

ABSTRACT
The endoribonuclease RNase E participates in mRNA degradation, rRNA processing, and tRNA maturation in Escherichia coli, but the precise reasons for its essentiality are unclear and much debated. The enzyme is most active on RNA substrates with a 5′-terminal monophosphate, which is sensed by a domain in the enzyme that includes residue R169; E. coli also possesses a 5′-pyrophosphohydrolase, RppH, that catalyzes conversion of 5′-terminal triphosphate to 5′-terminal monophosphate on RNAs. Although the C-terminal half (CTH), beyond residue approximately 500, of RNase E is dispensable for viability, deletion of the CTH is lethal when combined with an R169Q mutation or with deletion of rppH. In this work, we show that both these lethalities can be rescued in derivatives in which four or five of the seven rrn operons in the genome have been deleted. We hypothesize that the reduced stable RNA levels under these conditions minimize the need of RNase E to process them, thereby allowing for its diversion for mRNA degradation. In support of this hypothesis, we have found that other conditions that are known to reduce stable RNA levels also suppress one or both lethalities: (i) alterations in relA and spoT, which are expected to lead to increased basal ppGpp levels; (ii) stringent rpoB mutations, which mimic high intracellular ppGpp levels; and (iii) overexpression of DksA. Lethality suppression by these perturbations was RNase R dependent. Our work therefore suggests that its actions on the various substrates (mRNA, rRNA, and tRNA) jointly contribute to the essentiality of RNase E in E. coli.
IMPORTANCE The endoribonuclease RNase E is essential for viability in many Gram-negative bacteria, including Escherichia coli. Different explanations have been offered for its essentiality, including its roles in global mRNA degradation or in the processing of several tRNA and rRNA species. Our work suggests that, rather than its role in the processing of any one particular substrate, its distributed functions on all the different substrates (mRNA, rRNA, and tRNA) are responsible for the essentiality of RNase E in E. coli.
KEYWORDS: RNA processing and decay, RNase E, stable RNA expression, ppGpp, stringent rpoB mutants
INTRODUCTION
In all organisms, mRNA degradation has to be precisely regulated in order to regulate protein expression levels, to modulate protein expression according to the changing environment, and to recycle ribonucleotides for new RNA synthesis (1–3). mRNA degradation in Escherichia coli begins with progressive endonucleolytic cleavages by RNase E (with a 5′-to-3′ polarity) followed by exonucleolytic digestion of the resulting fragments by the 3′-5′ exoribonucleases polynucleotide phosphorylase (PNPase), RNase R, and RNase II. The 2- to 5-nucleotide (nt)-long oligonucleotides so generated are then converted to mononucleotides by an oligoribonuclease, Orn (4–6).
RNase E, which is essential for E. coli viability, is a crucial enzyme in mRNA decay, encoded by gene rne (7–9). It is a 1,061-amino-acid-long protein with catalytic activity residing in the N-terminal half of the protein (10); the C-terminal unstructured half of the polypeptide serves as a scaffold for binding of the proteins PNPase, RhlB helicase, and enolase, which together with RNase E, constitute the degradosome (11–15). The degradosome is proposed to assist in cleavage of structured RNAs (16, 17). RNase E also processes the precursors of stable RNAs (both rRNA and tRNA) and ssrA (encoding small stable RNA) to generate their mature and functional forms (18–21); it also degrades the stable RNAs in growing cells (22) and under starvation conditions (23). RNase E autoregulates its synthesis in response to changes in total cellular RNase E activity by cleaving its own mRNA in the 5′ untranslated region (UTR) (24–26).
The reasons for the essentiality of RNase E in E. coli are unclear, and several alternative models have been proposed. While some studies suggests that it is the processing of tRNA but not that of 9S RNA or mRNA decay that renders RNase E essential (19, 27–30), others suggest that degradation of around 100 distinct mRNA species contributes to its essential function (31). Studies with rne(Ts) alleles have also indicated that mRNA degradation is one of the essential functions of RNase E in Salmonella enterica (32). Furthermore, association of proper FtsZ levels, maintained by the processing of the ftsQAZ transcript by RNase E, with viability is also debated (33, 34).
Structural studies with its N-terminal catalytic half (residues 1 to 529) have shown that RNase E exists as a dimer of dimers, with each protomer possessing a large domain (residues 1 to 400), a small domain (residues 415 to 510), and a CPXCXGXG motif (residues 400 to 415) that coordinates a Zn2+ ion between two monomers (35). Each large domain carries the catalytic site as well as a “5′-sensor” pocket (which includes the residues R169 and T170) that can accommodate 5′-monophosphorylated substrate RNA (35, 36). The presence of the 5′-sensor region explains the preferential cleavage of 5′-monophosphorylated substrates by RNase E (4), which may be because of an increase in substrate binding affinity and/or in activity of the enzyme (6, 37); an R169Q mutation in RNase E abolishes this activation (35, 36).
All RNA synthesis is initiated with a 5′-terminal triphosphate moiety, and E. coli possesses a Nudix hydrolase, RppH, that acts to remove the pyrophosphate group from 5′-triphosphorylated RNA to generate the 5′-monophosphorylated product which is required for the first cleavage by RNase E to occur efficiently (38, 39). 5′-monophosphorylated RNAs are also generated by endonucleolytic cleavage of RNA, for example, by RNase E itself, by its paralog RNase G, or by enzymes such as RNase III. With respect to RNase E, while only the first cleavage of transcript is expected to be defective in the absence of RppH, all the cleavage reactions will be defective in strains with RNase E-R169Q (40, 41), which would explain why precursors of rRNA accumulate in the latter mutant (40, 41).
Neither deletion of RppH nor the R169Q mutation in RNase E's 5′-sensor domain is lethal, and it is only a subset of mRNAs (∼400) that become stabilized in a ΔrppH strain (39, 40). This suggests that the 5′-end-dependent pathway is not the sole pathway for RNA cleavage. Similarly, strains expressing RNase E variants with deletion of the C-terminal half (CTH) are viable but suffer defects in global mRNA decay (42), ribosome free mRNA degradation (43), and autoregulation (27, 44); these defects are likely contributed by one or more of several mechanisms, such as absence of degradosome assembly, reduced substrate binding affinity, loss of membrane interactions, and defective oligomerization. We (41) and others (40) have shown earlier that combining two mutations, namely, (i) deletion of rppH or the RNase E-R169Q mutation with (ii) deletion of the CTH (ΔCTH) of RNase E, imparts inviability to E. coli. We have interpreted these results to indicate the existence of two pathways for RNase E action (41). In this communication, we present evidence that the lethality associated with the R169Q-ΔCTH or ΔRppH-ΔCTH combination but not that associated with Δrne can be suppressed by various perturbations that are expected to reduce rRNA expression in the strains. We hypothesize that, by diverting RNase E from stable RNA processing to mRNA degradation, lowered stable RNA levels help in the optimal degradation of mRNA in strains with limiting RNase E activity. Our results therefore suggest that the distributed functions of RNase E contribute to its essentiality in E. coli.
RESULTS
Effect of basal ppGpp levels on RNase E-R169Q,ΔCTH and ΔRppH RNase E-ΔCTH double mutant lethalities.
We have reported earlier that RNase E-ΔCTH double mutants with either ΔRppH or RNase E-R169Q are inviable, presumably because of the combined loss of both the 5′-end-dependent and CTH-activated pathways of RNase E activity (41). In further studies, as described below, we have found that the inviabilities are apparently modulated by the basal ppGpp levels in the strains, as dictated by their genotypes at relA and spoT (the two genes involved in ppGpp synthesis [both relA and spoT] and degradation [spoT alone] [45, 46]).
For these studies, we employed the “shelter plasmid loss assay” as described in Materials and Methods, in which we scored for the ability of Δlac derivatives that had lost an unstable plasmid bearing lacZ+ and rne+ to remain viable and grow as white colonies (Lac−) on 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal)-supplemented plates. With this approach, we observed first that RNase E-R169Q,ΔCTH derivative of MG1655 were inviable on LB and viable on defined medium, while similar derivatives of MC4100 were viable on both media (Fig. 1A). The Lac− colonies obtained on defined medium from MG1655 derivative failed to purify on LB medium (Fig. 1B), confirming that these derivatives exhibit rich-medium lethality.
FIG 1.
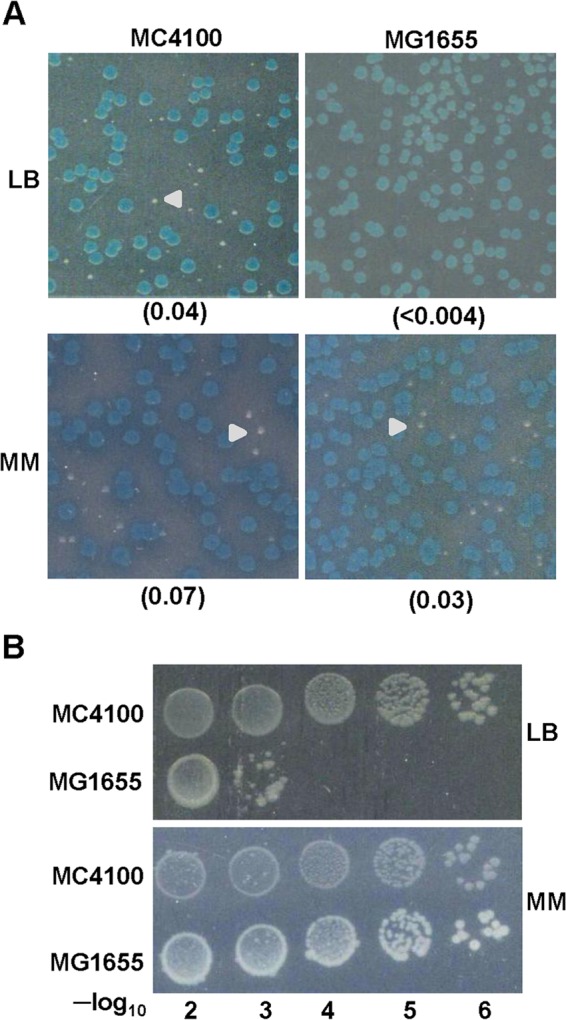
Growth characteristics of MC4100 and MG1655 derivatives of the rne-169Q,ΔCTH double mutant. (A) The GJ14067 (MC4100 Δrne) and GJ14033 [MG1655 lacZ(Am) Δrne] strains carrying the unstable plasmids pHYD1613 (rne+ lacZ+ Tpr) and pHYD2373 (rne-169Q,ΔCTH Kanr) were grown to stationary phase in LB with Tp and Kan, and suitable dilutions were plated on LB and MM (glucose-minimal A) with Kan and X-Gal (without Tp). Representative Lac− colonies are shown by arrowheads. The ratio of Lac− white colonies to total colonies is given in parentheses. (B) White colonies obtained on glucose-minimal A medium from GJ14067 and GJ14033 were grown in the same medium to stationary phase and were spotted at dilutions on LB and MM media.
Although the genotypes of strains MG1655 and MC4100 differ at several loci (47), we considered the possibility that it is the variations at relA and spoT between the two strains which are responsible for the observed phenotypic differences from the RNase E-R169Q,ΔCTH derivatives. Whereas MG1655 has the genotype relA+ spoT+, MC4100 has the genotype relA1 spoT1, with relA1 being associated with reduced ppGpp synthase activity and reduced ppGpp levels and spoT1 with reduced ppGpp hydrolase activity and increased ppGpp levels. Overall, basal ppGpp levels are higher in MC4100 than in MG1655 (48), and that growth rate is inversely correlated with intrinsic ppGpp levels (49, 50).
Accordingly, we constructed a set of isogenic strains with different relA (relA+, relA1, or ΔrelA) and spoT (spoT+, spoT1, or ΔspoT) alleles and tested them for viability with the RNase E-R169Q,ΔCTH and ΔRppH RNase E-ΔCTH combinations. These constructions were made in the genomic background of strain MC4100. The ΔrelA ΔspoT combination could not be reliably tested because of rapid accumulation of suppressors, many of which are presumably RNA polymerase mutants (51). The basal ppGpp levels in different derivatives have been inferred from their genotypes based on the earlier studies (49, 50, 52), and no direct ppGpp measurements were made in this work. Growth rates of the strains on LB medium (see Fig. S1 in the supplemental material) were also correlated with the inferred intrinsic ppGpp levels, as reported earlier (53).
The results, presented in Table 1, indicate that RNase E-R169Q,ΔCTH derivatives are inviable at low basal ppGpp levels (relA1 spoT+ and ΔrelA spoT+) on all media, viable only on defined medium at intermediate levels (relA+ spoT+), and viable on both LB and defined media at high ppGpp levels (relA+ spoT1 and relA1 spoT1) (Table 1). On the other hand, the ΔRppH RNase E-ΔCTH derivatives were inviable with all combinations of relA and spoT alleles tested (Table 1). With respect to control strains, the Δrne mutant was inviable with all the relA-spoT combinations (see Table S2, rows 11 to 15, in the supplemental material), while the RNase E-R169Q (Table S2, rows 1 to 5) and RNase E-ΔCTH (Table S2, rows 6 to 10) single mutants were viable under all conditions.
TABLE 1.
Effects of different combinations of relA and spoT alleles on the rne-169Q,ΔCTH and ΔrppH rne-ΔCTH double mutant lethalities and their suppression by rho and nusG mutationsa
| Genotypeb | No. of Lac− colonies/total (ratio) for the given mutations in the indicated strain and medium |
||||||||
|---|---|---|---|---|---|---|---|---|---|
|
rne-R169Q,ΔCTH |
ΔrppH rne-ΔCTH |
rne-R169Q,529Δ |
|||||||
| WT |
rho mutant, LB | nusG mutant, LB | WT, LB | rho mutant, LB | nusG mutant, LB | WT |
|||
| LB | MM | LB | MM | ||||||
| relA+ spoT1 | 8/198 (0.04) | 6/232 (0.03) | 12/116 (0.103) | 13/165 (0.07) | 0/261 (<0.003) | 16/164 (0.1) | 13/125 (0.10) | 48/288 (0.17) | 56/176 (0.31) |
| relA1 spoT1c | 6/226 (0.02) | 6/130 (0.04) | 16/164 (0.09) | 16/148 (0.11) | 0/240 (<0.004) | 14/197 (0.07) | 10/179 (0.05) | 16/176 (0.09) | 28/180 (0.15) |
| relA+ spoT+ | 0/199 (<0.005) | 6/192 (0.03) | 32/236 (0.13) | 0/176 (<0.005) | 0/203 (<0.004) | 16/148 (0.11) | 0/124 (<0.008) | 0/186 (<0/005) | 14/838 (0.02) |
| relA1 spoT+ | 0/136 (<0.007) | 0/109 (<0.009) | 0/199 (<0.005) | 0/208 (<0.005) | 0/153 (<0.006) | 0/128 (<0.005) | 0/112 (<0.009) | 0/284 (<0.003) | 0/480 (<0.002) |
| ΔrelA spoT+ | 0/108 (<0.009) | 0/96 (<0.01) | 0/206 (<0.004) | 0/213 (<0.004) | 0/106 (<0.009) | 0/176 (<0.005) | 0/168 (<0.005) | 0/160 (<0.006) | 0/520 (<0.001) |
The shelter plasmid loss assay was done as described in the legend to Fig. 1. Strains with the indicated genotypes (listed in Table S1 in the supplemental material) carry plasmids pHYD1613 and pHYD2373 (rne-R169Q,ΔCTH strains), plasmid pHYD1613 (ΔrppH rne-ΔCTH strains), or plasmids pHYD1613 and pRne-SG21 (rne-R169Q,529Δ strains) (a kind gift from George A. Mackie).
Genotypes are arranged in decreasing order of the strains' ppGpp levels as deduced from the earlier reports (49, 50, 52, 53).
The strain with this genotype gave plasmid-free white colonies in 4 out of 7 experiments, and we speculate that the presence of borderline ppGpp levels in this background is the cause of the fluctuating growth phenotype.
The RNase E C-terminal truncation employed in the experiments described above extends from residue 494, and hence the truncated form is also missing part of the enzyme's small domain, which could conceivably have affected its oligomerization. Accordingly, we also examined the effects of basal ppGpp status on the viability of an RNase E-R169Q,529Δ mutant, that is, where the R169Q mutation is borne on a protein with ΔCTH extending from residue 530; Garrey and Mackie have earlier reported that this is inviable in MG1655 (40).
In a comparison across isogenic strains, the rne-R169Q,529Δ double mutant was inviable on LB at intermediate levels of basal ppGpp (relA+ spoT+) (Table 1). It was also inviable at low basal ppGpp levels (relA1 spoT+ and ΔrelA spoT+) on both LB and minimal media but was viable on both media at high basal ppGpp levels (relA+ spoT1 and relA1 spoT1) (Table 1). The results therefore indicate that deletions of the CTH beyond residue 494 or 530 are equivalent in terms of their effects on compromising RNase E function.
Since ppGpp is known to regulate the expression of many genes (54), we have tested whether the reason for suppression at elevated ppGpp levels is an increase in RNase E expression and thereby its activity. For this, we measured the expression of β-galactosidase from a single-copy rne-lac fusion as described earlier (41), where β-galactosidase expression is inversely correlated with cellular RNase E levels (24). The results indicate that RNase E activity is unaffected by the basal ppGpp levels (Fig. 2A), which was also confirmed by immunoblot analysis of RNase E protein (Fig. 2A). Thus, increased RNase E expression at high basal ppGpp levels is not the cause of suppression.
FIG 2.
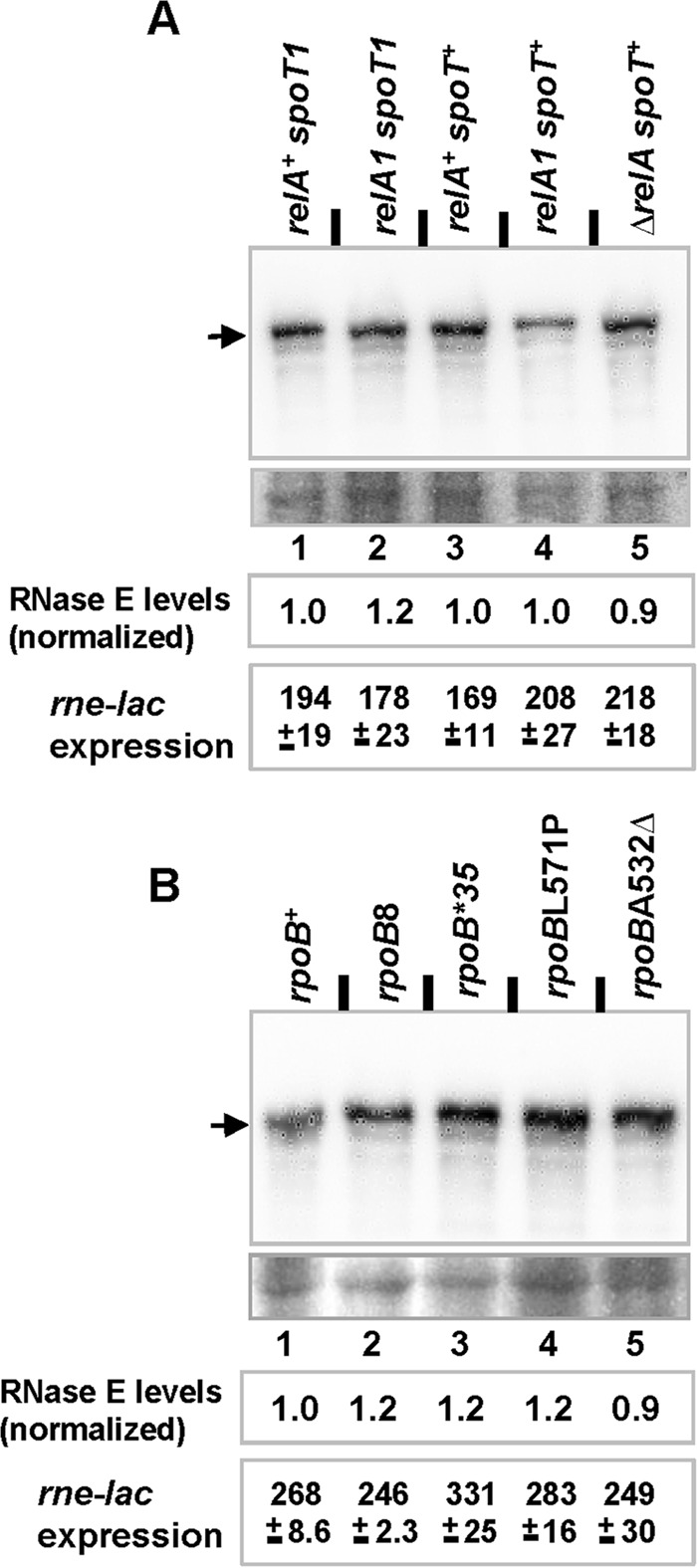
Effects of basal ppGpp levels and rpoB genotype on RNase E levels and rne-lac expression. RNase E levels were determined by immunoblotting following growth to mid-log phase of cultures with either various combinations of relA and spoT alleles (A) or different rpoB alleles (B). The upper image in each panel shows the results from probing with anti-RNase E antibody, and the band for RNase E is marked by an arrow; the lower image shows a representative area from the same blot after staining with amido black to serve as a loading and transfer control. For each of the strains, RNase E levels were quantitated (average from two independent experiments) as the ratio of densitometric intensities of the band for RNase E to that of a suitable band (common to all lanes) in the loading control; further, the values have also been normalized to the ratio calculated for lane 1 in each of the two panels. rne-lac expression values (expressed in Miller units) for strains of the indicated genotypes were determined in LB-grown cultures. The strains used in these experiments are listed in Table S1 in the supplemental material.
Suppression of ΔRppH RNase E-ΔCTH and RNase E-R169Q,ΔCTH lethalities by mutations in rho and nusG is also conditional on basal ppGpp levels.
In our earlier study (41), we had shown that missense mutations in rho and nusG, which adversely affect the process of Rho-dependent transcription termination, suppress the lethalities associated with both ΔRppH RNase E-ΔCTH and RNase E-R169Q,ΔCTH mutations. These findings were interpreted in the context of a model in which formation of excessive RNA-DNA hybrids (R-loops) in the rho and nusG mutants (55, 56) provides an alternative means of mRNA degradation in the RNase E-deficient strains.
In the present study, we have now tested the suppression ability of rho and nusG mutations in the different isogenic strain backgrounds with various basal ppGpp levels (Table 1). Our results indicate that for the ΔRppH RNase E-ΔCTH combination, which is inviable at all ppGpp levels, rho and nusG suppress lethality at high basal levels of ppGpp (relA+ spoT1 and relA1 spoT1) but not at the low ppGpp levels (relA1 spoT+ and ΔrelA spoT+) (Table 1). At the intermediate level of ppGpp (relA+ spoT+), only the rho mutation suppressed the inviability. For the RNase E-R169Q,ΔCTH combination, inviability in the relA+ spoT+ background alone (but not in the relA1 spoT+ or ΔrelA spoT+ background) was suppressed by the rho mutation (Table 1).
We also tested whether the transcription polarity relief phenotypes of rho and nusG mutants were affected by the basal ppGpp status of the strains. For this purpose, we determined the expression of lacZ downstream of the Rho-dependent terminator λ tR1 as previously described (57). The results indicate that the extent to which transcription termination is rendered defective by rho and nusG mutations is the same at high and low ppGpp levels (see Table S3 in the supplemental material). The missense rho and nusG single mutants were viable but sick at low ppGpp levels (see Table S2 [rows 16 to 25] and Fig. S2 in the supplemental material).
“Stringent” RNA polymerase (rpoB) mutations also suppress RNase E-R169Q,ΔCTH and ΔRppH RNase E-ΔCTH double mutant lethalities.
As we found that basal ppGpp levels modulate the growth of the strains with limiting RNase E activity, we also tested whether “stringent” rpoB mutations, which mimic the effect of high ppGpp levels (51, 58–60), could restore the viability of RNase E-R169Q,ΔCTH and ΔRppH RNase E-ΔCTH mutants. For this, we chose the stringent rpoB*35, rpoBL571P, and rpoBA532Δ RNA polymerase mutants and the nonstringent rpoB8 mutant (61) as a control. The stringent rpoB mutations alone suppressed lethalities of both RNase E-R169Q,ΔCTH (at both intermediate [relA+ spoT+] and low [relA1 spoT+] ppGpp levels) and ΔRppH RNase E-ΔCTH combinations (at high [relA1 spoT1] and low [relA1 spoT+] basal ppGpp levels) (Table 2). None of them suppressed Δrne lethality (Table 2).
TABLE 2.
Suppression of rne-169Q,ΔCTH and ΔrppH rne-ΔCTH double mutant lethalities by stringent RNA polymerase mutations and effect of deleting rnr in RNA polymerase mutantsa
| Genotype | No. of Lac− colonies/total (ratio) for the indicated mutation |
||||
|---|---|---|---|---|---|
| rpoB+ | rpoB8 | rpoB*35 | rpoBL571P | rpoBA532Δ | |
| rne-R169Q,ΔCTH (relA+ spoT+) | 0/241 (<0.001) | 0/206 (<0.004) | 8/184 (0.04) | 28/224 (0.13) | 36/216 (0.16) |
| rne-R169Q,ΔCTH (relA1 spoT+) | 0/208 (<0.004) | 0/152 (<0.006) | 6/162 (0.03) | 14/270 (0.05) | 16/402 (0.04) |
| ΔrppH rne-ΔCTH (relA1 spoT1) | 0/291 (<0.003) | 0/240 (<0.004) | 6/222 (0.03) | 24/224 (0.107) | 12/280 (0.04) |
| ΔrppH rne-ΔCTH (relA1 spoT+) | 0/167 (<0.003) | 0/128 (<0.007) | 32/188 (0.17) | 24/144 (0.17) | 40/184 (0.21) |
| Δrne (relA+ spoT1) | 0/140 (<0.007) | NDb | 0/220 (<0.004) | 0/282 (<0.003) | 0/312 (<0.003) |
| Δrne (relA1 spoT1) | 0/224 (<0.004) | ND | 0/264 (<0.003) | 0/253 (<0.003) | 0/160 (<0.006) |
| Δrnr rne-R169Q,ΔCTH (relA+ spoT+) | 0/148 (<0.006) | ND | 36/428 (0.08) | 18/157 (0.11) | 12/124 (0.09) |
| Δrnr ΔrppH rne-ΔCTH (relA1 spoT1) | ND | ND | 8/177 (0.04) | 16/180 (0.09) | 20/126 (0.15) |
| Δrnr ΔrppH rne-ΔCTH (relA1 spoT+) | ND | ND | 16/148 (0.11) | 16/244 (0.15) | 19/125 (0.07) |
The shelter plasmid loss assay for strains carrying either only pHYD1613 (rows 3, 4, 5, 6, 8, and 9) or pHYD1613 and pHYD2373 (rows 1, 2, and 7) was done as described in the legend to Fig. 1. The strains used are listed in Table S1 in the supplemental material.
ND, not determined, as the rpoB8 mutation was not suppressing the double mutant lethalities.
We also tested the stringent rpoB mutation rpoBA532Δ for its ability to suppress rne-R169Q,529Δ double mutant lethality, and the data indicated that viability was restored in these derivatives at levels of ppGpp that were intermediate (ratio of Lac− colonies to total = 0.26) or low (ratio of Lac− colonies to total = 0.27 and 0.13 in relA1 spoT+ and ΔrelA spoT+ backgrounds, respectively).
That increased RNase E expression is not the cause of suppression by stringent rpoB mutations was evident from both immunoblotting experiments and rne-lac expression assays; these measurements were performed in strains carrying either full-length RNase E (Fig. 2B) or the protein truncated for its CTH beyond residue 494 (see Fig. S4 in the supplemental material). We then studied whether the conditions conferring viability under situations of limiting RNase E activity, namely, presumptive basal high ppGpp levels or presence of stringent rpoB mutations, reduce rrn operon transcription, as measured with an rrnB P1-lacZ fusion in the appropriate strains (Table 3). This assay was performed in the MG1655 background in which the suppression phenotype is observed. As reported earlier (58, 60), β-galactosidase expression from the rrnB P1-lacZ fusion was reduced significantly (3-fold) in the stringent rpoB mutants compared to the rpoB+ strain and control rpoB8 mutant. Furthermore, there was a modest reduction in transcription from the rrnB promoter (1.2-fold) at high basal ppGpp levels (relA+ spoT1).
TABLE 3.
Effects of various rpoB alleles and high basal ppGpp levels on expression of the rrnB P1 promoter determined using rrnB P1-lacZ gene fusion
| Mutationa | rrnB P1-lacZ sp actb |
|---|---|
| rpoB+ | 1 |
| rpoB8 | 1.30 |
| rpoBA532Δ | 0.33 |
| rpoB*35 | 0.34 |
| rpoBL571P | 0.34 |
| relA+ spoT1 rpoB+ | 0.8 |
Except for the last row, the genotype of the strains is relA+ spoT+. From top to bottom, the strains used were RLG1350 (a kind gift from Richard L. Gourse), GJ14359, GJ14360, GJ14363, GJ14362, and GJ14365.
The specific activities of β-galactosidase of the strains with the indicated genotypes were determined after growth in LB medium, and the values were normalized to that of the wild-type strain. Values are averages from three independent experiments.
Taken together, these results suggest that reduction in stable RNA transcription is associated with the rescue of inviability under conditions of limiting RNase E activity and that when there is a small reduction in rrn transcription (as with high basal ppGpp levels), lethality of RNase E-R169Q,ΔCTH but not that of ΔRppH RNase E-ΔCTH is suppressed, while on the other hand when the reduction is more significant, both the lethalities are suppressed.
Inviability due to limiting RNase E generated by other means is also rescued by stringent rpoB mutations.
A strain in which RNase E is expressed from the Plac promoter is viable only in medium supplemented with ≥3 μM isopropyl-β-d-thiogalactopyranoside (IPTG) (62). We found in this study that stringent rpoB mutations enable such a strain to grow without IPTG (Fig. 3A). Suppression by the rpoBA532Δ mutation was most robust, followed by that by the rpoBL571P and rpoB*35 mutations.
FIG 3.
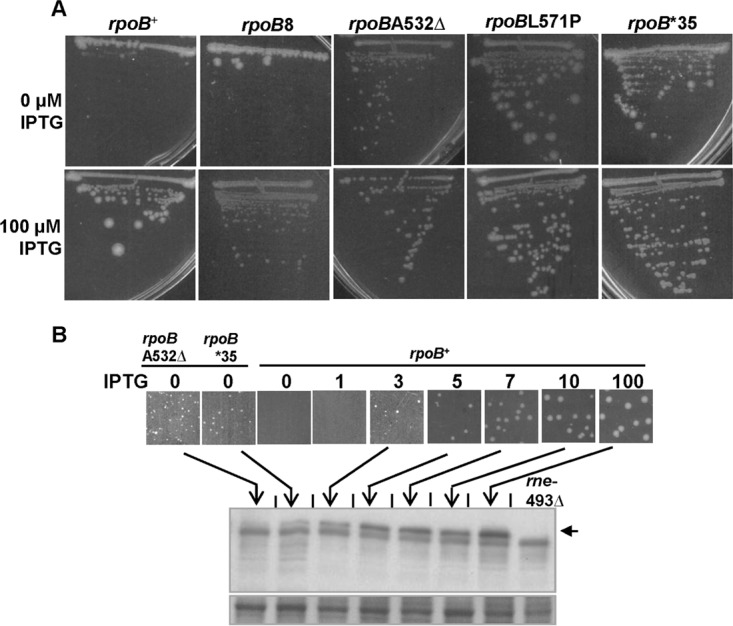
Stringent rpoB mutations rescue the inviability of a Plac-rne strain grown without inducer (IPTG). (A) Wild-type and rpoB mutant derivatives of Plac-rne strains were streaked on a pair of LB plates with the indicated micromolar IPTG concentrations and incubated at 37°C until the growth on plate without IPTG was clear (for 2 days). The strains used, from left to right, were GJ6974, GJ17402, GJ17403, GJ17404, and GJ17405. (B) Cultures of strains GJ17403, GJ17405, and GJ6974 were grown in LB with IPTG (100 μM), washed, and plated at suitable dilutions on LB plates with the indicated IPTG concentrations, and RNase E levels in the cells of colonies harvested from the plates after 48 h were determined by Western blotting. In the upper panel, the RNase E band is marked by the arrow; the lower panel shows a representative section of the same blot stained with amido black, which serves as a loading and transfer control. The band seen immediately below that of RNase E in this experiment was inferred to be a nonspecific band, since it is also observed in the lane for RNase E-ΔCTH (see also Fig. S4 in the supplemental material).
RNase E levels were determined by immunoblotting in colonies of Plac-rne strains carrying the stringent rpoB mutations that were grown on plates with 0 μM IPTG, and these levels were shown to be lower than the levels in colonies of the control rpoB+ strain grown on plates with 3 μM IPTG (Fig. 3B). These results indicate that suppression by the stringent rpoB mutations is not because of an IPTG-independent increase in RNase E expression in the Plac-rne strain (Fig. 3B).
Therefore, it appears that stringent rpoB mutations can suppress growth defects of limiting RNase E conditions that have arisen by any of the several different ways.
DksA dependence for viability of RNase E-R169Q,ΔCTH.
The protein DksA is required to potentiate the regulatory effects of ppGpp on transcription from rrn operons (reviewed in references 46 and 63). Therefore, we tested the requirement of DksA for viability of RNase E-R169Q,ΔCTH at high basal ppGpp levels.
Deletion of dksA from wild-type (WT), rho, and nusG derivatives of the RNase E-R169Q,ΔCTH mutant at high basal ppGpp levels (relA+ spoT1) resulted in inviability (Table 4), analogous to that observed for dksA+ at low ppGpp levels. On the other hand, viability conferred by the stringent rpoB mutation rpoBA532Δ was retained even in the ΔdksA derivative (ratio of Lac− colonies to total = 0.11); Vinella et al. (64) have also reported that the suppression by rpoBA532Δ of other low-[ppGpp] phenotypes is retained even in the absence of DksA.
TABLE 4.
Role of DksA and RNase R in the viability of the rne-R169Q,ΔCTH mutant at high basal ppGpp levels (relA+ spoT1)a
| Mutation | No. of Lac− colonies/total (ratio) for the indicated rne-R169Q,ΔCTH (relA+spoT1) strain |
||
|---|---|---|---|
| WT | rho mutant | nusG mutant | |
| WT | 12/218 (0.05) | 52/272 (0.19) | 30/166 (0.18) |
| ΔdksA | 0/144 (<0.006) | 6/136 (0.04)b | 11/159 (0.07)b |
| Δpnp | 14/262 (0.05) | NDc | ND |
| Δrnb | 6/154 (0.04) | ND | ND |
| Δrnr | 0/544 (<0.002) | 6/312 (0.02) | 9/320 (0.03) |
The shelter plasmid loss assay was done as described in the legend to Fig. 1. The following strains carrying plasmids pHYD1613 and pHYD2373 were employed: row 1, GJ15232, GJ15234, and GJ15235; row 2, GJ16159, GJ16157, and GJ16158; row 3, GJ15217; row 4, GJ15216; row 5, GJ15259, GJ15260, and GJ15261.
The Lac− colonies are considered inviable, as they did not grow or revive upon restreaking.
ND, not determined, as the corresponding wild-type strains were viable.
We also have checked whether overexpression of DksA can suppress the lethality associated with limiting RNase E at otherwise nonpermissive ppGpp levels, since Potrykus et al. (65) have demonstrated that such overexpression leads to inhibition of transcription initiation from the rrnBP1 promoter in a ppGpp-independent manner. Our results suggest that overexpression of DksA does suppress RNase E-R169Q,ΔCTH lethality at both intermediate and low ppGpp levels but that it does not suppress ΔRppH RNase E-ΔCTH lethality (Table 5).
TABLE 5.
Effect of overexpression of DksA on rne-R169Q,ΔCTH and ΔrppH rne-ΔCTH lethalitiesa
| Genotype | No. of Lac− colonies/total (ratio) for the indicated mutations |
|
|---|---|---|
| rne-R169Q,ΔCTH | ΔrppH rne-ΔCTH | |
| relA+ spoT+/pHR53 | 0/186 (<0.005) | 0/106 (<0.009) |
| relA+ spoT+/pJK537b | 17/138 (0.12) | 2/265 (0.007)c |
| relA1 spoT+/pHR53 | 0/203 (<0.005) | 0/180 (<0.005) |
| relA1 spoT+/pJK537b | 32/204 (0.14) | 14/320 (0.044)c |
The shelter plasmid loss assay for strains carrying pHYD1613 and pHYD2373 (rne-R169Q,ΔCTH) or only pHYD1613 (ΔrppH rne-ΔCTH) was done as described in the legend to Fig. 1. The strains carrying either pHR53 or pJK537 employed were as follows: rows 1 and 2, GJ15233 and GJ16022 for rne-R169Q,ΔCTH and ΔrppH rne-ΔCTH, respectively; rows 3 and 4, GJ14175 and GJ14157 for rne-R169Q,ΔCTH and ΔrppH rne-ΔCTH, respectively.
Overexpression of the dksA gene from this plasmid is brought about by the non-feedback-regulated promoter present upstream from the native dksA promoter (88).
The Lac− colonies are considered inviable, as they did not grow or revive upon restreaking.
Deletion of four or five rrn operons suppresses lethality associated with RNase E deficiency.
Based on the above observations, we considered the possibility that it is the reduction in the expression and levels of stable RNA that is associated with the rescue of lethality in strains with limiting RNase E activity. To test this hypothesis, we examined their viability in derivatives in which the rrn copy number (seven in the wild type) has been reduced (66).
We found that both combinations, ΔRppH RNase E-ΔCTH and RNase E-R169Q,ΔCTH, remain inviable when only one rrn operon is deleted (ΔrrnG) but are rendered viable upon deletion of four (ΔrrnGADE) or of five (ΔrrnGADEH) rrn operons (Table 6). Inviability of the rne-R169Q,529Δ double mutant also was suppressed upon deletion of four or five rrn operons (ratio of Lac− colonies to total = 0.05 and 0.18, respectively). These results strongly suggest that it is the lowered stable RNA levels brought about by various perturbations that can impart viability to strains with very low RNase E activity.
TABLE 6.
Reducing the number of rrn operons suppresses both rne-R169Q,ΔCTH and ΔrppH rne-ΔCTH lethalitiesa
| No. of rrn operons deleted (deleted operon[s]) | No. of Lac− colonies/total (ratio) for the indicated mutations |
|
|---|---|---|
| rne-169Q,ΔCTH | ΔrppH rne-ΔCTH | |
| 1 (ΔrrnG) | 0/62 (<0.02) | 0/160 (<0.006) |
| 4 (ΔrrnGADE) | 20/140 (0.14) | 52/252 (0.21) |
| 5 (ΔrrnGADEH) | 16/80 (0.20) | 21/141 (0.12) |
The shelter plasmid loss assay for strains carrying pHYD1613 and pHYD2373 (rne-169Q,ΔCTH) or only pHYD1613 (ΔrppH rne-ΔCTH) was done as described in the legend to Fig. 1. The strains used for the one-, four-, and five-operon deletions, respectively, were GJ16183, GJ16028, and GJ16030 (rne-169Q,ΔCTH) and GJ16184, GJ16124, and GJ16125 (ΔrppH rne-ΔCTH).
Rescue of lethality in strains with limiting RNase E is not because of a reduction in growth rate.
All conditions described above that restored viability to strains with limiting RNase E, namely, high basal ppGpp levels, stringent rpoB mutations, and decreased rrn copy number, are also associated with a decrease in the growth rate of the strains. We therefore tested whether a similar rescue of lethality could be enacted by any other perturbation which decreases the growth rate of the strains.
Deletion of neither crp nor hfq, both of which are associated with a reduction in growth rate (67, 68), rescued the lethality associated with limiting RNase E activity of the RNase E-R169Q,ΔCTH mutant (ratio of Lac− colonies to total = <0.003 and <0.002, respectively). Whereas the hfq mutant does exhibit a higher growth rate than the stringent rpoB mutants or the rrn operon-deleted strains, the crp mutant is as compromised for growth as is the rpoB*35 stringent mutant or the Δ4-rrn strain (see Table S4 in the supplemental material). These results indicate that a mere reduction in growth rate does not rescue the lethality associated with limiting RNase E.
Global repression of transcription does not rescue lethality caused by RNase E deficiency.
We then tested whether sublethal concentrations of rifampin would restore the viability to derivatives with RNase E-R169Q,ΔCTH (at intermediate and low basal ppGpp levels) or ΔRppH RNase E-ΔCTH (at high basal ppGpp levels). The lethalities could not be rescued at any of the rifampin concentration tested (see Table S5 in the supplemental material), suggesting that a global reduction in transcription is not equivalent to the suppressors described above, such as elevated basal ppGpp levels, stringent rpoB mutations, or decrease in rrn copy number. Sublethal rifampin also did not rescue the inviability of the Plac-rne strain on plates with 0 μM IPTG (data not shown).
Role of RNase R in viability of strains with limiting RNase E activity.
As mentioned above, three exoribonucleases, PNPase, RNase II, and RNase R, are involved in mRNA turnover (13, 69–71) following endonucleolytic cleavage by RNase E, and we tested their role, if any, in the suppression of lethalities associated with limiting RNase E at high basal ppGpp levels or with stringent rpoB mutations.
Depletion of RNase R (but not of PNPase or RNase II) resulted in inviability of RNase E-R169Q,ΔCTH at high basal (relA+ spoT1) ppGpp levels (Table 4, compare Δrnr with WT). However, deletion of rnr in the presence of rho or nusG mutations (for RNase E-R169Q,ΔCTH at high ppGpp levels) (Table 4) and stringent rpoB mutations (for both the mutants) (Table 2) did not lead to loss of viability.
We also found that depletion of RNase R from the Plac-rne derivative with high basal ppGpp levels (relA1 spoT1) resulted in a 100-fold reduction in CFU at 2 μM IPTG (see Fig. S3 in the supplemental material), confirming the need for RNase R to support growth under conditions of low RNase E activity and high ppGpp. The possible role of RNase R during RNase E-limited growth is discussed below.
DISCUSSION
RNase E is essential for viability and plays important roles in both mRNA degradation and stable RNA processing. Combined perturbations of both its 5′-end-dependent pathway (occurring in ΔRppH or RNase E-R169Q strains) and CTH-activated pathway (in RNase E ΔCTH) lead to deficiency in RNase E activity that results in growth inhibition (40, 41). In this work we report that high basal ppGpp levels, rpoB mutations that encode stringent RNA polymerases, or reduced rrn copy numbers alleviate the growth defects associated with limiting RNase E activity. We propose that reduced rrn transcription under these conditions results in diversion of the enzyme activity to achieve efficient mRNA turnover as a consequence of the lowered requirement of RNase E for processing of stable RNA, since the cellular levels of the latter are now reduced.
Evidence for suppression of inviability associated with limiting RNase E activity by the reduction of stable RNA levels.
Three observations support the hypothesis that reduced stable RNA levels result in the suppression of inviability associated with RNase E-R169Q,ΔCTH and RNase E-ΔCTH ΔRppH double mutants that have limiting RNase E activity. First, all rpoB mutations (rpoBA532Δ, rpoB*35, and rpoBL571P) that suppressed both the lethalities also conferred reduced transcription from the rrnB P1 promoter (Table 3). Other groups have also shown earlier that stringent rpoB mutations reduce transcription from the rrnB P1 promoter in vivo and in vitro (58, 60).
We also obtained a modest reduction (1.2-fold) in transcription from the rrnB P1 promoter at high basal ppGpp levels (where RNase E-R169Q,ΔCTH is viable) compared to that at intermediate ppGpp levels (relA+ spoT+) where the strain is inviable. Ryals et al. (72) had reported a small increase in rrn transcription in relA mutants during exponential growth relative to that in relA+ strains, with this difference becoming more pronounced during the stringent response. Likewise, Barker et al. (73) had also shown that rrn transcription in ΔrelA ΔspoT mutants is only marginally elevated compared to that in the wild-type strain.
The second line of evidence in support of our hypothesis is that DksA overexpression suppressed RNase E-R169Q,ΔCTH lethality at both intermediate and low ppGpp levels (Table 5). Overproduction of DksA reduces transcription initiation from the rrnB P1 promoter in either the presence or absence of ppGpp (65). Conversely, deletion of dksA is associated with an increase in transcription from the rrnB P1 promoter during steady-state growth (65, 74). Consistent with these reports, we find that the RNase E-R169Q,ΔCTH double mutant, like its rho and nusG derivatives, is inviable upon DksA depletion even at high basal ppGpp levels (Table 4).
The third and most important finding to support the notion that reduction of stable RNA levels is the cause of suppression is that deletion of four or five rrn operons suppressed both RNase E-R169Q,ΔCTH and RNase E-ΔCTH ΔRppH lethalities (Table 6). The decrease in rrn operon copy number is correlated with decreases both in the RNA/protein ratio and in growth rates in nutrient-rich media (75, 76). At the same time, we have also shown that it is not a reduction in growth rate per se which is responsible for suppression of inviability in strains with limiting RNase E.
Why do perturbations expected to lower stable RNA levels rescue strains with limiting RNase E activity?
Stable RNA constitutes the majority of total RNA in the cell (77), and the requirement of RNase E for processing of stable RNA is high compared to that needed for degradation of mRNAs; it is estimated that in each generation RNase E makes around 400,000 and 90,000 cleavages, respectively, for processing of rRNA/tRNA and for degradation of mRNA (5). We therefore speculate that in the mutants with limiting RNase E activity, a reduction in stable RNA levels leads to diversion of the enzyme activity to degrade mRNA and thus to restore viability.
The reduction in rrn transcript levels is greater with stringent rpoB mutations than it is with high basal ppGpp levels (Table 3). It is perhaps for this reason that RNase R is important at high basal ppGpp levels for the suppression of RNase E-R169Q,ΔCTH lethality (Table 4) but is dispensable in the presence of the stringent rpoB mutations (Table 2). Based on both our earlier studies (41) and this work, it also appears that RNase E-ΔCTH ΔRppH is more defective in RNA metabolism than is RNase E-R169Q,ΔCTH, so that the former's viability is not restored at high ppGpp or high DksA levels.
Distributed functions of RNase E as the reason for its essentiality.
Given that RNase E participates in processing/degradation of numerous substrates, the reason for its essentiality has remained unclear and several alternative models have been proposed in this regard, as mentioned above (19, 27, 29–31). On the basis of our present studies, we hypothesize that there is an optimal threshold level of the enzyme that is required for each of the substrate families (mRNA, rRNA, and tRNA) and that inviability can arise if there is a perturbation in fulfilling any one or more of these requirements. We believe that this model can also satisfactorily account for the discrepant observations and conclusions from other groups (19, 27–32) to explain RNase E's essentiality.
Role of RNase R in strains with limiting RNase E.
Taking these findings together with our earlier results, it would appear that the viability of strains with limiting RNase E activity can be restored if sufficient mRNA turnover can be achieved in the cells. Thus, mutations in rho and nusG, which are postulated to provide R-loop-mediated RNA degradation (41), and a decrease in stable RNA content, which allows diversion of RNase E for mRNA turnover (this study), confer viability on strains with limiting RNase E activity.
In this context, our finding that suppression of RNase E-R169Q,ΔCTH is abolished in rnr mutants can perhaps be explained as follows. RNase R is reported to degrade full-length mRNA in various situations, such as in stationary phase (78) and under RNase E-deficient conditions (79). The levels of RNase R are also elevated in different rne mutants (80). Accordingly, we speculate that RNase R also contributes to mRNA degradation, especially in cells with limiting RNase E activity.
MATERIALS AND METHODS
Growth media, bacterial strains, and plasmids.
LB medium and glucose-minimal A (MM) medium were routinely used as rich and defined media (81) with appropriate concentrations of antibiotics, namely, ampicillin (Amp) (40 μg/ml for plasmid and 20 μg/ml for chromosomal marker), kanamycin (Kan) (25 μg/ml), chloramphenicol (Cm) (30 μg/ml), trimethoprim (Tp) (60 μg/ml), and tetracycline (Tet) (15 μg/ml), wherever necessary. X-Gal was used at 50 μg/ml, and the isopropyl-β-d-thiogalactoside (IPTG) concentration varied from 2 μM to 100 μM according to the requirement of the experiment. Strains were routinely grown at 37°C
The E. coli K-12 strains used in this work are listed in Table S1 in the supplemental material. The C-terminal half truncated derivative of RNase E that is used in our work is 493 amino acids long and is designated RNase E-ΔCTH. The RNase E-R169Q,ΔCTH double mutant carries RNase E-ΔCTH with an R169Q mutation, and the ΔRppH RNase E-ΔCTH double mutant is devoid of RppH and carries RNase E-ΔCTH.
Several plasmids used in this study are derivatives of pMU575, which is an unstable single-copy-number plasmid with an IncW replicon and Tpr antibiotic marker: pHYD1613 (rne+) (41), pHYD2411 (Salmonella enterica serovar Typhimurium rho+ gene) (55), and pHYD2385 (Salmonella Typhimurium nusG+) (J. Krishna Leela, unpublished data). Other plasmids (with salient features in parentheses) employed include pHYD2373 (pWSK129 derivative carrying rne-R169Q,ΔCTH [encoding RNase E-R169Q,ΔCTH], the pSC101 replicon, Kanr, and Cmr) (41), pRne-SG21 (pWSK129 derivative carrying rne-R169Q,529Δ [encoding RNase E-R169Q,529Δ], the pSC101 replicon, and Kanr) (40), pCP20 (pSC101-based Ts replicon encoding Flp recombinase, Cmr, and Ampr) (82), pJK537 (1.8-kb fragment carrying dksA cloned in pBR322, pMB1, and Ampr) (83), and pHR53 (pJK537 derivative carrying truncated dksA gene, pMB1, and Ampr) (84).
Screening methodology for synthetic lethal phenotypes.
A “shelter plasmid loss assay” using an unstable single-copy-number plasmid (85) based on vector pMU575, which is Tpr and lacZ+, was routinely employed as described previously to check whether particular mutant combinations in strains were lethal (41). For example, a Δlac but otherwise wild-type strain carrying plasmid pHYD1613 (a pMU575 derivative carrying rne+) yields ∼10% plasmid-free white colonies on X-Gal-containing medium, whereas a Δrne strain fails to yield viable white colonies. Serial dilutions of stationary-phase cultures carrying the pMU575 derivative grown in LB-Tp were plated on LB with X-Gal (without Tp) to obtain ∼200 colonies, and the ratio of white to total colonies was determined.
Immunoblot analysis of in vivo RNase E levels.
Procedures for lysate preparation, electrophoresis through sodium dodecyl sulfate-polyacrylamide gels, electroblotting to a polyvinylidene difluoride (PVDF) membrane, and staining with amido black (to determine the quantity of protein loading in different lanes) were as described earlier (41). Blocking of PVDF membrane was done with 5% fat-free milk, and the membrane was probed with primary anti-RNase E antibody (rabbit, polyclonal; a gift from A. J. Carpousis), washed, and probed with secondary antibody (anti-rabbit IgG raised in goat) conjugated to horseradish peroxidase as described previously (86). The membrane was then washed and probed for antibody-reactive bands with the aid of a chemiluminescence detection system according to the manufacturers' protocols (Amersham ECL Prime and Sigma Chemical Co., St. Louis, MO).
Other techniques.
Procedures for P1 transduction (81), in vitro DNA manipulations and transformation (86), and β-galactosidase assays (81) were as described previously. Quantitation of the extent of polarity relief in rho and nusG mutants by using Plac-lacZ and Plac-tR1-lacZ (where tR1 is a Rho-dependent terminator from lambdoid phage H19B) fusion construct pairs was performed as reported previously (57). Derivatives with an autoregulated rne-lac fusion were used to determine the status of cellular RNase E activity as described previously (41). An rrnB P1-lacZ construct was used for measurements of transcriptional expression and regulation of the rrn operons (58). The dksA, rnr, pnp, rnb, crp, hfq, and rppH gene deletions were sourced from the Keio collection (87).
Supplementary Material
ACKNOWLEDGMENTS
We thank R. L. Gourse, C. L. Squires, H. Mori, George A. Mackie, and J. Krishna Leela for strains and plasmids used in this study and A. J. Carpousis for RNase E antibody. We thank J. Gowrishankar, R. Harinarayanan, Abhijit A Sardesai, and J. Krishna Leela for advice.
P.H. was supported by a Research Associate Fellowship from the Centre of Excellence in Microbial Biology, Department of Biotechnology, Government of India. This work was supported by a Centre of Excellence in Microbial Biology research grant (BT/01/COE/07/02-II) from the Department of Biotechnology, Government of India.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00724-16.
REFERENCES
- 1.Ludwig H, Homuth G, Schmalisch M, Dyka FM, Hecker M, Stulke J. 2001. Transcription of glycolytic genes and operons in Bacillus subtilis: evidence for the presence of multiple levels of control of the gapA operon. Mol Microbiol 41:409–422. doi: 10.1046/j.1365-2958.2001.02523.x. [DOI] [PubMed] [Google Scholar]
- 2.Mäder U, Hennig S, Hecker M, Homuth G. 2004. Transcriptional organization and posttranscriptional regulation of the Bacillus subtilis branched-chain amino acid biosynthesis genes. J Bacteriol 186:2240–2252. doi: 10.1128/JB.186.8.2240-2252.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marincola G, Schafer T, Behler J, Bernhardt J, Ohlsen K, Goerke Wolz CC. 2012. RNase Y of Staphylococcus aureus and its role in the activation of virulence genes. Mol Microbiol 85:817–832. doi: 10.1111/j.1365-2958.2012.08144.x. [DOI] [PubMed] [Google Scholar]
- 4.Mackie GA. 1998. Ribonuclease E is a 5′-end-dependent endonuclease. Nature 395:720–723. doi: 10.1038/27246. [DOI] [PubMed] [Google Scholar]
- 5.Mackie GA. 2013. RNase E: at the interface of bacterial RNA processing and decay. Nat Rev Microbiol 11:45–57. doi: 10.1038/nrmicro2930. [DOI] [PubMed] [Google Scholar]
- 6.Jourdan SS, McDowall KJ. 2008. Sensing of 5′ monophosphate by Escherichia coli RNase G can significantly enhance association with RNA and stimulate the decay of functional mRNA transcripts in vivo. Mol Microbiol 67:102–115. [DOI] [PubMed] [Google Scholar]
- 7.Coburn GA, Mackie GA. 1999. Degradation of mRNA in Escherichia coli: an old problem with some new twists. Prog Nucleic Acid Res Mol Biol 62:55–108. [DOI] [PubMed] [Google Scholar]
- 8.Grunberg-Manago M. 1999. Messenger RNA stability and its role in control of gene expression in bacteria and phages. Annu Rev Genet 33:193–227. doi: 10.1146/annurev.genet.33.1.193. [DOI] [PubMed] [Google Scholar]
- 9.Régnier P, Arraiano CM. 2000. Degradation of mRNA in bacteria: emergence of ubiquitous features. Bioessays 22:235–244. [DOI] [PubMed] [Google Scholar]
- 10.McDowall KJ, Cohen SN. 1996. The N-terminal domain of the rne gene product has RNase E activity and is non-overlapping with the arginine-rich RNA-binding site. J Mol Biol 255:349–355. doi: 10.1006/jmbi.1996.0027. [DOI] [PubMed] [Google Scholar]
- 11.Miczak A, Kaberdin VR, Wei CL, Lin-Chao S. 1996. Proteins associated with RNase E in a multicomponent ribonucleolytic complex. Proc Natl Acad Sci U S A 93:3865–3869. doi: 10.1073/pnas.93.9.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vanzo NF, Li YS, Py B, Blum E, Higgins CF, Raynal LC, Krisch HM, Carpousis AJ. 1998. Ribonuclease E organizes the protein interactions in the Escherichia coli RNA degradosome. Genes Dev 2:2770–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deutscher MP. 2006. Degradation of RNA in bacteria: comparison of mRNA and stable RNA. Nucleic Acids Res 34:659–666. doi: 10.1093/nar/gkj472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carpousis AJ, Luisi BF, McDowall KJ. 2009. Endonucleolytic initiation of mRNA decay in Escherichia coli. Prog Mol Biol Transl Sci 85:91–135. doi: 10.1016/S0079-6603(08)00803-9. [DOI] [PubMed] [Google Scholar]
- 15.Arraiano CM, Andrade JM, Domingues S, Guinote IB, Malecki M, Matos RG, Moreira RN, Pobre V, Reis FP, Saramago M, Silva IJ, Viegas SC. 2010. The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol Rev 34:883–923. doi: 10.1111/j.1574-6976.2010.00242.x. [DOI] [PubMed] [Google Scholar]
- 16.Py B, Higgins CF, Krisch HM, Carpousis AJ. 1996. A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature 381:169–172. doi: 10.1038/381169a0. [DOI] [PubMed] [Google Scholar]
- 17.Coburn GA, Miao X, Briant DJ, Mackie GA. 1999. Reconstitution of a minimal RNA degradosome demonstrates functional coordination between a 3′ exonuclease and a DEAD-box RNA helicase. Genes Dev 13:2594–2603. doi: 10.1101/gad.13.19.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Z, Pandit S, Deutscher MP. 1999. RNase G (CafA protein) and RNase E are both required for the 5′-maturation of 16S ribosomal RNA. EMBO J 18:2878–2885. doi: 10.1093/emboj/18.10.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ow MC, Kushner SR. 2002. Initiation of tRNA maturation by RNase E is essential for cell viability in E. coli. Genes Dev 16:1102–1115. doi: 10.1101/gad.983502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Z, Deutscher MP. 2002. RNase E plays an essential role in the maturation of Escherichia coli tRNA precursors. RNA 8:97–109. doi: 10.1017/S1355838202014929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin-Chao S, Wei Chia-Li, Lin Yi-Tzu. 1999. RNase E is required for the maturation of ssrA RNA and normal ssrA RNA peptide-tagging activity. Proc Natl Acad Sci U S A 96:12406–12411. doi: 10.1073/pnas.96.22.12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bessarab DA, Kaberdin VR, Wei CL, Liou GG, Lin-Chao S. 1998. RNA components of Escherichia coli degradosome: evidence for rRNA decay. Proc Natl Acad Sci U S A 95:3157–3161. doi: 10.1073/pnas.95.6.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sulthana S, Basturea GN, Deutscher MP. 2016. Elucidation of pathways of ribosomal RNA degradation: as essential role for RNase E. RNA 22:1163–1171. doi: 10.1261/rna.056275.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jain C, Belasco JG. 1995. RNase E autoregulates its synthesis by controlling the degradation rate of its own mRNA in Escherichia coli: unusual sensitivity of the rne transcript to RNase E activity. Genes Dev 9:84–96. doi: 10.1101/gad.9.1.84. [DOI] [PubMed] [Google Scholar]
- 25.Sousa S, Marchand I, Dreyfus M. 2001. Autoregulation allows Escherichia coli RNase E to adjust continuously its synthesis to that of its substrates. Mol Microbiol 42:867–878. [DOI] [PubMed] [Google Scholar]
- 26.Schuck A, Diwa A, Belasco JG. 2009. RNase E autoregulates its synthesis in Escherichia coli by binding directly to a stem-loop in the rne5′-untranslated region. Mol Microbiol 72:470–478. doi: 10.1111/j.1365-2958.2009.06662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ow MC, Liu Q, Kushner SR. 2000. Analysis of mRNA decay and rRNA processing in Escherichia coli in the absence of RNase E-based degradosome assembly. Mol Microbiol 38:854–866. doi: 10.1046/j.1365-2958.2000.02186.x. [DOI] [PubMed] [Google Scholar]
- 28.Deana A, Belasco JG. 2004. The function of RNase G in Escherichia coli is constrained by its amino and carboxyl termini. Mol Microbiol 51:1205–1217. doi: 10.1046/j.1365-2958.2003.03905.x. [DOI] [PubMed] [Google Scholar]
- 29.Perwez T, Hami D, Maples VF, Min Z, Wang BC, Kushner SR. 2008. Intragenic suppressors of temperature-sensitive rne mutations lead to the dissociation of RNase E activity on mRNA and tRNA substrates in Escherichia coli. Nucleic Acids Res 36:5306–5318. doi: 10.1093/nar/gkn476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung DH, Min Z, Wang BC, Kushner SR. 2010. Single amino acid changes in the predicted RNase H domain of Escherichia coli RNase G lead to complementation of RNase E deletion mutants. RNA 16:1371–1385. doi: 10.1261/rna.2104810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee K, Bernstein JA, Cohen SN. 2002. RNase G complementation of rne null mutation identifies functional interrelationships with RNase E in Escherichia coli. Mol Microbiol 43:1445–1456. doi: 10.1046/j.1365-2958.2002.02848.x. [DOI] [PubMed] [Google Scholar]
- 32.Hammarlöf DL, Bergman JM, Garmendia E, Hughes D. 2015. Turnover of mRNAs is one of the essential functions of RNase E. Mol Microbiol 98:34–45. doi: 10.1111/mmi.13100. [DOI] [PubMed] [Google Scholar]
- 33.Takada A, Nagai K, Wachi M. 2005. A decreased level of FtsZ is responsible for inviability of RNase E-deficient cells. Genes Cells 10:733–741. doi: 10.1111/j.1365-2443.2005.00872.x. [DOI] [PubMed] [Google Scholar]
- 34.Tamura M, Lee K, Miller CA, Moore CJ, Shirako Y, Kobayashi M, Cohen SN. 2006. RNase E maintenance of proper FtsZ/FtsA ratio required for nonfilamentous growth of Escherichia coli cells but not for colony forming ability. J Bacteriol 188:5145–5152. doi: 10.1128/JB.00367-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Callaghan AJ, Marcaida MJ, Stead JA, McDowall KJ, Scott WG, Luisi BF. 2005. Structure of Escherichia coli RNase E catalytic domain and implications for RNA turnover. Nature 437:1187–1191. doi: 10.1038/nature04084. [DOI] [PubMed] [Google Scholar]
- 36.Koslover DJ, Callaghan AJ, Marcaida MJ, Garman EF, Martick M, Scott WG, Luisi BF. 2008. The crystal structure of the Escherichia coli RNase E apoprotein and a mechanism for RNA degradation. Structure 16:1238–1244. doi: 10.1016/j.str.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang X, Belasco JG. 2004. Catalytic activation of multimeric RNase E and RNase G by 5′-monophsophorylated RNA. Proc Natl Acad Sci U S A 101:9211–9216. doi: 10.1073/pnas.0401382101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Celesnik H, Deana A, Belasco JG. 2007. Initiation of RNA decay in Escherichia coli by 5′ pyrophosphate removal. Mol Cell 27:79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deana A, Celesnik H, Belasco JG. 2008. The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature 451:355–358. doi: 10.1038/nature06475. [DOI] [PubMed] [Google Scholar]
- 40.Garrey SM, Mackie GA. 2011. Roles of the 5′-phosphate sensor domain in RNase E. Mol Microbiol 80:1613–1624. doi: 10.1111/j.1365-2958.2011.07670.x. [DOI] [PubMed] [Google Scholar]
- 41.Anupama K, Leela JK, Gowrishankar J. 2011. Two pathways for RNase E action in Escherichia coli in vivo and bypass of its essentiality in mutants defective for Rho-dependent transcription termination. Mol Microbiol 82:1330–1348. doi: 10.1111/j.1365-2958.2011.07895.x. [DOI] [PubMed] [Google Scholar]
- 42.Bernstein JA, Lin PH, Cohen SN, Lin-Chao S. 2004. Global analysis of Escherichia coli RNA degradosome function using DNA microarrays. Proc Natl Acad Sci U S A 101:2758–2763. doi: 10.1073/pnas.0308747101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lopez PJ, Marchand I, Joyce SA, Dreyfus M. 1999. The C-terminal half of RNase E, which organizes the Escherichia coli degradosome, participates in mRNA degradation but not rRNA processing in vivo. Mol Microbiol 33:188–199. doi: 10.1046/j.1365-2958.1999.01465.x. [DOI] [PubMed] [Google Scholar]
- 44.Leroy A, Vanzo NF, Sousa S, Dreyfus M, Carpousis AJ. 2002. Function in Escherichia coli of the non-catalytic part of RNase E: role in the degradation of ribosome-free mRNA. Mol Microbiol 45:1231–1243. doi: 10.1046/j.1365-2958.2002.03104.x. [DOI] [PubMed] [Google Scholar]
- 45.Dalebroux ZD, Swanson MS. 2012. ppGpp: magic beyond RNA polymerase. Nat Rev Microbiol 10:203–212. doi: 10.1038/nrmicro2720. [DOI] [PubMed] [Google Scholar]
- 46.Hauryliuk V, Atkinson GC, Murakami KS, Tenson T, Gerdes K. 2015. Recent functional insights into the role of (p) ppGpp in bacterial physiology. Nat Rev Microbiol 13:298–309. doi: 10.1038/nrmicro3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ferenci T, Zhou Z, Betteridge T, Ren Y, Liu Y, Feng L, Reeves PR, Wang L. 2009. Genomic sequencing reveals regulatory mutations and recombinational events in the widely used MC4100 lineage of Escherichia coli K-12. J Bacteriol 191:4025–4029. doi: 10.1128/JB.00118-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spira B, Hu X, Ferenci T. 2008. Strain variation in ppGpp concentration and RpoS levels in laboratory strains of Escherichia coli K-12. Microbiology 154:2887–2895. doi: 10.1099/mic.0.2008/018457-0. [DOI] [PubMed] [Google Scholar]
- 49.Laffler T, Gallant JA. 1974. spoT, a new genetic locus involved in the stringent response in E. coli. Cell 1:27–30. doi: 10.1016/0092-8674(74)90151-2. [DOI] [Google Scholar]
- 50.Sarubbi E, Rudd KE, Cashel M. 1988. Basal ppGpp level adjustment shown by new spoT mutants affect steady state growth rates and rrnA ribosomal promoter regulation in Escherichia coli. Mol Gen Genet 213:214–222. doi: 10.1007/BF00339584. [DOI] [PubMed] [Google Scholar]
- 51.Murphy H, Cashel M. 2003. Isolation of RNA polymerase suppressors of a (p)ppGpp deficiency. Methods Enzymol 371:596–601. doi: 10.1016/S0076-6879(03)71044-1. [DOI] [PubMed] [Google Scholar]
- 52.Lazzarini RA, Cashel M, Gallant J. 1971. On the regulation of guanosine tetraphosphate levels in stringent and relaxed strains of Escherichia coli. J Biol Chem 246:4381–4385. [PubMed] [Google Scholar]
- 53.Madison KE, Jones-Foster EN, Vogt A, Kirtland Turner S, North SH, Nakai H. 2014. Stringent response processes suppress DNA damage sensitivity caused by deficiency in full-length translation initiation factor 2 or PriA helicase. Mol Microbiol 92:28–46. doi: 10.1111/mmi.12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Durfee T, Hansen AM, Zhi H, Blattner FR, Jin DJ. 2008. Transcription profiling of the stringent response in Escherichia coli. J Bacteriol 190:1084–1096. doi: 10.1128/JB.01092-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leela JK, Syeda AH, Anupama K, Gowrishankar J. 2013. Rho-dependent transcription termination is essential to prevent excessive genome-wide R-loops in Escherichia coli. Proc Natl Acad Sci U S A 110:258–263. doi: 10.1073/pnas.1213123110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harinarayanan R, Gowrishankar J. 2003. Host factor titration by chromosomal R-loops as a mechanism for runaway plasmid replication in transcription termination defective mutants of Escherichia coli. J Mol Biol 332:31–46. doi: 10.1016/S0022-2836(03)00753-8. [DOI] [PubMed] [Google Scholar]
- 57.Saxena S, Gowrishankar J. 2011. Modulation of Rho-dependent transcription termination in Escherichia coli by the H-NS family of proteins. J Bacteriol 193:3832–3841. doi: 10.1128/JB.00220-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou YN, Jin DJ. 1998. The rpoB mutants destabilizing initiation complexes at stringently controlled promoters behave like “stringent” RNA polymerases in Escherichia coli. Proc Natl Acad Sci U S A 95:2908–2913. doi: 10.1073/pnas.95.6.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cashel M, Gentry DR, Hernandez VJ, Vinella D. 1996. The stringent response, p 1458–1496. In Neidhardt FC, Curtiss R III, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE (ed), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed, vol 1 ASM Press, Washington, DC. [Google Scholar]
- 60.Barker MM, Gaal T, Gourse RL. 2001. Mechanism of regulation of transcription initiation by ppGpp. II. Models for positive control based on properties of RNAP mutants and competition for RNAP. J Mol Biol 305:689–702. [DOI] [PubMed] [Google Scholar]
- 61.Jin DJ, Gross CA. 1991. RpoB8, a rifampicin-resistant termination-proficient RNA polymerase, has an increased K, for purine nucleotides during transcription elongation. J Biol Chem 266:14478–14465. [PubMed] [Google Scholar]
- 62.Jain C, Deana A, Belasco JG. 2002. Consequences of RNase E scarcity in Escherichia coli. Mol Microbiol 43:1053–1064. doi: 10.1046/j.1365-2958.2002.02808.x. [DOI] [PubMed] [Google Scholar]
- 63.Ross W, Sanchez-Vazquez P, Chen AY, Lee JH, Burgos HL, Gourse RL. 2016. ppGpp binding to a site at the RNAP-DksA interface accounts for its dramatic effects on transcription initiation during the stringent response. Mol Cell 62:811–823. doi: 10.1016/j.molcel.2016.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vinella D, Potrykus K, Murphy H, Cashel M. 2012. Effects on growth by changes of the balance between GreA, GreB, and DksA suggest mutual competition and functional redundancy in Escherichia coli. J Bacteriol 194:261–273. doi: 10.1128/JB.06238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Potrykus K, Vinella D, Murphy H, Szalewska-Palasz A, D'Ari R, Cashel M. 2006. Antagonistic regulation of Escherichia coli ribosomal RNA rrnB P1 promoter activity by GreA and DksA. J Biol Chem 281:15238–15248. doi: 10.1074/jbc.M601531200. [DOI] [PubMed] [Google Scholar]
- 66.Quan S, Skovgaard O, McLaughlin RE, Buurman ET, Squires CL. 2015. Markerless Escherichia coli rrn deletion strains for genetic determination of ribosomal binding sites. G3 (Bethesda) 5:2555–2557. doi: 10.1534/g3.115.022301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Perrenoud A, Sauer U. 2005. Impact of global transcriptional regulation by ArcA, ArcB, Cra, Crp, Cya, Fnr, and Mlc on glucose catabolism in Escherichia coli. J Bacteriol 187:3171–3179. doi: 10.1128/JB.187.9.3171-3179.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsui HC, Leung HC, Winkler ME. 1994. Characterization of broadly pleiotropic phenotypes caused by an hfq insertion mutation in Escherichia coli K-12. Mol Microbiol 13:35–49. doi: 10.1111/j.1365-2958.1994.tb00400.x. [DOI] [PubMed] [Google Scholar]
- 69.Mohanty BK, Kushner SR. 2000. Polynucleotide phosphorylase, RNase II and RNase E play different roles in the in vivo modulation of polyadenylation in Escherichia coli. Mol Microbiol 36:982–994. doi: 10.1046/j.1365-2958.2000.01921.x. [DOI] [PubMed] [Google Scholar]
- 70.Mohanty BK, Kushner SR. 2003. Genomic analysis in Escherichia coli demonstrates differential roles for polynucleotide phosphorylase and RNase II in mRNA abundance and decay. Mol Microbiol 50:645–658. doi: 10.1046/j.1365-2958.2003.03724.x. [DOI] [PubMed] [Google Scholar]
- 71.Zuo Y, Deutscher MP. 2001. Exoribonuclease superfamilies: structural analysis and phylogenetic distribution. Nucleic Acids Res 29:1017–1026. doi: 10.1093/nar/29.5.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ryals J, Little R, Bremer H. 1982. Control of rRNA and tRNA syntheses in Escherichia coli by guanosine tetraphosphate. J Bacteriol 151:1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Barker MM, Gaal T, Josaitis CA, Gourse RL. 2001. Mechanism of regulation of transcription initiation by ppGpp. I. Effects of ppGpp on transcription initiation in vivo and in vitro. J Mol Biol 305:673–688. [DOI] [PubMed] [Google Scholar]
- 74.Paul BJ, Barker MM, Ross W, Schneider DA, Webb C, Foster JW, Gourse RL. 2004. DksA: a critical component of the transcription initiation machinery that potentiates the regulation of rRNA promoters by ppGpp and the initiating NTP. Cell 118:311–322. doi: 10.1016/j.cell.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 75.Condon C, French S, Squires C, Squires CL. 1993. Depletion of functional ribosomal RNA operons in Escherichia coli causes increased expression of the remaining intact copies. EMBO J 12:4305–4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Asai T, Condon C, Voulgaris J, Zaporojets D, Shen B, Al-Omar M, Squires C, Squires CL. 1999. Construction and initial characterization of Escherichia coli strains with few or no intact chromosomal rRNA operons. J Bacteriol 181:3803–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bremer H, Dennis PP. 1996. Modulation of chemical composition and other parameters of the cell by growth rate, p 1553–1569. In Neidhardt FC, Curtiss R III, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE (ed), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed, vol 2 ASM Press, Washington, DC. [Google Scholar]
- 78.Andrade JM, Cairrão F, Arraiano CM. 2006. RNase R affects gene expression in stationary phase: regulation of ompA. Mol Microbiol 60:219–228. doi: 10.1111/j.1365-2958.2006.05092.x. [DOI] [PubMed] [Google Scholar]
- 79.Andrade JM, Hajnsdorf E, Régnier P, Arraiano CM. 2009. The poly(A)-dependent degradation pathway of rpsO mRNA is primarily mediated by RNase R. RNA 15:316–326. doi: 10.1261/rna.1197309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cairrão F, Arraiano CM. 2006. The role of endoribonucleases in the regulation of RNase R. Biochem Biophys Res Commun 343:731–737. doi: 10.1016/j.bbrc.2006.03.040. [DOI] [PubMed] [Google Scholar]
- 81.Miller JH. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 82.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kang PJ, Craig EA. 1990. Identification and Characterization of a New Escherichia coli gene that is a dosage-dependent suppressor of a dnaK deletion mutation. J Bacteriol 172:2055–2064. doi: 10.1128/jb.172.4.2055-2064.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nazir A, Harinarayanan R. 2015. Inactivation of cell division protein FtsZ by SulA makes Lon indispensable for the viability of a ppGpp0 strain of Escherichia coli. J Bacteriol 198:688–700. doi: 10.1128/JB.00693-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bernhardt TG, de Boer PA. 2004. Screening for synthetic lethal mutants in Escherichia coli and identification of EnvC (YibP) as a periplasmic septal ring factor with murein hydrolase activity. Mol Microbiol 52:1255–1269. doi: 10.1111/j.1365-2958.2004.04063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 87.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chandrangsu P, Wang L, Choi SH, Gourse RL. 2012. Suppression of a dnaKJ deletion by multicopy dksA results from non-feedback-regulated transcripts that originate upstream of the major dksA promoter. J Bacteriol 194:1437–1446. doi: 10.1128/JB.06726-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.