

Summary
Abnormalities in lungs caused by emphysema might alter their response to sepsis and the occurrence of acute lung injury (ALI). This study compared the extension of ALI in response to intraperitoneal lipopolysaccharide (LPS) injection in Wistar rats with and without emphysema induced by elastase. Adult male Wistar rats were randomized into four groups: control, emphysema without sepsis, normal lung with sepsis and emphysema with sepsis. Sepsis was induced, and 24 h later the rats were euthanised. The following analysis was performed: blood gas measurements, bronchoalveolar lavage (BAL), lung permeability and histology. Animals that received LPS showed significant increase in a lung injury scoring system, inflammatory cells in bronchoalveolar lavage (BAL) and IL‐6, TNF‐α and CXCL2 mRNA expression in lung tissue. Animals with emphysema and sepsis showed increased alveolocapillary membrane permeability, demonstrated by higher BAL/serum albumin ratio. In conclusion, the presence of emphysema induced by elastase increases the inflammatory response in the lungs to a systemic stimulus, represented in this model by the intraperitoneal injection of LPS.
Keywords: acute lung injury, acute respiratory distress syndrome, lipopolysaccharides, pancreatic elastase, pulmonary emphysema, sepsis
Chronic obstructive pulmonary disease (COPD) is a global health problem and a leading cause of morbidity and mortality, giving rise to economic and social burdens (Landis et al. 2014). It is characterized by persistent airflow limitation, that is usually progressive, and that is determined by a combination of airway disease (obstructive bronchitis and bronchiolitis) and parenchymal destruction (emphysema), both due to an enhanced inflammatory response to noxious particles and gases (GOLD, 2015). The airway component of COPD is associated with a high risk of acute exacerbations, which are usually precipitated by infections and are characterized by an increase in bronchial inflammation and a worsening in patient′s symptoms (Burgel et al. 2009).
Emphysema, the other major component of COPD, is characterized by alveolar wall destruction as a result of inflammation and ineffective repair (Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2015). Although patients with predominance of emphysema do not seem to experience acute exacerbations frequently (Fujimoto et al. 2006; Han et al. 2009), they show a chronic pattern of inflammation involving increased numbers of cytotoxic lymphocytes, neutrophils and macrophages. These cells produce proteases involved in tissue destruction also release local and systemic inflammatory mediators, which amplify the inflammation and the structural disruption (Finkelstein et al. 1995; Retamales et al. 2001).
The lungs are also injured in acute systemic inflammatory conditions, such as sepsis, resulting in complex pathologic changes, clinically manifested as the acute respiratory distress syndrome (ARDS) (Ware & Matthay 2000). Epidemiological studies have demonstrated that COPD is a risk factor for ARDS (Veeravagu et al. 2013, 2014; Rincon et al. 2014), and one can speculate that the lung structural and inflammatory abnormalities caused by the emphysema in the lungs might alter the way they respond to sepsis and thus influence the incidence and severity of acute lung injury.
Experimental studies have shown that the presence of emphysema increases the inflammatory response to a new inhaled insult, such as bacterial or viral infection (Pang et al. 2008; Tokairin et al. 2008). However, as far as we know, no study has assessed the response of emphysematous lungs to a systemic insult, when the injury first reaches the endothelium of the alveolar–capillary barrier, as it occurs with sepsis. Therefore, in this study, we aimed to assess the extension and severity of acute lung injury (ALI) in response to intraperitoneal injection of LPS, which simulates sepsis, in an animal model of emphysema induced by elastase.
Methods
Animal preparation
Twenty‐four adult male Wistar rats (weighing 307.6 ± 25.9 g) were obtained from the Reproduction Biology Center, Federal University of Juiz de Fora vivarium (Brazil). Over the week prior to the experiment, groups of three animals were housed in clear plastic cages with stainless steel wire lids and pinewood shavings as bedding. Additionally, the rats were kept in an animal room with controlled environmental conditions (12‐h light/12‐h dark cycle, temperature 22°C) on closed ventilated shelves. The animals were fed with rat chow pellets (an average of 25 g daily) and received water at libitum.
Experimental protocol
The animals were anesthetized with an intraperitoneal bolus of ketamine (80 mg/kg) and xylazine (8 mg/kg), and then were randomly assigned to two main groups: control (C) and emphysema (E). In group E, they received intratracheal administration of porcine pancreatic elastase (PPE) (Calbiochem, USA), 12 U dissolved in 0.15 ml of phosphate‐buffered solution (PBS), while group C received 0.15 ml of PBS.
After 3 weeks, the animals of group E and group C were randomized to receive either Escherichia coli lipopolysaccharide (LPS serotype 055:B5, purified by phenol extraction, Sigma‐Aldrich, Israel), 10 mg/kg dissolved in 0.5 ml of 0.9% saline solution, or an equivalent amount of saline, both intraperitoneally (Figure 1).
Figure 1.
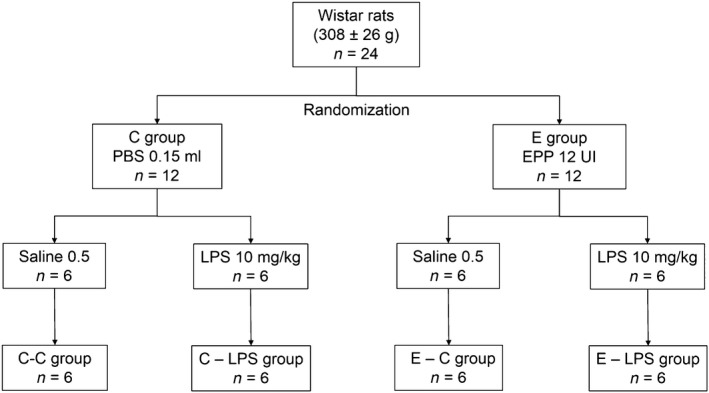
Group allocation and time line of the study design. PPE, pancreatic porcine elastase; LPS, lipopolysaccharide; i.t., intratracheal; i.p., intraperitoneal; C‐C, control; C‐LPS, normal lung with sepsis; E‐C, emphysema without sepsis; E‐LPS, emphysema with sepsis.
After 24 h, the animals were anesthetized as previously described, and the trachea was cannulated with a 14‐gauge tube. A right carotid line was inserted into sample blood for arterial blood gas analysis (ABL 90 flex, Radiometer, Copenhagen, Denmark). The rats were then paralysed (rocuronium 1 mg/kg, intravenously) and mechanically ventilated (Inspira ASV, Harvard Apparatus, USA) with the following parameters: tidal volume (VT) = 6 ml/kg, respiratory rate (RR) = 80 breaths/min, inspiratory‐to‐expiratory ratio = 1:2, FIO2 = 1.0 and positive end‐expiratory pressure (PEEP) = 5 cmH2O. After 10 min of ventilation, an arterial blood gas analysis was obtained. Thereafter, a laparotomy was performed and the animals were euthanized by exsanguination through section of the abdominal aorta. The trachea was clamped at end‐inspiration, and the lungs were removed for further analysis.
Lung histology
Lungs were removed in blocks, and the lower right lobes were separated, fixed in 10% buffered formaldehyde and then processed for paraffin embedding. Four‐micrometre‐thick slices were cut and stained with haematoxylin–eosin. Morphological examinations were performed by one lung pathologist blinded to experimental details, with a conventional light microscope (Zeiss, Hallbergmoos, Germany).
Morphological quantification of emphysema was performed by measuring the mean linear intercept (L m) in 10 fields per animal (Dunnill 1962). Briefly, a grid composed of 100 points and 50 lines was superimposed over a conventional light microscope field at a magnification of 100×, and L m was measured as the following formula:
where L tot is the total length of the lines and L i is the number of times the alveolar walls are intercepted by the lines. For each animal, the final result was the average of the 10 fields analysed.
Lung injury was quantified using a weighted scoring system, as described elsewhere (Matute‐Bello et al. 2011). Briefly, 20 random fields at a magnification of 400× were independently scored. Values of zero, one or two were used to represent the severity based on the following findings: neutrophils in the alveolar space, neutrophils in the interstitial space, hyaline membranes, proteinaceous debris filling the airspaces and alveolar septal thickening. To generate a lung injury score, the sum of the five variables was weighted according to the relevance ascribed to each one. The resulting score was a continuous value between zero (normal) and one (the most severe injury). Additionally, the extent of each lung injury score component was calculated based on the sum of the values (zero, one or two) of each of the 20 analysed fields.
Bronchoalveolar lavage (BAL)
The left lung was washed three times via a tracheal tube with 4 ml of phosphate‐buffered saline solution containing ethylenediamine tetraacetic acid (10 nM). The fluid was centrifuged at 1500 g‐force for 10 min to separate the cellular from the non‐cellular elements. Total leucocyte numbers were measured in a Neubauer chamber under light microscopy after diluting the samples in Turk solution (2% acetic acid). The cell pellet was resuspended in PBS and stained with May–Grunwald–Giemsa for differential cell counts, which were performed with a minimum of 300 cells.
IL‐6, TNF‐α and CXCL2 mRNA expressions
Quantitative real‐time RT‐PCR was performed to measure the expression of IL‐6, TNF‐α and CXCL2 genes. Right middle lobe was cut and transferred to microcentrifuge flex tubes with TRIzol® reagent for total RNA extraction by standard procedure.
cDNA synthesis
RNA concentration and purity were determined on a spectrophotometer. cDNA synthesis was carried out using a two‐step cDNA synthesis kit (Promega, USA). One microgram of RNA was reverse‐transcribed into cDNA using GoScriptTM Reverse Transcriptase (Promega, USA) according to the manufacturer's protocol using a total reaction of 20 μl. Real‐time quantitative polynucleotide chain reaction (RT‐qPCR) was performed using 5 μl of concentrate SybrGreen for a final volume of 10 μl containing 50 ng of cDNA. For the determination of the initial relative quantity of cDNA, samples were amplified with IL‐6, TNF‐α, CXCL2 and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) primers. Reactions were run on an Applied Biosystems 7500 RT‐qPCR machine (Applied Biosystems, USA). An internal standard (GAPDH) was set up to normalize the relative gene expression level, and this standard was run with each different experiment. Melt curves analyses were performed for all genes, and the specificity as well as integrity of the PCR products was confirmed by the presence of a single peak. Relative expression was calculated from the differences in cycle time of an internal standard (GAPDH) compared with the target mRNA. Duplicate cycle thresholds (CT) values were analysed in Microsoft Excel (Microsoft) using the comparative CT (2‐∆∆CT) method (Applied Biosystems, USA).
Lung permeability
To assess lung permeability abnormalities, albumin concentration was measured in BAL and serum using the bromocresol green method (Labtest Diagnostics, Brazil), and the ratio BAL/serum was determined.
Statistical analysis
The normality of the data was analysed by the Kolmogorov–Smirnov test. Parametric data were expressed as mean ± standard deviation, and nonparametric data were expressed as median (interquartile range). Parametric data were analysed using anova followed by Tukey test when required. Nonparametric data were analysed with the Kruskal–Wallis test followed by Mann–Whitney U‐test when required. Adjustments for repeated measures were made according to the Bonferroni correction. A P < 0.05 was considered significant. All statistical analysis was performed using spss 17.0 for Windows (SPSS Inc., Illinois, USA).
Ethical approval statement
This study was approved by the Ethical Committee for Animal Handling of Federal University of Juiz de Fora, Minas Gerais, Brazil (protocol number 066/2013).
Results
Two animals died within the first hour after intratracheal instillation of PPE and were replaced. There were no other deaths throughout the 21‐day period after intratracheal instillation of PPE or PBS, or the 24‐h period after intraperitoneal instillation of LPS or saline. We did not find significant differences between the groups in body weight during the study.
Development of emphysema
In the emphysema groups (E‐LPS and E‐C), the L m was greater compared with the control groups (C‐LPS and C‐C). There was no significant difference between the two groups that received PPE in the L m, showing that, as expected, the intraperitoneal administration of LPS did not influence the development of emphysema (Figure 2). Airspace enlargement and alveolar wall destruction were seen more frequently in the emphysema groups than in the control ones (Figure 2).
Figure 2.
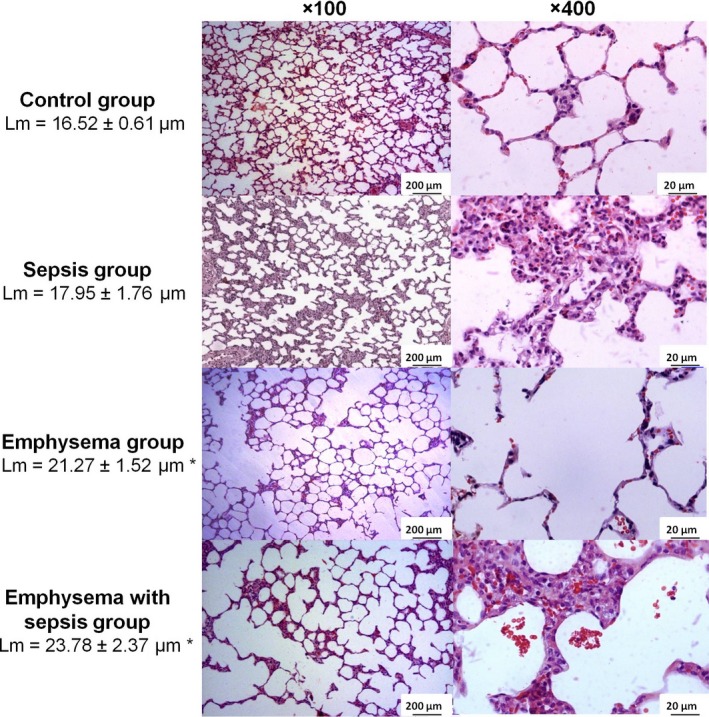
Photomicrographs of lung parenchyma stained with haematoxylin–eosin. Note that sepsis group showed alveolar wall thickening, neutrophils in the interstitium and in the airspace, and proteinaceous debris in the airspace. Emphysema group showed airspace enlargement and alveolar wall destruction. Emphysema with sepsis showed emphysematous abnormalities (airspace enlargement and alveolar wall destruction) combined to acute lung injury findings (alveolar wall thickening, neutrophils in the interstitium and in the airspace, and proteinaceous debris in the airspace). The mean linear intercept (Lm) values are shown as means and standard deviation. Statistical analysis was performed using one‐way anova followed by Tukey test. *P < 0.05 compared with control and sepsis groups.
Effects of LPS injection
Histology
Twenty‐four hours after intraperitoneal injection of LPS, histological analysis of lung specimens revealed alveolar wall thickening, neutrophil infiltration in the interstitium and alveoli and proteinaceous debris in the airspaces (Figure 2).
Lung injury scoring system
The animals that received intraperitoneal LPS (E‐LPS and C‐LPS) showed a higher ALI score compared with those that did not receive (C‐LPS and C‐C). The analysis of each component of the score demonstrated that E‐LPS and C‐LPS, compared with C‐LPS and C‐C, had greater alveolar and interstitial neutrophil infiltration, as well as greater amount of alveolar proteinaceous debris. No significant difference was observed between the two groups that received LPS (E‐LPS and C‐LPS), neither in the ALI score, nor in each of its individual components (Table 1).
Table 1.
Acute lung injury score
| Score | Groups | |||
|---|---|---|---|---|
| C‐C | C‐LPS | E‐C | E‐LPS | |
| Overall Score | 0.11 (0.09) | 0.62 (0.19)* , # | 0.15 (0.05) | 0.59 (0.13)* , # |
| Alveolar neutrophils | 0.50 (2.50) | 25.00 (12.25)* , # | 1.00 (2.00) | 25.50 (12.25)* , # |
| Interstitial neutrophils | 11.00 (8.00) | 39.50 (2.00)* , # | 16.50 (4.00) | 40.00 (0)* , # |
| Proteinaceous debris | 2.83 ± 2.23 | 18.50 ± 9.16* , # | 5.50 ± 5.75 | 15.67 ± 7.66* , # |
| Hyaline membrane | 0 (0) | 1.00 (2.00) | 0 (0) | 0 (0.50) |
| Septal thickening | 14.67 ± 13.26 | 23.00 ± 7.10 | 8.00 ± 9.51 | 22.00 ± 9.76 |
Acute lung injury score and its components. *P < 0.05 compared with C‐C; # P < 0.05 compared with E‐C. C‐C, control; C‐LPS, normal lung with sepsis; E–C, emphysema without sepsis; E‐LPS, emphysema with sepsis. Values are expressed as mean ± standard deviation or median (interquartile range) for normally and non‐normally distributed data respectively. Statistical analysis was performed using one‐way anova followed by Tukey test or by Kruskal–Wallis test followed by Mann–Whitney U‐test, for normally and non‐normally distributed data respectively. Adjustments for repeated measures were made according to the Bonferroni correction.
Bronchoalveolar lavage
Intratracheal instillation of PPE induced an inflammatory lung response, as shown by the increased number of total cells, neutrophils and macrophages in the BAL of the E‐C group, compared with the C‐C group. The E‐C group also showed a greater macrophage count in the BAL compared with the C‐LPS and E‐LPS groups.
Intraperitoneal LPS injection led to a further increase in the BAL cellularity, as demonstrated by the greater number of total cells and neutrophils in the E‐LPS group compared with the E‐C and the C‐C groups, and in the C‐LPS group compared with the C‐C group. When both groups that received intraperitoneal LPS were compared, the E‐LPS group showed a greater total cell and neutrophil counts in the BAL (Figure 3).
Figure 3.
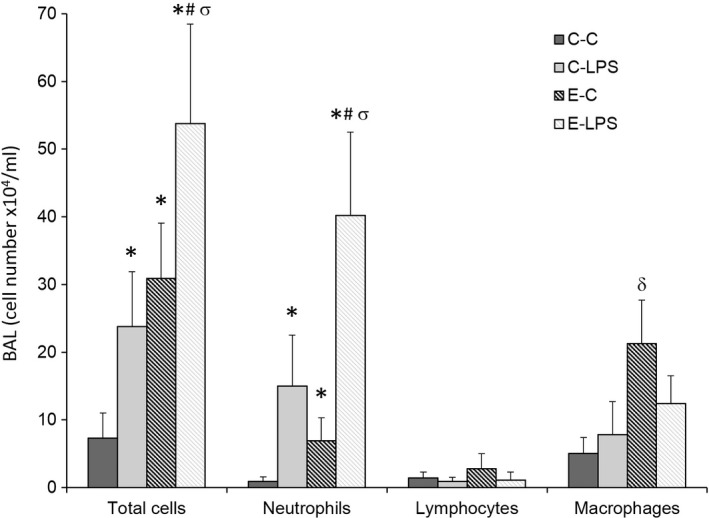
Cell count in bronchoalveolar lavage (cell number x104/ml). C‐C, control; C‐LPS, normal lung with sepsis; E‐C, emphysema without sepsis; E‐LPS, emphysema with sepsis; values are shown as means and standard deviation. Statistical analysis was performed using one‐way anova followed by Tukey test for total cells and neutrophils, and Kruskal–Wallis test followed by Mann–Whitney U‐test for neutrophils and lymphocytes. Adjustments for repeated measures were made according to the Bonferroni correction. *P < 0.05 compared with C‐C; # P < 0.05 compared with E‐C; σ P < 0.05 compared with C‐LPS; δ P < 0.05 compared with E‐LPS, C‐LPS, C‐C.
IL‐6, TNF‐α and CXCL2 mRNA expressions
The IL‐6, TNF‐α and CXCL2 mRNA expressions were higher in the animals that received intraperitoneal LPS (C‐LPS and E‐LPS) compared with the control groups (C‐C and E‐C). Between the two LPS groups, with or without emphysema, there were no differences regarding these molecule expressions (Figure 4).
Figure 4.
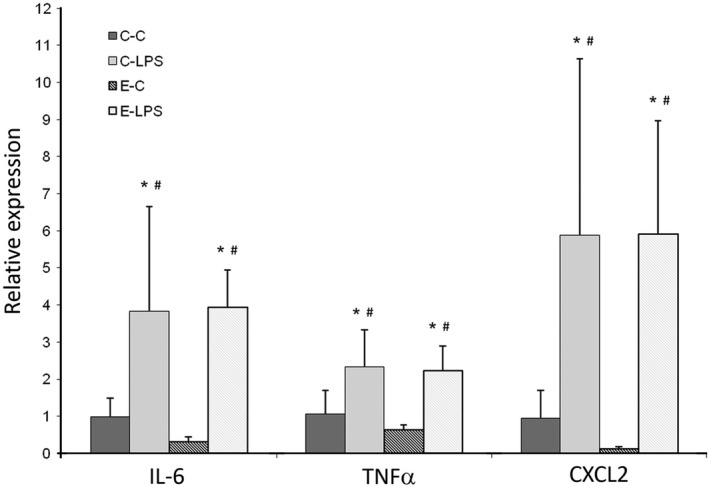
Relative expression of IL‐6, TNF‐α and CXCL2 mRNA in right upper lobe. GAPDH was used as internal standard for normalization. C‐C: control; C‐LPS: normal lung with sepsis; E‐C: emphysema without sepsis; E‐LPS: emphysema with sepsis. Values are shown as means and standard deviation. Statistical analysis was performed using Kruskal–Wallis test followed by Mann–Whitney U‐test. Adjustments for repeated measures were performed according to the Bonferroni correction.*P < 0.05 when compared with C‐C group. # P < 0.05 when compared with E‐C group.
Alveolocapillary membrane permeability
Animals that received intratracheal PPE and intraperitoneal LPS showed increased alveolocapillary membrane permeability, which was demonstrated by higher BAL/serum albumin ratio compared with E‐C and C‐C groups (Figure 5).
Figure 5.
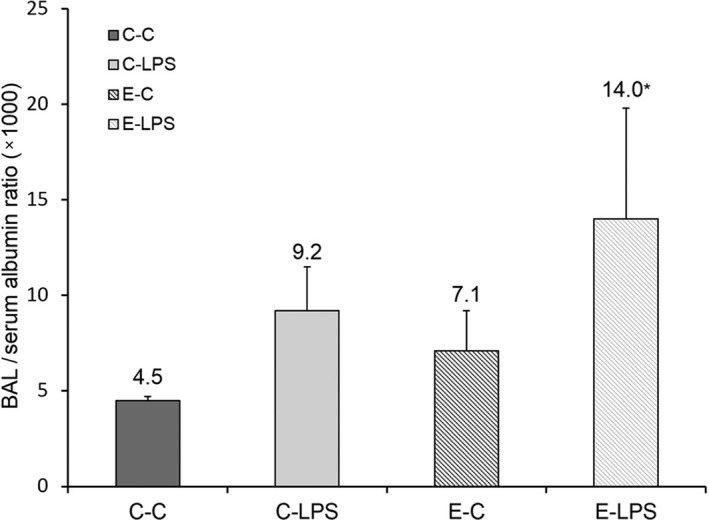
Bronchoalveolar lavage/serum albumin ratio (1000). C‐C, control; C‐LPS, normal lung with sepsis; E‐C, emphysema without sepsis; E‐LPS, emphysema with sepsis. Values are shown as means and standard deviation. Statistical analysis was performed using Kruskal–Wallis test followed by Mann–Whitney U‐test. Adjustments for repeated measures were made according to the Bonferroni correction. *P < 0.05 compared with C‐C.
Arterial blood gases
No significant differences were observed in the PaO2 and PaCO2 among all the groups. Intraperitoneal injection of LPS induced metabolic acidosis, and these animals showed lower levels of HCO3 (Table 2).
Table 2.
Arterial blood gas parameters
| Score | Groups | |||
|---|---|---|---|---|
| C‐C | C‐LPS | E‐C | E‐LPS | |
| pH | 7.25 ± 0.12 | 7.13 ± 0.07 | 7.23 ± 0.02 | 7.15 ± 0.10 |
| PaO2 | 372 ± 41 | 343 ± 88 | 345 ± 27 | 363 ± 25 |
| PaCO2 | 51 (38) | 50 (8) | 54 (4) | 46 (21) |
| HCO3 | 22.0 ± 1.0 | 16.0 ± 2.1* , # | 20.4 ± 0.9 | 16.0 ± 1.8* , # |
Arterial blood gas parameters. *P < 0.05 compared with C‐C; # P < 0.05 compared with E‐C.C‐C: control; C‐LPS: normal lung with sepsis; E‐C: emphysema without sepsis; E‐LPS: emphysema with sepsis. Values are expressed as mean ± SD or median (interquartile range) for normally and non‐normally distributed data respectively. Statistical analysis was performed using one‐way anova followed by Tukey test or by Kruskal–Wallis test followed by Mann–Whitney U‐test, for normally and non‐normally distributed data respectively. Adjustments for repeated measures were made according to the Bonferroni correction.
Discussion
In our study intraperitoneal LPS injection led to inflammatory ALI in rats with and without emphysema, as shown by a higher ATS score, increased neutrophil count in the BAL and higher mRNA expression of inflammatory mediators in these animals compared with their controls. Emphysema was associated with a greater lung inflammation in response to intraperitoneal LPS injection, characterized by increased neutrophil count in the BAL, and to a higher alveolocapillary permeability, assessed in this study by BAL/serum albumin ratio.
In the present study, emphysema was induced by a single intratracheal instillation of PPE, a model that leads to histological and morphological characteristics compatible with those of the panacinar emphysema (Yokoyama et al. 1987). The animals that received PPE, compared with the control group, showed greater L m, proving the model's effectiveness. Interestingly, we also observed that PPE instillation induced an inflammatory response, demonstrated by a higher number of total cells, neutrophils and macrophages in the BAL of the E‐C group. In keeping with our findings, increased number of inflammatory cells has also been observed in lung specimens of patients with emphysema (Retamales et al. 2001). Previous studies have demonstrated that elastase instillation is followed by acute inflammation characterized by injury to the alveolar–capillary membrane and histological changes that resemble ALI. This acute phase seems to last about 1 week. After this period, only histological features of emphysema such as airspace enlargement and alveolar wall destruction are left (Vecchiola et al. 2011). Our results showed that 3 weeks after elastase exposure, there was still enhanced lung inflammation, expressed by increased cellularity in the BAL although with no histological features of ALI. We speculate that this persistent inflammation might justify a more intense response of the emphysematous lungs, when exposed to another noxious stimulus, in this model represented by intraperitoneal LPS.
No significant changes in arterial blood gases were observed despite the occurrence of lung morphological changes induced by the emphysema. This is in agreement with other experimental study of emphysema, which showed dissociation between the degree of tissue loss and blood gas impairment (Bates et al. 2007).
The intraperitoneal injection of LPS induced an inflammatory pulmonary response characterized by higher total cell and neutrophil counts in the BAL, and increased mRNA expressions of IL‐6, TNF‐α and CXCL2, when compared to the animals that received intraperitoneal saline. This inflammation was associated with the occurrence of ALI, demonstrated by higher values of the ALI score, due to increased alveolar and interstitial neutrophil infiltration, and to the presence of proteinaceous debris filling the airspaces, despite the absence of impairment on either gas exchange, or alveolocapillary permeability. Similar findings have been described with experimental animal models of extrapulmonary injury induced by intraperitoneal or intravenous LPS injections, in which the lung injury is always mild and with minimal or no physiological dysfunction (Kabir et al. 2002; Silva et al. 2013).
In this study, the presence of emphysema potentiated the inflammatory pulmonary response to the intraperitoneal injection of LPS. Among the animals that received LPS, those with emphysema showed higher total cell and neutrophil counts in the BAL when compared to those without emphysema. Moreover, the E‐LPS group showed higher BAL/serum albumin ratio, which is a marker of alveolar–capillary membrane injury, compared with the other groups. This higher inflammatory response observed in the animals with emphysema after LPS exposure did not occur with the mediators that were evaluated. The proinflammatory cytokines IL‐6 and TNF‐α, and the neutrophil chemoattractant CXCL2 mRNA expressions were evenly increased in both groups that received LPS. It is possible that other inflammatory pathways that were not assessed in this study are involved in the inflammatory response to intraperitoneal LPS in emphysematous lungs.
These results corroborate the findings of other experimental studies that evaluated the interaction between emphysema and the inflammatory response to a second noxious stimulus, such as infection by bacteria or virus. In these studies, the presence of emphysema was associated with a more intense inflammatory response, although, paradoxically, with lower capacity to eradicate the pathogens involved (Pang et al. 2008; Tokairin et al. 2008; Sajjan et al. 2009; Wang et al. 2010). In these studies, the overexpression of TLR and the impairment in bacterial clearance could have explained the more expressive inflammation in emphysematous lungs. Our study differs from these due to the nature of the second injury, whose origin was extrapulmonary. However, it seems that in the same way, the presence of emphysema may have primed the lung, by increasing the number of inflammatory cells, thus amplifying the response to the second insult.
As far as we are concerned, this is the first experimental study that evaluated the impact of emphysema on the response against the intraperitoneal injection of LPS, which simulates extrapulmonary sepsis. Clinically, this correlation has been speculated in studies in which patients with COPD showed a higher risk to develop ARDS after some stimulus, such as severe neurologic injury, trauma and multiple transfusions (Calfee et al. 2011; Toy et al. 2012; Veeravagu et al. 2013, 2014; Rincon et al. 2014). Knowing this interaction is important because in the presence of patients with COPD and of risk factors for lung injury measures to minimize the risk of ARDS, such as the adoption of protective ventilation strategies should be taken as well as an effort to early diagnosis. Moreover, the inflammatory mediators we assessed could not explain our findings; thus, further studies might clarify the pathways by which these conditions can potentiate each other, and test therapeutic interventions.
Nevertheless, there are some limitations in this study that should be considered. This model of emphysema does not encompass the chronic bronchitis component of COPD; thus, it does not represent the most common phenotype in clinical practice. Moreover, as the emphysema was induced by a single intratracheal instillation of PPE, extrapolation of the results to the slowly progressive disease seen in humans should be made very cautiously. The ALI was induced by intraperitoneal injection of LPS, which is known as a model of mild and transient lung injury. However, we must be also careful about extrapolatimg the results from this aspect of the study, since in clinical situations patients usually develop ALI in a setting of several triggers rather than in this way.
Conclusion
In conclusion, the presence of emphysema increases the inflammatory pulmonary response to a systemic stimulus, represented in this model by the intraperitoneal injection of LPS. The chronic inflammation present in the emphysematous lungs may be the reason for this exaggerated reaction. Consequently, one should be aware of this possibility when patients with COPD are exposed to risk factors for ARDS, in order to avoid further insults that can potentiate the risk and in order to make an early diagnosis of a serious complication.
Conflict of interests
The authors declare that they have no conflict of interests.
Acknowledgments
This study was supported by a research grant from Rede Mineira TOXIFAR, FAPEMIG and the Centre of Reproductive Biology.
References
- Bates J.H., Davis G.S., Majumdar A., Butnor K.J. & Suki B. (2007) Linking parenchymal disease progression to changes in lung mechanical function by percolation. Am. J. Respir. Crit. Care Med. 176, 617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgel P.R., Nesme‐Meyer P., Chanez P. et al (2009) Cough and sputum production are associated with frequent exacerbations and hospitalizations in COPD subjects. Chest 135, 975–982. [DOI] [PubMed] [Google Scholar]
- Calfee C.S., Matthay M.A., Eisner M.D. et al (2011) Active and passive cigarette smoking and acute lung injury after severe blunt trauma. Am. J. Respir. Crit. Care Med. 183, 1660–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunnill M.S. (1962) Quantitative methods in the study of pulmonary pathology. Thorax 17, 320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein R., Fraser R.S., Ghezzo H. & Cosio M.G. (1995) Alveolar inflammation and its relation to emphysema in smokers. Am. J. Respir. Crit. Care Med. 152, 1666–1672. [DOI] [PubMed] [Google Scholar]
- Fujimoto K., Kitaguchi Y., Kubo K. & Honda T. (2006) Clinical analysis of chronic obstructive pulmonary disease phenotypes classified using high‐resolution computed tomography. Respirology 11, 731–740. [DOI] [PubMed] [Google Scholar]
- Global Initiative for Chronic Obstructive Lung Disease (GOLDz) (2015) ‐Global Strategy for the Diagnosis, Management and Prevention of COPD. http://www.goldcopd.org/guidelines-global-strategy-for-diagnosis-management.html/ (accessed 12.06.15).
- Han M.K., Bartholmai B., Liu L.X. et al (2009) Clinical significance of radiologic characterizations in COPD. COPD 6, 459–467. [DOI] [PubMed] [Google Scholar]
- Kabir K., Gelinas J.P., Chen M. et al (2002) Characterization of a murine model of endotoxin‐induced acute lung injury. Shock 17, 300–303. [DOI] [PubMed] [Google Scholar]
- Landis S.H., Muellerova H., Mannino D.M. et al (2014) Continuing to Confront COPD International Patient Survey: methods, COPD prevalence, and disease burden in 2012‐2013. Int. J. Chron. Obstruct. Pulmon. Dis. 9, 597–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute‐Bello G., Downey G., Moore B.B. et al (2011) An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Respir Cell Mol Biol 44, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang B., Hong W., West‐Barnette S.L., Kock N.D. & Swords W.E. (2008) Diminished ICAM‐1 expression and impaired pulmonary clearance of nontypeable Haemophilus influenzae in a mouse model of chronic obstructive pulmonary disease/emphysema. Infect. Immun. 76, 4959–4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retamales I., Elliott W.M., Meshi B. et al (2001) Amplification of inflammation in emphysema and its association with latent adenoviral infection. Am. J. Respir. Crit. Care Med. 164, 469–473. [DOI] [PubMed] [Google Scholar]
- Rincon F., Maltenfort M., Dey S. et al (2014) The prevalence and impact of mortality of the acute respiratory distress syndrome on admissions of patients with ischemic stroke in the United States. J. Intensive Care Med. 29, 357–364. [DOI] [PubMed] [Google Scholar]
- Sajjan U., Ganesan S., Comstock A.T. et al (2009) Elastase‐ and LPS‐exposed mice display altered responses to rhinovirus infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 297, L931–L944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva P.L., Moraes L., Santos R.S. et al (2013) Recruitment maneuvers modulate epithelial and endothelial cell response according to acute lung injury etiology. Crit. Care Med. 41, e256–e265. [DOI] [PubMed] [Google Scholar]
- Tokairin Y., Shibata Y., Sata M. et al (2008) Enhanced immediate inflammatory response to Streptococcus pneumoniae in the lungs of mice with pulmonary emphysema. Respirology 13, 324–332. [DOI] [PubMed] [Google Scholar]
- Toy P., Gajic O., Bacchetti P. et al (2012) Transfusion‐related acute lung injury: incidence and risk factors. Blood 119, 1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecchiola A., de la Llera J.F., Ramirez R., Olmos P., Herrera C.I. & Borzone G. (2011) Differences in acute lung response to elastase instillation in two rodent species may determine differences in severity of emphysema development. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R148–R158. [DOI] [PubMed] [Google Scholar]
- Veeravagu A., Jiang B., Rincon F., Maltenfort M., Jallo J. & Ratliff J.K. (2013) Acute respiratory distress syndrome and acute lung injury in patients with vertebral column fracture(s) and spinal cord injury: a nationwide inpatient sample study. Spinal Cord 51, 461–465. [DOI] [PubMed] [Google Scholar]
- Veeravagu A., Chen Y.R., Ludwig C. et al (2014) Acute lung injury in patients with subarachnoid hemorrhage: a nationwide inpatient sample study. World Neurosurg. 82, e235–e241. [DOI] [PubMed] [Google Scholar]
- Wang D., Wang Y. & Liu Y.N. (2010) Experimental pulmonary infection and colonization of Haemophilus influenzae in emphysematous hamsters. Pulm. Pharmacol. Ther. 23, 292–299. [DOI] [PubMed] [Google Scholar]
- Ware L.B. & Matthay M.A. (2000) The acute respiratory distress syndrome. N. Engl. J. Med. 342, 1334–1349. [DOI] [PubMed] [Google Scholar]
- Yokoyama E., Nambu Z., Uchiyama I. & Kyono H. (1987) An emphysema model in rats treated intratracheally with elastase. Environ Res J 42(2), 340–352. [DOI] [PubMed] [Google Scholar]