelling induced by hypoxia in rodents (Yu et al. 2005; Mrabat et al. 2009). Because vascular atherosclerosis is a typical VSMC proliferative lesion, it is reasonable to hypothesize that PNP could ameliorate vascular remodelling in atherosclerosis‐prone mice, such as ApoE−/−.
Some authors have suggested that the protective role of LMWHs in inhibiting the proliferative lesion in vessels was not related to its anti‐coagulant property in a mouse model of transluminal injury, but to its anti‐proliferative properties and the consequent reduction in neo‐intima formation (Zhao et al. 2011). Other researchers have demonstrated that preventing growth factors from binding to their receptors decreases oncogene expression at the injured site of artery wall (Hibino et al. 2005; Ariyoshi et al. 2008; Relini et al. 2008). However, mechanisms by which LMWHs work as an inhibitor against arterial remodelling still remain unknown.
The passage into the pathological intimal thickening, that is the initial recognized form of an atherosclerotic lesion, is correlated with modifications in the morphological and biochemical features of the VSMCs and of their extracellular matrix, which leads to the release of hydrolytic enzymes, cytokines, chemokines and inflammatory factors such as tumour necrosis factor‐α (TNF‐α), which is the principal factor involved in the pathogenesis of atherosclerosis (Cao et al. 2015). Therefore, the use of molecules, such as PNP, that may inhibit TNF‐α‐induced proliferation and migration of VSMCs could be advantageous in discovering drugs full in treatment that could be use for vascular disease, such as atherosclerosis.
Critical players in regulating VSMC proliferation and migration are TSP‐1 and PAI‐1 which may represent targets for clinical therapeutic interventions.
On the basis of the experimental and clinical findings, increased levels of PAI‐1 could be related to repair processes that modulate the local proteolytic potential, resulting in cell migration, and then could be a marker of a tissue proliferative repair processes and of atherosclerosis progression (Schiffrin et al. 2007). PAI‐1 can also have a role in the regulation of the local inflammation.
Some authors have demonstrated the involvement of TSP‐1 in plaque progression and, in particular, that this protein expression may be regulated by inflammatory cells. Indeed, TSP‐1 expression was demonstrated in the wall of atherosclerosis‐prone vessels and not in vessels in physiologic conditions.
Our findings could demonstrate that LMWHs, in particular PNP, exert an anti‐inflammatory effect, by regulating cytokine expression and monocyte recruitment to endothelium (Christopherson et al. 2002; Kereiakes 2003).
Collectively, our data could suggest that PNP reduced higher levels of TSP‐1, PAI‐1, inflammatory markers and iNOS expression in atherosclerotic vessels and that PNP might play a previously unrecognized role in preventing neo‐intima formation in atherosclerosis‐prone mice vessels. This is the first evidence that PNP prevents the development of advanced atherosclerotic plaques. If PNP does inhibit abnormal vascular remodelling, and alter oxidative stress and inflammation, then its protective effect may be useful not only in advancing our understanding of mechanism but also as an adjunct in therapy. Even if PNP itself does not prove to be the therapeutic modality, technological advances now permit structural modifications of heparin, and it may be possible to synthesise variants which have stronger anti‐proliferative and less anti‐coagulant activity.
Thus the present research has clinical significance, and suggests that more work in vitro be performed to elucidate the underlying mechanism by which heparin, and in particular PNP, could prohibit VSMC proliferation.
Conflict of interest
The authors declare no conflict of interest.
Funding source
This study was supported by institutional grant (ex‐60%) of the University of Brescia.
Acknowledgements
We sincerely thank Miss Lorena Giugno and Miss Castrezzati Stefania for their technical support.
References
- Ariyoshi W., Takahashi T., Kanno T. et al (2008) Heparin inhibits osteoclastic differentiation and function. J. Cell. Biochem. 103, 1707–1717. [DOI] [PubMed] [Google Scholar]
- Artico M., De Santis S., Tranquilli Leali F.M., Cavallotti D., Celestini A. & Cavallotti C. (2004) Diabetic rats treated by low molecular weight heparin OP 2123/parnaparin: morphological changes in the kidney and heart. J. Diabetes Complications 18, 119–125. [DOI] [PubMed] [Google Scholar]
- Artico M., Riganò R., Buttari B. et al (2011) Protective role of parnaparin in reducing systemic inflammation and atherosclerotic plaque formation in ApoE−/− mice. Int. J. Mol. Med. 27, 561–565. [DOI] [PubMed] [Google Scholar]
- Bonomini F., Tengattini S., Fabiano A., Bianchi R. & Rezzani R. (2008) Atherosclerosis and oxidative stress. Histol. Histopathol. 23, 381–390. [DOI] [PubMed] [Google Scholar]
- Bonomini F., Filippini F., Hayek T. et al (2010) Apolipoprotein E and its role in aging and survival. Exp. Gerontol. 45, 149–157. [DOI] [PubMed] [Google Scholar]
- Breen D.M., Dolinsky V.W., Zhang H. et al (2012) Resveratrol inhibits neointimal formation after arterial injury through an endothelial nitric oxide synthase‐dependent mechanism. Atherosclerosis 222, 375–381. [DOI] [PubMed] [Google Scholar]
- Cao Q., Jiang Y., Shi J. et al (2015) Artemisinin inhibits the proliferation, migration, and inflammatory reaction induced by tumor necrosis factor‐α in vascular smooth muscle cells through nuclear factor kappa B pathway. J. Surg. Res. 194, 667–678. [DOI] [PubMed] [Google Scholar]
- Christopherson K.W. 2nd, Campbell J.J., Travers J.B. & Hromas R.A. (2002) Low‐molecular‐weight heparins inhibit CCL21‐induced T cell adhesion and migration. J. Pharmacol. Exp. Ther. 302, 290–295. [DOI] [PubMed] [Google Scholar]
- Clark N.P. (2008) Low‐molecular‐weight heparin use in the obese, elderly, and in renal insufficiency. Thromb. Res. 123(Suppl 1), S58–S61. [DOI] [PubMed] [Google Scholar]
- Coleman R., Hayek T., Keidar S. & Aviram M. (2006) A mouse model for human atherosclerosis: long‐term histopathological study of lesion development in the aortic arch of apolipoprotein E‐deficient (E0) mice. Acta Histochem. 108, 415–424. [DOI] [PubMed] [Google Scholar]
- Desai A., Zhao Y. & Warren J.S. (2008) Development of atherosclerosis in Balb/c apolipoprotein E‐deficient mice. Cardiovasc. Pathol. 17, 233–240. [DOI] [PubMed] [Google Scholar]
- Dettori A.G. (1995) Parnaparin: a review of its pharmacological profile and clinical application. Drugs Today 31, 19–35. [Google Scholar]
- Dettori A.G. & Babbini M. (1992) Human pharmacology of a low molecular weight heparin (Alfa‐LMWH): an update. Med. Res. Rev. 12, 373–389. [DOI] [PubMed] [Google Scholar]
- Frampton J.E. & Faulds D. (1994) Parnaparin. A review of its pharmacology and clinical application in the preventive and treatment of thromboembolic and other vascular disorders. Drugs 47, 652–676. [DOI] [PubMed] [Google Scholar]
- Hibino S., Shibuya M., Hoffman M.P. et al (2005) Laminin alpha5 chain metastasis‐ and angiogenesis‐inhibiting peptide blocks fibroblast growth factor 2 activity by binding to the heparan sulfate chains of CD44. Cancer Res. 65, 10494–10501. [DOI] [PubMed] [Google Scholar]
- Hirsh J. & Raschke R. (2004) Heparin and low‐molecular‐weight heparin: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 126(Suppl), 188S–203S. [DOI] [PubMed] [Google Scholar]
- Karki R., Jeon E.R. & Kim D.W. (2013) Nelumbo nucifera leaf extract inhibits neointimal hyperplasia through modulation of smooth muscle cell proliferation and migration. Nutrition 29, 268–275. [DOI] [PubMed] [Google Scholar]
- Kereiakes D.J. (2003) Adjunctive pharmacotherapy before percutaneous coronary intervention in non‐ST‐elevation acute coronary syndromes: the role of modulating inflammation. Circulation 108(Suppl 1), III22–III27. [DOI] [PubMed] [Google Scholar]
- Macchi M. & Maggiore L. (1987) OP‐2123. Drugs Future 12, 1111–1112. [Google Scholar]
- Marchi E.G. (1992) Alfa Wassermann low molecular weight heparin In: Low Molecular Weight Heparins in Clinical Practice, pp. 237–244 (ed. Doutremepuich C.), New York, Basel, Hong Kong: Marcel Dekker Inc. [Google Scholar]
- Mrabat H., Garg H.G. & Hales C.A. (2009) Growth inhibition of bovine pulmonary artery smooth muscle cells following long‐term heparin treatment. J. Cell. Physiol. 221, 603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musumeci G., Coleman R., Imbesi R. et al (2014a) ADAM‐10 could mediate cleavage of N‐cadherin promoting apoptosis in human atherosclerotic lesions leading to vulnerable plaque: a morphological and immunohistochemical study. Acta Histochem. 116, 1148–1158. [DOI] [PubMed] [Google Scholar]
- Musumeci G., Imbesi R., Magro G. et al (2014b) N‐cadherin has a protective role in stable human atherosclerotic plaques: a morphological and immunohistochemical study. J. Histol. Histopathol. 1, 4. [Google Scholar]
- Napoli C., de Nigris F., Williams‐Ignarro S., Pignalosa O., Sica V. & Ignarro L.J. (2006) Nitric oxide and atherosclerosis: an update. Nitric Oxide 15, 265–279. [DOI] [PubMed] [Google Scholar]
- Relini A., De Stefano S., Torrassa S. et al (2008) Heparin strongly enhances the formation of beta2‐microglobulin amyloid fibrils in the presence of type I collagen. J. Biol. Chem. 283, 4912–4920. [DOI] [PubMed] [Google Scholar]
- Rodella L.F., Bonomini F., Rezzani R. et al (2007) Atherosclerosis and the protective role played by different proteins in apolipoprotein E‐deficient mice. Acta Histochem. 109, 45–51. [DOI] [PubMed] [Google Scholar]
- Rodella L.F., Rossini C., Favero G., Foglio E., Loreto C. & Rezzani R. (2012) Nicotine‐induced morphological changes in rat aorta: the protective role of melatonin. Cells Tissues Organs 195, 252–259. [DOI] [PubMed] [Google Scholar]
- Schiffrin E.L., Lipman M.L. & Mann J.F.E. (2007) Chronic kidney disease: effects on the cardiovascular system. Circulation 116, 85–97. [DOI] [PubMed] [Google Scholar]
- Takeda R., Suzuki E., Satonaka H. et al (2005) Blockade of endogenous cytokines mitigates neointimal formation in obese Zucker rats. Circulation 111, 1398–1406. [DOI] [PubMed] [Google Scholar]
- Tian J., Pei H., James J.C. et al (2005) Circulating adhesion molecules in apoE‐deficient mouse strains with different atherosclerosis susceptibility. Biochem. Biophys. Res. Commun. 329, 1102–1107. [DOI] [PubMed] [Google Scholar]
- Verhoeff F.H. (1908) Some new staining methods of wide applicability including a rapid differential stain for elastic tissue. JAMA 50, 876–877. [Google Scholar]
- Wong G.C. & Giugliano R.P. (2003) Low‐molecular‐weight heparins for the treatment of acute coronary syndromes. Semin. Vasc. Med. 3, 391–402. [DOI] [PubMed] [Google Scholar]
- Yu L., Quinn D.A., Garg H.G. & Hales C.A. (2005) Cyclin‐dependent kinase inhibitor p27Kip1, but not p21WAF1/Cip1, is required for inhibition of hypoxia‐induced pulmonary hypertension and remodeling by heparin in mice. Circ. Res. 97, 937–945. [DOI] [PubMed] [Google Scholar]
- Zhao G., Shaik R.S., Zhao H., Beagle J., Kuo S. & Hales C.A. (2011) Low molecular weight (LMW) heparin inhibits injury‐induced femoral artery remodeling in mouse via upregulating CD44 expression. J. Vasc. Surg. 53, 1359–1367. [DOI] [PubMed] [Google Scholar]