

ABSTRACT
Bacterial microcompartments (MCPs) are extremely large proteinaceous organelles that consist of an enzymatic core encapsulated within a complex protein shell. A key question in MCP biology is the nature of the interactions that guide the assembly of thousands of protein subunits into a well-ordered metabolic compartment. In this report, we show that the N-terminal 37 amino acids of the PduB protein have a critical role in binding the shell of the 1,2-propanediol utilization (Pdu) microcompartment to its enzymatic core. Several mutations were constructed that deleted short regions of the N terminus of PduB. Growth tests indicated that three of these deletions were impaired MCP assembly. Attempts to purify MCPs from these mutants, followed by gel electrophoresis and enzyme assays, indicated that the protein complexes isolated consisted of MCP shells depleted of core enzymes. Electron microscopy substantiated these findings by identifying apparently empty MCP shells but not intact MCPs. Analyses of 13 site-directed mutants indicated that the key region of the N terminus of PduB required for MCP assembly is a putative helix spanning residues 6 to 18. Considering the findings presented here together with prior work, we propose a new model for MCP assembly.
IMPORTANCE Bacterial microcompartments consist of metabolic enzymes encapsulated within a protein shell and are widely used to optimize metabolic process. Here, we show that the N-terminal 37 amino acids of the PduB shell protein are essential for assembly of the 1,2-propanediol utilization microcompartment. The results indicate that it plays a key role in binding the outer shell to the enzymatic core. We propose that this interaction might be used to define the relative orientation of the shell with respect to the core. This finding is of fundamental importance to our understanding of microcompartment assembly and may have application to engineering microcompartments as nanobioreactors for chemical production.
KEYWORDS: microcompartment; carboxysome; 1,2-propanediol; Salmonella; vitamin B12
INTRODUCTION
Hundreds of species of bacteria produce complex proteinaceous organelles known as bacterial microcompartments (MCPs) (1–8). The function of MCPs is to optimize metabolic pathways having intermediates that are toxic or poorly retained by the cell envelope (5, 7, 9). They consist of sequentially acting metabolic enzymes (an enzymatic core) encapsulated within a protein shell that controls the diffusion of enzyme substrates and products while confining toxic or volatile intermediates (10–14). MCPs are among the largest multiprotein complexes known. They are typically 100 to 150 nm in diameter and up to a gigadalton or more in mass and are built from thousands of protein subunits of 10 to 20 different types. A key question of MCP architecture, and the subject of this report, is the protein-protein interactions that guide the assembly of thousands of protein subunits into a functioning bacterial organelle with a defined higher-order structure.
The first bacterial MCP identified was the carboxysome, which is used to enhance autotrophic CO2 fixation (15, 16). Subsequent studies showed that MCPs are involved in the catabolism of 1,2-propanediol (1,2-PD), ethanolamine, choline, glycerol, rhamnose, fucose, and fucoidan (11, 17–22). Based on bioinformatics analyses, MCPs are produced by about 20% of bacteria distributed across 11 kingdom-level taxa and have a number of variant forms with uncertain physiological roles (1, 2, 6, 8). Notably, MCPs have been linked to bacterial pathogenesis, as well as to heart disease, via their metabolic role in the gut microbiome, and they are a promising basis for the development of engineered nanocompartments (5, 23–29). A remarkable feature of bacterial MCPs is that the outer shells of diverse metabolic types are built primarily from the same family of proteins known as bacterial microcompartment (BMC) domain proteins (26, 30, 31). BMC domain family members have a flat hexagonal quaternary structure and tile edge to edge into extended protein sheets (30). Many are functionally diversified, and a typical MCP shell is composed of 4 to 10 different BMC domain proteins (5, 7). The edge-to-edge interactions that drive shell protein tessellation are thought to be conserved among varied types of BMC domain proteins, allowing the formation of complex shells (31, 32). Another key aspect of MCP architecture is the use of short sequence extensions on the N and C termini of both lumen enzymes and structural proteins to guide assembly (33–37). Many enzymes that localize to the lumen of bacterial MCPs have N-terminal extensions that are absent from homologs that are not associated with MCPs (35, 37), and in a number of cases, these extensions (typically about 20 amino acids in length) are necessary and sufficient for the encapsulation of enzymes into MCP shells (25, 33–37). In one case, an N-terminal-targeting sequence was found to interact with short C-terminal α-helix conserved on several shell proteins, and this binding interaction is thought to guide encapsulation (38, 39). In addition, a short C-terminal sequence of the CcmN protein is essential for carboxysome formation and is proposed to bind the CcmK2 shell protein (37).
Perhaps the best understood MCP is the 1,2-PD utilization (Pdu) MCP of Salmonella enterica serovar Typhimurium LT2. The Pdu MCP encapsulates enzymes used for the B12-dependent degradation of 1,2-PD, which is an important carbon and energy source, particularly in anaerobic environments (40, 41). Initially, 1,2-PD diffuses into the lumen of the MCP, where it is converted to propionaldehyde by coenzyme B12-dependent diol dehydratase (40, 42). Propionaldehyde is then converted to propionyl coenzyme A (propionyl-CoA) by CoA-dependent aldehyde dehydrogenase (PduP) or to 1-propanol by the PduQ alcohol dehydrogenase (43, 44). The PduL enzyme converts propionyl-CoA to propionyl-PO4, which exits the MCP and enters central metabolism via the methyl citrate pathway (45–48). The protein shell of the Pdu MCP confines propionaldehyde, minimizing its toxicity and diffusive loss (14, 49). This shell is composed of seven BMC domain proteins (PduA, PduB, PduB′, PduJ, PduK, PduT, and PduU) and a bacterial microcompartment vertex (BMV) protein (PduN) and may also include PduM, but the function of this protein is uncertain (50–52). The most abundant shell proteins of the Pdu MCP are PduB and PduB′ (51). Together, they make up about 50% of the shell and almost 25% of the total MCP protein (51). PduB and PduB′ are expressed from overlapping genes and are identical in sequence, except that PduB has a 37-amino-acid N-terminal extension (51, 53). In this report, we investigate the role of the 37-amino-acid N-terminal extension of PduB, and based on our results, we present a new model for MCP assembly.
RESULTS
Multiple-sequence analysis and secondary structure prediction for the N-terminal region of PduB.
To investigate the function of the N-terminal region of PduB, we constructed a multiple-sequence alignment to look for conserved sequence features. PSI-BLAST (54) was used to identify PduB homologs in GenBank, and Clustal Omega (55) was used to construct the alignment. Amino acid residues L6 to V18 of the PduB N-terminal region were highly conserved across diverse genera, but residues A18 to E35 were much more divergent, particularly when distantly related genera were compared (Fig. 1). Analyses with the small-peptide structure prediction software PEP-FOLD (56) suggested that the N terminus of PduB is composed of an α-helix (L6 to V18), followed by a disordered region (A18 to E35). Thus, the N-terminal extension that distinguishes PduB from PduB′ consists of a highly conserved predicted α-helix and a less conserved disordered region connected to the two tandem BMC domains that comprise the core structure of both PduB and PduB′ (57).
FIG 1.

Multiple-sequence alignment of the first 53 amino acids of the PduB/PduB′ coding region, with their respective translation start sites indicated. The PduB protein has a 37-amino-acid N-terminal extension not present in PduB′ but is otherwise identical in amino acid sequence. Blue-highlighted residues have >80% amino acid sequence identity across the sequences shown, whose GI numbers are (top to bottom) 409994423, 545166260, 737633100, 490278627, 507082779, 506359047, 738157884, 696233319, 746121973, and 496088740.
N-terminal deletion mutants.
To further investigate the role of the N-terminal extension of PduB, a series of scarless mutants were constructed by Sac-Cat recombineering and verified by DNA sequencing (Fig. 2): in the PduB Δ1–37 mutant, the entire N-terminal extension that distinguishes PduB from PduB′ is deleted; in the PduB Δ6–12 mutant, the N-terminal half of the highly conserved predicted α-helix is removed; in the PduB Δ11–25 mutant, the C-terminal half of the same α-helix and part of the linker region are removed; in the PduB Δ27–32 mutant, a somewhat unusual proline-rich area of the linker region is deleted; and in the PduB M38A mutant, the start codon for PduB′ is changed to CGC (alanine) such that PduB M38A is produced but PduB′ is not (verified as described below).
FIG 2.
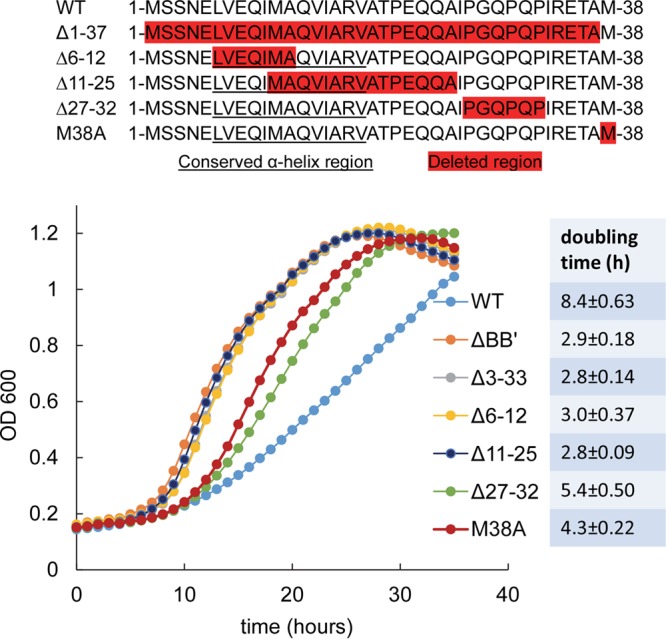
Short N-terminal deletions in PduB result in faster growth of Salmonella on 1,2-propanediol minimal medium. The top panel is a schematic of N-terminal deletions that were tested. Residues highlighted in red indicate the deleted regions. Underlined residues correspond to a highly conserved putative α-helix. The pduBB′ deletion mutant is a control known to have a very high growth rate due to an MCP assembly defect. Other PduB deletion mutants are indicated in the legend. The M38A mutant changes the start codon of PduB′ and blocks its translation; thus, it is essentially a ΔpduB′ mutant. The table to the right shows the doubling time for each strain, and the error shown is one standard deviation determined from the results from three or more independent experiments. The wild type (WT) is Salmonella enterica LT2 serovar Typhimurium. OD600, optical density at 600 nm.
Effect of N-terminal PduB deletions on MCP assembly.
Our initial analysis of the PduB deletions was to test their effects on MCP assembly in vivo (Fig. 2). Prior studies showed that Salmonella mutants unable to correctly assemble the Pdu MCP grow substantially faster than the wild type on 1,2-PD minimal medium with limiting B12 (49). This phenotype, which is due to increased access of the MCP lumen enzymes to their substrates, is a reliable test of the integrity/permeability of the Pdu MCP (49, 50, 58). The deletion mutants described above showed a range of growth rates on 1,2-PD at limiting B12 (Fig. 2). The PduB Δ1–37, PduB Δ6–12, and PduB Δ11–25 mutants grew much faster than the wild type on minimal 1,2-PD medium, with limiting B12 indicating an assembly defect. Indeed, the growth rates resulting from these short deletions were similar to that observed for a full-length pduBB′ deletion, which was previously shown to produce no MCPs (50). On the other hand, the PduB Δ27–32 mutation and the PduB M38A mutation (which prevents translation of the PduB′ protein by replacement of its start codon) resulted in intermediate growth phenotypes indicative of a partial assembly defect (Fig. 2). We consider it unlikely that any of the mutations described above had substantial effects on PduB folding, since prior crystallographic studies showed that PduB lacking 37 N-terminal amino acids (PduB′) folds normally (57) and, below, we show that these mutant proteins are still incorporated into the shell of the Pdu MCP. Hence, the studies described above indicate that the N-terminal region of PduB plays a role in assembly of the Pdu MCP.
To further investigate the role of the N terminus of PduB in MCP assembly, MCPs from each of the mutants described above were purified and analyzed. MCP yield (the total protein content of the purified MCPs/gram of cells) relative to the wild type was used to evaluate MCP stability during purification. SDS-PAGE was used to examine protein content, and enzyme assays were used to determine the relative abundance of two major lumen enzymes, B12-dependent diol dehydratase (PduCDE) and aldehyde dehydrogenase (PduP). The PduB M38A and PduB Δ27–32 mutant MCPs were purified with somewhat reduced yields compared to the wild type, at 66% and 75%, respectively, consistent with the partially impaired assembly/stability indicated by growth tests (Fig. 3). However, the protein content of these mutant MCPs was similar to the wild type by SDS-PAGE, with the exception of the changes due to the mutations (Fig. 3). For the PduB Δ27–32 mutant, the truncated PduB protein ran just above the PduD band. For the PduB M38A mutant, the PduB′ band is missing, consistent with prior studies that indicated that M38 is the start of PduB′ (51, 53). All other major proteins in MCPs purified from these mutants were present at levels similar to the wild type. Some minor MCP proteins (PduN, PduK, and PduL) are difficult to observe by SDS-PAGE; hence, their abundance in the mutant MCPs is uncertain. We also measured the PduCDE (diol dehydratase) activity of the purified mutant MCPs. The activities of the PduB M38A (25.5 ± 0.9 μmol · min−1 · mg−1) and PduB Δ27–32 (25.6 ± 0.9 μmol · min−1 · mg−1) mutants were similar to that of the wild type (26.0 ± 1.3 μmol · min−1 · mg−1), indicating normal encapsulation of PduCDE within the MCP (Fig. 3). For PduP aldehyde dehydrogenase, activities were 9.3 ± 0.6, 10.2 ± 0.8, and 10.3 ± 0.08 μmol · min−1 · mg−1 for the wild type, PduB Δ27–32 mutant, and PduB M38A mutant, respectively (Fig. 3). Overall, analyses of purified MCPs suggest that the PduB Δ27–32 and PduB M38A mutants have a defect that partially impairs MCP assembly or stability, leading to a reduced yield during purification; however, this defect does not have a large effect on the composition of purified MCPs.
FIG 3.
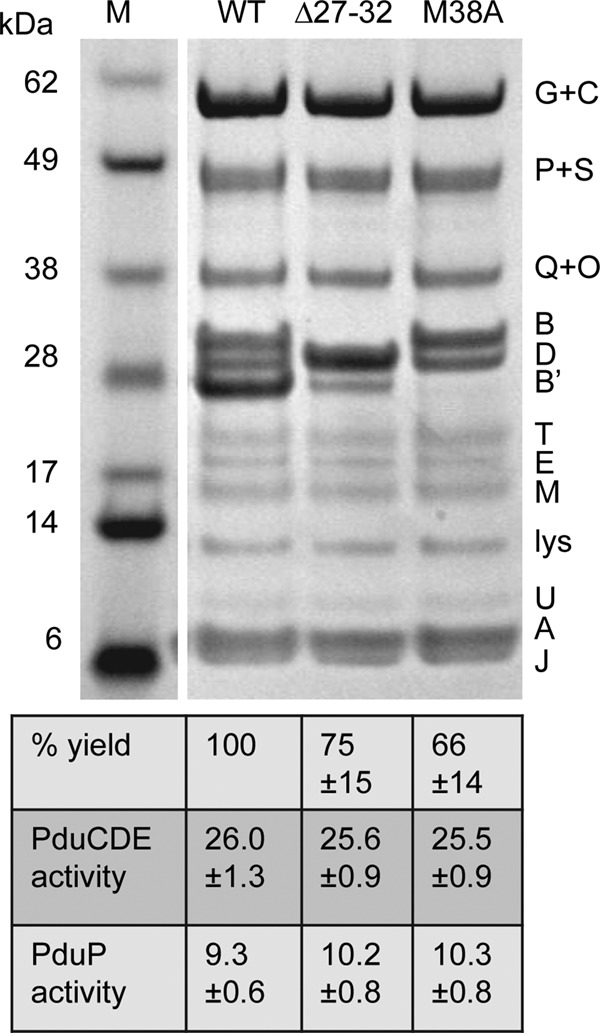
SDS-PAGE of MCPs purified from the PduB Δ27–32 mutant, the PduB M38A mutant, and the wild type (WT). Letters on the right indicate the Pdu protein represented by each band. Lysozyme (lys) is not an MCP component but was used to lyse cells. The percent yield of purified MCPs indicates grams of protein/gram of cells relative to the wild type. The specific activities of the major lumen enzymes, the PduCDE diol dehydratase and the PduP aldehyde dehydrogenase, are shown in micromoles per minute per milligram. Ten micrograms of protein was loaded in each lane. Gels were stained using a Coomassie-based protocol. The enzyme assay methods are described in Materials and Methods. The error shown is one standard deviation determined from the results from three or more independent experiments. M, molecular mass markers.
The other N-terminal deletion mutants analyzed (PduB Δ1–37, PduB Δ11–25, and PduB Δ6–12) all had growth phenotypes indicative of a more severe MCP assembly defect (Fig. 2). In addition, yields of MCPs from these mutants (total protein obtained in the MCP fraction/gram of cells) were drastically lower than that from the wild type, as only 11%, 27%, and 30% relative recovery was obtained, respectively, which is indicative of a substantial assembly defect (Fig. 4). We also analyzed the protein content of the MCP fractions purified from the PduB Δ1–37, PduB Δ11–25, and PduB Δ6–12 mutants by SDS-PAGE (Fig. 4). Prior studies have shown that the MCP purification protocol used here allows the isolation of partially assembled and misassembled MCP complexes (32, 50). The complexes obtained from the PduB Δ1–37, PduB Δ11–25, and PduB Δ6–12 mutants had a substantially altered protein composition compared to wild-type MCPs (Fig. 4). These complexes included the major shell proteins of the Pdu MCP, PduA, PduB′, and PduJ, as well as truncated versions of PduB resulting from the mutagenesis and the minor shell proteins PduM, PduU, and PduT. Interestingly, however, these mutants appeared to have substantially reduced levels of the major lumen enzymes (PduG, PduC, PduP, PduQ plus PduO, PduD, and PduE) (Fig. 4). Enzyme assays substantiated that the MCP complexes isolated from the PduB Δ1–37, PduB 6–12, and PduB Δ11–25 mutants contained much lower levels of the PduCDE diol dehydratase and the PduP aldehyde dehydrogenase than the wild type (Fig. 4). We propose that these mutations impaired the association of lumen enzymes with the MCP shell but had lesser effects on interactions among the major shell proteins, although more complex explanations are possible. It is notable that the mutant forms of PduB analyzed here were incorporated into the MCP shell complexes that were isolated, supporting their normal folding. Furthermore, the yields of purified “empty shells” were underestimated, since the enzymatic core constitutes about 50% of the total MCP protein (Fig. 4). Hence, analysis of the MCPs purified from the PduB Δ1–37, PduB Δ11–25, and PduB Δ6–12 mutants substantiates a key role for the N terminus of PduB in MCP assembly.
FIG 4.
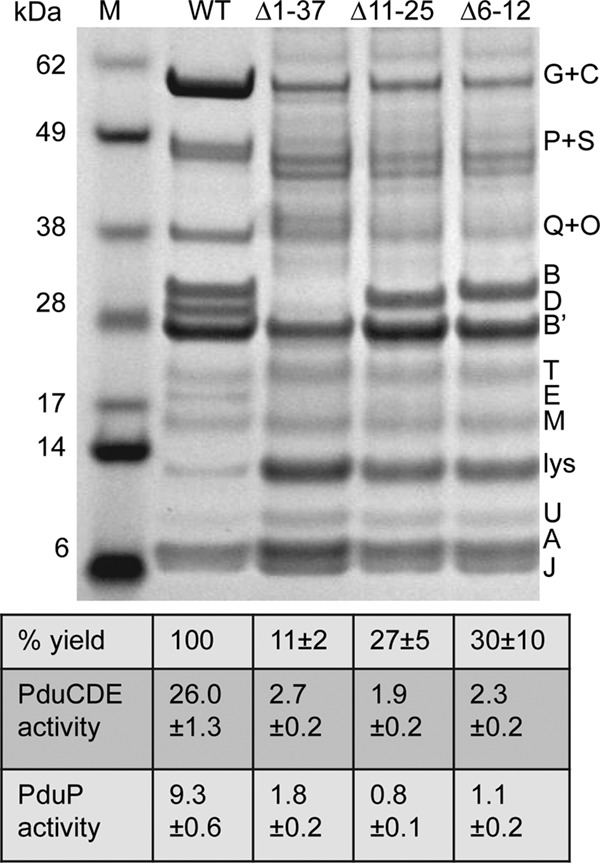
SDS-PAGE of MCPs purified from pduB deletion mutants with MCP assembly defects. WT, wild type. Letters on the right indicate the Pdu protein represented by each band. Lysozyme (lys) is not an MCP component but was used to lyse cells. The percent yield of purified MCPs indicates grams of protein/gram of cells relative to the wild type. The specific activities of the major lumen enzymes, the PduCDE diol dehydratase and the PduP aldehyde dehydrogenase, are shown in micromoles per minute per milligram. The error shown is one standard deviation determined from the results from three or more independent experiments. Ten micrograms of protein was loaded in each lane. Gels were stained using a Coomassie-based protocol. Enzyme assays are described in Materials and Methods.
Point mutations in the N-terminal α-helix of PduB.
To further define the role of the N terminus of PduB in MCP assembly, the highly conserved predicted α-helical region (residues L6 to V18) was examined. Thirteen scarless chromosomal mutations were constructed (L6T, V7T, E8A, Q9A, I10T, M11S, A12S, Q13A, V14T, I15T, R17A, and V18T), such that each amino acid side chain along the helix was changed from polar to nonpolar or vice versa. All mutations were verified by DNA sequencing. The effect of each mutation on MCP integrity was first examined using growth tests. As described above, fast growth of Salmonella on 1,2-PD at low B12 indicates a compromised MCP structure (49, 50). Strains having PduB L6T, A12S, and I15T mutations grew similarly to the wild type on 1,2-PD minimal medium with low B12. This suggested that these residues do not play a critical role in MCP assembly. Strains with PduB E8A, Q9A, Q13A, R17A, and V18T mutations grew slightly slower than the wild type on 1,2-PD medium, but the effect was small and not clearly significant. More interesting were strains with PduB V7T, I10T, M11S, and V14T mutations, which showed significantly faster growth than the wild type (Fig. 5). This suggests that V7, I10, M11, and V14 of PduB have important functional roles in assembly of the Pdu MCP.
FIG 5.
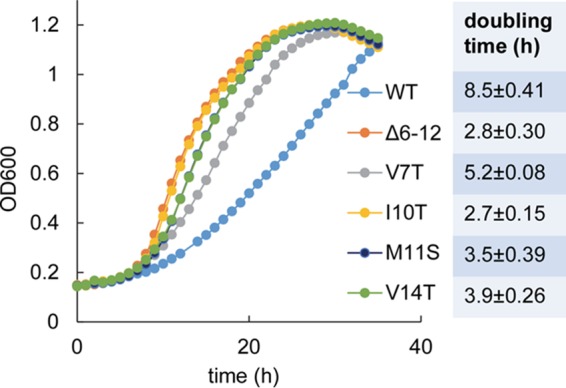
Individual point mutations along the highly conserved putative α-helix on the N terminus of PduB result in fast growth on 1,2-propanediol minimal medium. WT, wild type. The PduB Δ6–12 mutant is a control strain known to have a very high growth rate due to an MCP assembly defect. Four pduB mutants with site-directed mutations (V7T, I19T, M11S, and V14T) were also tested. The table to the right shows the doubling time for each strain, and the error shown is one standard deviation determined from the results from three or more independent experiments. Growth curves were determined with a microplate reader, as described in Materials and Methods.
To further investigate the effects of the PduB V7T, M11S, V14T, and I10T mutations on MCP assembly, MCPs were purified and analyzed from each mutant strain. Purification yields were 80%, 90%, 70%, and 25% relative to the wild type, respectively, indicating that each mutation reduced MCP stability, with I10T having the largest effect. When we examined the protein content of MCPs purified from each mutant by SDS-PAGE, the expected shell proteins (PduB, PduB′, PduA, PduJ, PduT, PduU, and PduM) were present in each case (Fig. 6). Notably, however, MCPs purified from the PduB I10T mutant had substantially reduced levels of lumen enzymes compared to the wild type, including PduCDE diol dehydratase and PduP aldehyde dehydrogenase (PduP), and the other mutant MCPs appeared to have somewhat reduced PduCDE levels (Fig. 6). To better quantify the lumen enzymes present, we measured the PduCDE and PduP enzyme activities in MCPs purified from each mutant (Fig. 6). PduCDE diol dehydratase activity was lower than the wild type in each mutant MCP, with the I10T mutant showing the biggest differential (∼18% activity compared to the wild type) (Fig. 6). The specific activity of PduP (the second most abundant lumen enzyme) was reduced in the I10T mutant MCPs (∼35% activity) but not in the other mutant MCPs assayed. Thus, analyses of purified MCPs supported the growth studies that indicated a role for PduB V7, I10, M11, and V14 in proper MCP assembly. They also tentatively suggested that these residues have particular importance in the association of the PduCDE diol dehydratase with the Pdu MCP.
FIG 6.
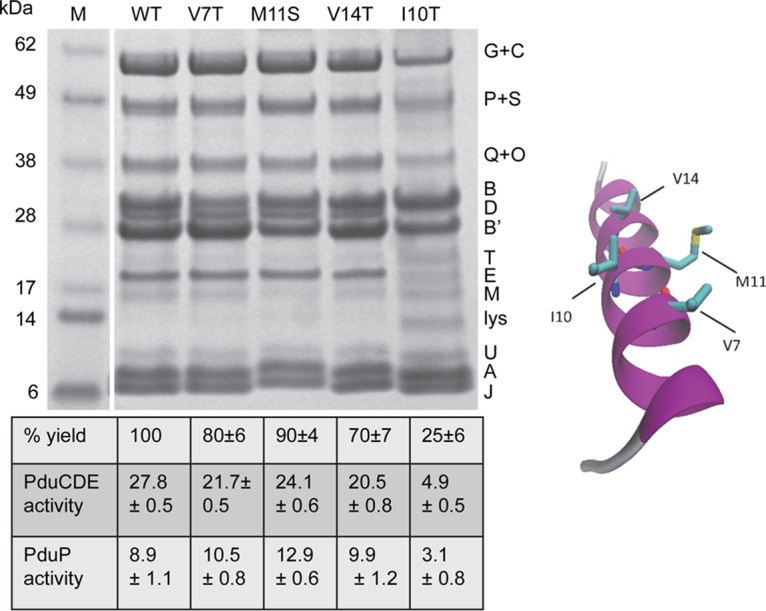
SDS-PAGE of MCPs purified from PduB mutants with site-directed mutations in the conserved N-terminal α-helix. WT, wild type. Letters on the right indicate the Pdu protein represented by each band. Lysozyme (lys) is not an MCP component but was used to lyse cells. The percent yield of purified MCPs indicates grams of protein/gram of cells relative to the wild type. The specific activities of the major lumen enzymes, the PduCDE diol dehydratase and the PduP aldehyde dehydrogenase, are given in micromoles per minute per milligram. The error shown is one standard deviation determined from the results from three or more independent experiments. Ten micrograms of protein was loaded in each lane. Gels were stained using a Coomassie-based protocol. Enzyme assays are described in Materials and Methods. On the right is a model that maps the residues of the PduB N terminus that were altered.
Transmission electron microscopy of purified MCPs from selected mutants.
Transmission electron microscopy (TEM) was used to evaluate the MCPs/partially assembled MCPs purified from selected mutants, including the PduB M38A, PduB Δ1–37, and PduB Δ6–12 mutants (Fig. 7). TEM showed that the PduB M38A mutant formed MCPs similar in appearance to the wild type, which supported the SDS-PAGE analyses and enzyme assays that indicated a normal protein content (Fig. 3). On the other hand, TEM of the MCP fraction purified from the PduB Δ1–37 mutant revealed primarily structures larger than wild-type MCPs, with an appearance similar to empty MCP shells described earlier (33). This was supportive of the SDS-PAGE results, which showed that the MCP fraction purified from the PduB Δ1–37 mutant consisted mainly of shell proteins and only a small amount lumen enzymes (Fig. 4). Similarly, TEM indicated that the PduB Δ6–12 mutant formed mainly large (216 ± 66 nm) empty MCP shells, which was also consistent with SDS-PAGE results (Fig. 4). For both deletion mutants (PduB Δ1–37 and Δ6–12), substantial proteinaceous debris was observed in the purified MCP fraction. Given that SDS-PAGE indicated that these fractions consisted mainly of shell proteins (Fig. 4), we consider it likely that the debris is shell protein aggregates. Hence, TEM results support the idea that the N-terminal region of PduB is needed for proper MCP assembly.
FIG 7.

Transmission electron microscopy images of MCPs and MCP subcomplexes isolated from selected pduB deletion mutants. The wild-type strain is Salmonella enterica serovar Typhimurium LT2. MCP/MCP subcomplexes were negatively stained, as described previously (32, 43). Arrowheads indicate MCPs or putative empty MCP shells.
DISCUSSION
Bacterial MCPs are extremely large multiprotein complexes that are widely used as metabolic organelles. A key question about MCP morphogenesis is the nature of the interactions that direct the assembly of thousands of protein subunits into a highly ordered functioning organelle. From a broad perspective, bacterial MCPs consist of a protein core composed of sequentially acting enzymes encapsulated within a selectively permeable protein shell. Their assembly requires interactions specific to the shell and the core, as well as associations that tether the shell to the core. In this report, we present several lines of evidence that indicate that the N-terminal region of PduB plays a crucial role in binding the shell of the Pdu MCP to its enzymatic core. Various mutations in the N-terminal region of PduB resulted in a growth phenotype indicative of impaired MCP assembly (fast growth on 1,2-PD at low B12). Purification of MCPs from several strains carrying mutations in the N terminus of PduB (Δ6–12, Δ11–25, and Δ1–37 mutants) led to the isolation of MCPs depleted in the core enzymes, as measured by SDS-PAGE and enzyme assays, but with the expected content of major shell proteins as assessed by SDS-PAGE. TEM provided further support, as MCPs purified from two different mutants revealed structures that appeared to be large empty MCP shells. Putting this in context, prior studies showed that deletion of any individual shell protein of the Pdu MCP (other than PduB) did not prevent shell-core interactions, since MCPs purified from these mutants had the expected complement of core enzymes and shell proteins (32, 50, 52, 59). Furthermore, we showed that an M38A mutation (which prevents translation of PduB′) also had relatively little effect on the association of the shell with the enzymatic core (Fig. 3). Thus, studies encompassing the roles of every Pdu shell protein in MCP assembly indicate that the N-terminal region of PduB mediates the major interaction that binds the shell of the Pdu MCP to its enzymatic core.
A theme in the assembly of bacterial MCPs is that short extensions on the N or C terminus of both structural proteins and core enzymes play a key role in determining higher-order structure (4). This idea developed from studies that showed the N-terminal 18 amino acids of the PduP core enzyme function as a targeting sequence that is necessary and sufficient for encapsulation of proteins within the Pdu MCP, as well as an extensive bioinformatic analysis that predicted that analogous short extensions are widely used in MCP assembly (35). Further work showed that short N-terminal extensions are used to target enzymes to diverse types of MCPs, both native and recombinant (27, 33, 34, 36, 39, 60–62). Mechanistic studies found that the N-terminal targeting sequences of PduP form an α-helix that binds to a short C-terminal helix of the PduA, PduJ, and/or PduK shell proteins to mediate encapsulation (38, 39). Similarly, the targeting sequences of the EutC and EutE enzymes of the ethanolamine MCP bind a corresponding helix on the EutS shell protein (although the helix is internal rather than terminal, due to circular permutation of the EutS shell protein) (62). Thus, it is generally accepted that binding interactions between enzyme-targeting sequences and shell proteins drive the encapsulation of enzymes into the lumen of diverse bacterial MCPs. However, the question of specificity with regard to the binding of the enzymatic core to the shell is unresolved and somewhat paradoxical. Studies indicate that a single N-terminal enzyme-targeting sequence can bind multiple shell proteins (38, 39) and vice versa, i.e., that a single shell protein can bind multiple core enzymes through targeting sequence associations (59, 62, 63). Recent findings have also shown that targeting sequences from diverse MCP systems (Eut and Grp) can direct the encapsulation of proteins into the Pdu MCP via a conserved hydrophobic motif (36). Thus, the seemingly low specificity observed for core-shell interactions raises the question of whether MCP cores and shells (each of which are composed of 4 to 10 different proteins) have defined relative molecular orientations. Above, we presented data indicating that the N-terminal region of PduB plays the primary role in binding the shell of the Pdu MCP to its enzymatic core. Hence, we propose a model for assembly of the Pdu MCP in which binding of the N-terminal region of PduB fixes the position of the shell relative to the core.
In this study, we also conducted a bioinformatic analysis of the N terminus of the PduB shell protein and found that it consists of a highly conserved putative α-helix and a less conserved coiled region. Prior studies indicated that the targeting sequences of core enzymes are short α-helices that bind to short α-helices in shell proteins (38, 62). Binding occurs along conserved surfaces and mediates enzyme encapsulation (36, 38, 60, 62). Site-directed mutagenesis of PduB α-helix at residues 6 to 18 indicated that a conserved hydrophobic patch formed by residues V7, I10, M11, and V14 was crucial to MCP assembly. This patch is reminiscent of surfaces present in other MCP assembly sequences found primarily at the termini of shell proteins and lumen enzymes. Presumably, this hydrophobic surface interacts with a core enzyme of the Pdu MCP, possibly a targeting sequence. However, further work will be needed to find the binding partner(s) of PduB.
A number of labs are developing bacterial MCPs as nanobioreactors for the production of renewable chemicals and pharmaceuticals (24, 26, 27, 64). Spatial organization of enzymes by encapsulation within protein shells (as exemplified in MCP systems) has the ability to accelerate catalysis, prevent side reactions, and minimize the harmful effects of toxic intermediates. A key advance in this field has been the production of empty MCP shells which were then filled with heterologous cargo by fusing targeting/encapsulation sequences to desired enzymes/proteins (33, 36, 60–62, 64–68). Further development of this approach for efficient encapsulation of multiple enzymes with defined stoichiometries will likely require the use of various types of targeting sequences, as well as parameterization of the molecular interactions that define encapsulation. Here, we have defined a new terminal sequence (the N-terminal region of the PduB shell protein) that mediates a major interaction between the enzymatic core and the shell of the Pdu MCP. This sequence might prove useful for developing additional systems that mediate protein encapsulation into engineered MCPs.
The findings reported here also relate to two views of MCP biogenesis that have been proposed. Studies of the α-carboxysome suggest that the shell and the core assemble more or less simultaneously (69), while studies of the β-carboxysome suggest that the enzymatic core assembles first and then is rapidly enveloped by the shell (although the factors that delay association of the shell and core have not been determined) (70, 71). Given the key role of the N terminus of PduB in fastening the shell of the Pdu MCP to its enzymatic core, it is likely to play a prominent role in biogenesis, perhaps even making the first point of contact. We also observed that the essentially empty MCP shells produced by certain mutants were much larger than wild-type MCPs. This suggests that interactions between the shell and the core may help to impart curvature to the shell and define MCP size, but certainly this is not the only factor involved, and indeed, prior studies have pointed out the key role of vertex proteins in guiding shell curving (72, 73). Hence, the findings presented here may help with future studies on the dynamics and mechanisms of MCP assembly.
MATERIALS AND METHODS
Chemicals and reagents.
Antibiotics, coenzyme B12 (Ado-B12), vitamin B12 (CN-B12), NADH, and NAD+ were from Sigma Chemical Company (St. Louis, MO). Choice Taq Blue master mix was from Denville Scientific (Holliston, MA). KOD Hot Start master mix was from EMD Millipore (Billerica, MA). Bacterial protein extraction reagent (B-PERII), NuPAGE Bis-Tris gels, SimplyBlue SafeStain, and other reagents were from Thermo Fisher Scientific (Pittsburgh, PA).
Bacterial strains and growth conditions.
The bacterial strains used in this study are listed in Table 1. The rich media used were lysogeny broth (LB), also known as Luria-Bertani/Lennox medium (Becton, Dickinson and Company, Franklin Lakes, NJ) (74), and Terrific broth (MP Biomedicals, Solon, OH). The minimal medium was no-carbon-E (NCE) medium (75). Growth curves were determined using a Synergy HT microplate reader (BioTek, Winooski, VT), as previously described (32). NCE minimal medium was supplemented with 0.6% 1,2-PD, 0.3 mM each Val, Ile, Leu, Thr, Phe, Met, and Cys, 0.1 mM each Tyr and Trp, 20 μM calcium pantothenate, 50 μM iron(III) citrate, and CN-B12, as indicated in Results.
TABLE 1.
Strains used in this study
| Straina | Description | Source |
|---|---|---|
| BE293 | Carries pKD46 | Lab collection |
| SS23 | ΔpduBB′ | Lab collection |
| BL55 | ΔpduB (P27–P32)::pblaP3-sacB-cat/pKD46 | This study |
| BL56 | ΔpduB (P27–P32) | This study |
| BL62 | ΔpduB (M1–A37)::pblaP3-sacB-cat/pKD46 | This study |
| BL137 | pduB M38A | This study |
| BL144 | ΔpduB (M11–A25) | This study |
| BL155 | ΔpduB (L6–A12)::pblaP3-sacB-cat/pkD46 | This study |
| BL156 | ΔpduB (L6–A12) | This study |
| BL157 | pduB (L6T) | This study |
| BL158 | pduB (E8A) | This study |
| BL159 | pduB (L10T) | This study |
| BL160 | pduB (A12S) | This study |
| BL179 | pduB (V7T) | This study |
| BL180 | pduB (M11S) | This study |
| BL181 | pduB (V14T) | This study |
| BL182 | pduB (I15T) | This study |
| BL183 | pduB (Q9A) | This study |
| BL184 | pduB (Q13A) | This study |
| BL232 | ΔpduB (M1–A37) (RBS = AGGA for pduB′) | This study |
| BL234 | pduB (R17A) | This study |
| BL235 | pduB (V18T) | This study |
All strains are derivatives of S. enterica serovar Typhimurium LT2 which contain scarless mutations that change the amino acid/nucleotide sequence as indicated. RBS, ribosome binding site.
Construction of chromosomal mutations.
Scarless chromosomal deletions and point mutations were made by Sac-Cat recombineering using the lambda Red recombinase, as described previously (32, 76). The oligonucleotides used for recombineering are listed in Table S1. All mutations were verified by DNA sequencing.
MCP purification and electron microscopy.
MCP purification was performed by detergent extraction and differential centrifugation, as described previously (52), with the modification that growth medium was supplemented with 50 μM Fe(III) citrate. The amount of purified MCPs obtained (in grams of protein) per gram of cells was used to calculate the percent yield. For electron microscopy, purified MCPs were fixed to carbon-coated copper grids and negatively stained, as described previously (32, 43). Grids were viewed under a JEOL 2100 scanning transmission electron microscope (JEOL USA, Inc., Peabody, MA).
Enzyme assays and protein methods.
Coenzyme B12-dependent diol dehydratase (PduCDE) activities were determined by a linked continuous spectrophotometric assay, as described previously (59). PduP aldehyde dehydrogenase activities were measured by following the conversion of NAD+ to NADH spectrophotometrically at 340 nm, as described previously (44). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was carried out using NuPAGE 4 to 12% Bis-Tris gels, Novex mini cells (Invitrogen), and SimplyBlue SafeStain (Thermo Fisher Scientific). Protein was measured using Bio-Rad protein assay reagent, which is based on the method of Bradford, with bovine serum albumin as the standard.
ACKNOWLEDGMENTS
This work was supported by grant AI081146 from the National Institutes of Health to T.A.B.
We thank the ISU DNA Sequencing and Synthesis Facility for assistance with DNA analyses, the ISU Microscopy and Nanoimaging Facility for help with electron microscopy, and members of the Bobik laboratory, especially Sharmistha Sinha, for helpful discussions.
REFERENCES
- 1.Abdul-Rahman F, Petit E, Blanchard JL. 2013. The distribution of polyhedral bacterial microcompartments suggests frequent horizontal transfer and operon reassembly. J Phylogen Evol Biol 1:118. doi: 10.4172/2329-9002.1000118. [DOI] [Google Scholar]
- 2.Axen SD, Erbilgin O, Kerfeld CA. 2014. A taxonomy of bacterial microcompartment loci constructed by a novel scoring method. PLoS Comput Biol 10:e1003898. doi: 10.1371/journal.pcbi.1003898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bobik TA. 2006. Polyhedral organelles compartmenting bacterial metabolic processes. Appl Microbiol Biotechnol 70:517–525. doi: 10.1007/s00253-005-0295-0. [DOI] [PubMed] [Google Scholar]
- 4.Bobik TA, Lehman BP, Yeates TO. 2015. Bacterial microcompartments: widespread prokaryotic organelles for isolation and optimization of metabolic pathways. Mol Microbiol 98:193–207. doi: 10.1111/mmi.13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chowdhury C, Sinha S, Chun S, Yeates TO, Bobik TA. 2014. Diverse bacterial microcompartment organelles. Microbiol Mol Biol Rev 78:438–468. doi: 10.1128/MMBR.00009-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jorda J, Lopez D, Wheatley NM, Yeates TO. 2013. Using comparative genomics to uncover new kinds of protein-based metabolic organelles in bacteria. Protein Sci 22:179–195. doi: 10.1002/pro.2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rae BD, Long BM, Badger MR, Price GD. 2013. Functions, compositions, and evolution of the two types of carboxysomes: polyhedral microcompartments that facilitate CO2 fixation in cyanobacteria and some proteobacteria. Microbiol Mol Biol Rev 77:357–379. doi: 10.1128/MMBR.00061-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zarzycki J, Erbilgin O, Kerfeld CA. 2015. Bioinformatic characterization of glycyl radical enzyme-associated bacterial microcompartments. Appl Environ Microbiol 81:8315–8329. doi: 10.1128/AEM.02587-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kerfeld CA, Heinhorst S, Cannon GC. 2010. Bacterial microcompartments. Annu Rev Microbiol 64:391–408. doi: 10.1146/annurev.micro.112408.134211. [DOI] [PubMed] [Google Scholar]
- 10.Dou Z, Heinhorst S, Williams EB, Murin CD, Shively JM, Cannon GC. 2008. CO2 fixation kinetics of Halothiobacillus neapolitanus mutant carboxysomes lacking carbonic anhydrase suggest the shell acts as a diffusional barrier for CO2. J Biol Chem 283:10377–10384. doi: 10.1074/jbc.M709285200. [DOI] [PubMed] [Google Scholar]
- 11.Penrod JT, Roth JR. 2006. Conserving a volatile metabolite: a role for carboxysome-like organelles in Salmonella enterica. J Bacteriol 188:2865–2874. doi: 10.1128/JB.188.8.2865-2874.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rondon MR, Horswill AR, Escalante-Semerena JC. 1995. DNA polymerase I function is required for the utilization of ethanolamine, 1,2-propanediol, and propionate by Salmonella Typhimurium LT2. J Bacteriol 177:7119–7124. doi: 10.1128/jb.177.24.7119-7124.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rondon MR, Kazmierczak R, Escalante-Semerena JC. 1995. Glutathione is required for maximal transcription of the cobalamin biosynthetic and 1,2-propanediol utilization (cob/pdu) regulon and for the catabolism of ethanolamine, 1,2-propanediol, and propionate in Salmonella Typhimurium LT2. J Bacteriol 177:5434–5439. doi: 10.1128/jb.177.19.5434-5439.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sampson EM, Bobik TA. 2008. Microcompartments for B12-dependent 1,2-propanediol degradation provide protection from DNA and cellular damage by a reactive metabolic intermediate. J Bacteriol 190:2966–2971. doi: 10.1128/JB.01925-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Price GD, Badger MR. 1989. Isolation and characterization of high CO2-requiring-mutants of the cyanobacterium Synechococcus PCC7942: two phenotypes that accumulate inorganic carbon but are apparently unable to generate CO2 within the carboxysome. Plant Physiol 91:514–525. doi: 10.1104/pp.91.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shively JM, Ball F, Brown DH, Saunders RE. 1973. Functional organelles in prokaryotes: polyhedral inclusions (carboxysomes) of Thiobacillus neapolitanus. Science 182:584–586. doi: 10.1126/science.182.4112.584. [DOI] [PubMed] [Google Scholar]
- 17.Craciun S, Balskus EP. 2012. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc Natl Acad Sci U S A 109:21307–21312. doi: 10.1073/pnas.1215689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erbilgin O, McDonald KL, Kerfeld CA. 2014. Characterization of a planctomycetal organelle: a novel bacterial microcompartment for the aerobic degradation of plant saccharides. Appl Environ Microbiol 80:2193–2205. doi: 10.1128/AEM.03887-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jameson E, Fu T, Brown IR, Paszkiewicz K, Purdy KJ, Frank S, Chen Y. 2016. Anaerobic choline metabolism in microcompartments promotes growth and swarming of Proteus mirabilis. Environ Microbiol 18:2886–2898. doi: 10.1111/1462-2920.13059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martínez-del Campo A, Bodea S, Hamer HA, Marks JA, Haiser HJ, Turnbaugh PJ, Balskus EP. 2015. Characterization and detection of a widely distributed gene cluster that predicts anaerobic choline utilization by human gut bacteria. mBio 6(2):e00042-15. doi: 10.1128/mBio.00042-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sriramulu DD, Liang M, Hernandez-Romero D, Raux-Deery E, Lunsdorf H, Parsons JB, Warren MJ, Prentice MB. 2008. Lactobacillus reuteri DSM 20016 produces cobalamin-dependent diol dehydratase in metabolosomes and metabolizes 1,2-propanediol by disproportionation. J Bacteriol 190:4559–4567. doi: 10.1128/JB.01535-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Talarico TL, Casas IA, Chung TC, Dobrogosz WJ. 1988. Production and isolation of reuterin, a growth inhibitor produced by Lactobacillus reuteri. Antimicrob Agents Chemother 32:1854–1858. doi: 10.1128/AAC.32.12.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Conner CP, Heithoff DM, Julio SM, Sinsheimer RL, Mahan MJ. 1998. Differential patterns of acquired virulence genes distinguish Salmonella strains. Proc Natl Acad Sci U S A 95:4641–4645. doi: 10.1073/pnas.95.8.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frank S, Lawrence AD, Prentice MB, Warren MJ. 2013. Bacterial microcompartments moving into a synthetic biological world. J Biotechnol 163:273–279. doi: 10.1016/j.jbiotec.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 25.Held M, Quin MB, Schmidt-Dannert C. 2013. Eut bacterial microcompartments: insights into their function, structure, and bioengineering applications. J Mol Microbiol Biotechnol 23:308–320. doi: 10.1159/000351343. [DOI] [PubMed] [Google Scholar]
- 26.Kerfeld CA, Erbilgin O. 2015. Bacterial microcompartments and the modular construction of microbial metabolism. Trends Microbiol 23:22–34. doi: 10.1016/j.tim.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 27.Kim EY, Tullman-Ercek D. 2013. Engineering nanoscale protein compartments for synthetic organelles. Curr Opin Biotechnol 24:627–632. doi: 10.1016/j.copbio.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 28.Thiennimitr P, Winter SE, Winter MG, Xavier MN, Tolstikov V, Huseby DL, Sterzenbach T, Tsolis RM, Roth JR, Baumler AJ. 2011. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc Natl Acad Sci U S A 108:17480–17485. doi: 10.1073/pnas.1107857108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsai SJ, Yeates TO. 2011. Bacterial microcompartments insights into the structure, mechanism, and engineering applications. Prog Mol Biol Transl Sci 103:1–20. doi: 10.1016/B978-0-12-415906-8.00008-X. [DOI] [PubMed] [Google Scholar]
- 30.Kerfeld CA, Sawaya MR, Tanaka S, Nguyen CV, Phillips M, Beeby M, Yeates TO. 2005. Protein structures forming the shell of primitive bacterial organelles. Science 309:936–938. doi: 10.1126/science.1113397. [DOI] [PubMed] [Google Scholar]
- 31.Yeates TO, Jorda J, Bobik TA. 2013. The shells of BMC-type microcompartment organelles in bacteria. J Mol Microbiol Biotechnol 23:290–299. doi: 10.1159/000351347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sinha S, Cheng S, Sung YW, McNamara DE, Sawaya MR, Yeates TO, Bobik TA. 2014. Alanine scanning mutagenesis identifies an asparagine-arginine-lysine triad essential to assembly of the shell of the Pdu microcompartment. J Mol Biol 426:2328–2345. doi: 10.1016/j.jmb.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choudhary S, Quin MB, Sanders MA, Johnson ET, Schmidt-Dannert C. 2012. Engineered protein nano-compartments for targeted enzyme localization. PLoS One 7:e33342. doi: 10.1371/journal.pone.0033342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fan C, Bobik TA. 2011. The N-terminal region of the medium subunit (PduD) packages adenosylcobalamin-dependent diol dehydratase (PduCDE) into the Pdu microcompartment. J Bacteriol 193:5623–5628. doi: 10.1128/JB.05661-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fan C, Cheng S, Liu Y, Escobar CM, Crowley CS, Jefferson RE, Yeates TO, Bobik TA. 2010. Short N-terminal sequences package proteins into bacterial microcompartments. Proc Natl Acad Sci U S A 107:7509–7514. doi: 10.1073/pnas.0913199107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jakobson CM, Kim EY, Slininger MF, Chien A, Tullman-Ercek D. 2015. Localization of proteins to the 1,2-propanediol utilization microcompartment by non-native signal sequences is mediated by a common hydrophobic motif. J Biol Chem 290:24519–24533. doi: 10.1074/jbc.M115.651919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kinney JN, Salmeen A, Cai F, Kerfeld CA. 2012. Elucidating essential role of conserved carboxysomal protein CcmN reveals common feature of bacterial microcompartment assembly. J Biol Chem 287:17729–17736. doi: 10.1074/jbc.M112.355305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fan C, Cheng S, Sinha S, Bobik TA. 2012. Interactions between the termini of lumen enzymes and shell proteins mediate enzyme encapsulation into bacterial microcompartments. Proc Natl Acad Sci U S A 109:14995–15000. doi: 10.1073/pnas.1207516109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lawrence AD, Frank S, Newnham S, Lee MJ, Brown IR, Xue WF, Rowe ML, Mulvihill DP, Prentice MB, Howard MJ, Warren MJ. 2014. Solution structure of a bacterial microcompartment targeting peptide and its application in the construction of an ethanol bioreactor. ACS Synth Biol 3:454–465. doi: 10.1021/sb4001118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bobik TA, Havemann GD, Busch RJ, Williams DS, Aldrich HC. 1999. The propanediol utilization (pdu) operon of Salmonella enterica serovar Typhimurium LT2 includes genes necessary for formation of polyhedral organelles involved in coenzyme B12-dependent 1,2-propanediol degradation. J Bacteriol 181:5967–5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Obradors N, Badia J, Baldoma L, Aguilar J. 1988. Anaerobic metabolism of the l-rhamnose fermentation product 1,2-propanediol in Salmonella Typhimurium. J Bacteriol 170:2159–2162. doi: 10.1128/jb.170.5.2159-2162.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bobik TA, Xu Y, Jeter RM, Otto KE, Roth JR. 1997. Propanediol utilization genes (pdu) of Salmonella Typhimurium: three genes for the propanediol dehydratase. J Bacteriol 179:6633–6639. doi: 10.1128/jb.179.21.6633-6639.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng S, Fan C, Sinha S, Bobik TA. 2012. The PduQ enzyme is an alcohol dehydrogenase used to recycle NAD+ internally within the Pdu microcompartment of Salmonella enterica. PLoS One 7:e47144. doi: 10.1371/journal.pone.0047144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leal NA, Havemann GD, Bobik TA. 2003. PduP is a coenzyme-A-acylating propionaldehyde dehydrogenase associated with the polyhedral bodies involved in B12-dependent 1,2-propanediol degradation by Salmonella enterica serovar Typhimurium LT2. Arch Microbiol 180:353–361. doi: 10.1007/s00203-003-0601-0. [DOI] [PubMed] [Google Scholar]
- 45.Horswill AR, Escalante-Semerena JC. 1999. Salmonella Typhimurium LT2 catabolizes propionate via the 2-methylcitric acid cycle. J Bacteriol 181:5615–5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Y, Jorda J, Yeates TO, Bobik TA. 2015. The PduL phosphotransacylase is used to recycle coenzyme A within the Pdu microcompartment. J Bacteriol 197:2392–2399. doi: 10.1128/JB.00056-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y, Leal NA, Sampson EM, Johnson CL, Havemann GD, Bobik TA. 2007. PduL is an evolutionarily distinct phosphotransacylase involved in B12-dependent 1,2-propanediol degradation by Salmonella enterica serovar Typhimurium LT2. J Bacteriol 189:1589–1596. doi: 10.1128/JB.01151-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Palacios S, Starai VJ, Escalante-Semerena JC. 2003. Propionyl coenzyme A is a common intermediate in the 1,2-propanediol and propionate catabolic pathways needed for expression of the prpBCDE operon during growth of Salmonella enterica on 1,2-propanediol. J Bacteriol 185:2802–2810. doi: 10.1128/JB.185.9.2802-2810.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Havemann GD, Sampson EM, Bobik TA. 2002. PduA is a shell protein of polyhedral organelles involved in coenzyme B12-dependent degradation of 1,2-propanediol in Salmonella enterica serovar Typhimurium LT2. J Bacteriol 184:1253–1261. doi: 10.1128/JB.184.5.1253-1261.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng S, Sinha S, Fan C, Liu Y, Bobik TA. 2011. Genetic analysis of the protein shell of the microcompartments involved in coenzyme B12-dependent 1,2-propanediol degradation by Salmonella. J Bacteriol 193:1385–1392. doi: 10.1128/JB.01473-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Havemann GD, Bobik TA. 2003. Protein content of polyhedral organelles involved in coenzyme B12-dependent degradation of 1,2-propanediol in Salmonella enterica serovar Typhimurium LT2. J Bacteriol 185:5086–5095. doi: 10.1128/JB.185.17.5086-5095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sinha S, Cheng S, Fan C, Bobik TA. 2012. The PduM protein is a structural component of the microcompartments involved in coenzyme B12-dependent 1,2-propanediol degradation by Salmonella enterica. J Bacteriol 194:1912–1918. doi: 10.1128/JB.06529-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parsons JB, Dinesh SD, Deery E, Leech HK, Brindley AA, Heldt D, Frank S, Smales CM, Lunsdorf H, Rambach A, Gass MH, Bleloch A, McClean KJ, Munro AW, Rigby SE, Warren MJ, Prentice MB. 2008. Biochemical and structural insights into bacterial organelle form and biogenesis. J Biol Chem 283:14366–14375. doi: 10.1074/jbc.M709214200. [DOI] [PubMed] [Google Scholar]
- 54.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. 2013. Analysis tool Web Services from the EMBL-EBI. Nucleic Acids Res 41:W597–W600. doi: 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maupetit J, Derreumaux P, Tuffery P. 2009. PEP-FOLD: an online resource for de novo peptide structure prediction. Nucleic Acids Res 37:W498–W503. doi: 10.1093/nar/gkp323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pang A, Liang M, Prentice MB, Pickersgill RW. 2012. Substrate channels revealed in the trimeric Lactobacillus reuteri bacterial microcompartment shell protein PduB. Acta Crystallogr D Biol Crystallogr 68:1642–1652. doi: 10.1107/S0907444912039315. [DOI] [PubMed] [Google Scholar]
- 58.Chowdhury C, Chun S, Pang A, Sawaya MR, Sinha S, Yeates TO, Bobik TA. 2015. Selective molecular transport through the protein shell of a bacterial microcompartment organelle. Proc Natl Acad Sci U S A 112:2990–2995. doi: 10.1073/pnas.1423672112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chowdhury C, Chun S, Sawaya MR, Yeates TO, Bobik TA. 2016. The function of the PduJ microcompartment shell protein is determined by the genomic position of its encoding gene. Mol Microbiol 101:770–783. doi: 10.1111/mmi.13423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim EY, Tullman-Ercek D. 2014. A rapid flow cytometry assay for the relative quantification of protein encapsulation into bacterial microcompartments. Biotechnol J 9:348–354. doi: 10.1002/biot.201300391. [DOI] [PubMed] [Google Scholar]
- 61.Lee MJ, Brown IR, Juodeikis R, Frank S, Warren MJ. 2016. Employing bacterial microcompartment technology to engineer a shell-free enzyme-aggregate for enhanced 1,2-propanediol production in Escherichia coli. Metab Eng 36:48–56. doi: 10.1016/j.ymben.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Quin MB, Perdue SA, Hsu SY, Schmidt-Dannert C. 2016. Encapsulation of multiple cargo proteins within recombinant Eut nanocompartments. Appl Microbiol Biotechnol 100:9187–9200. doi: 10.1007/s00253-016-7737-8. [DOI] [PubMed] [Google Scholar]
- 63.Jorda J, Liu Y, Bobik TA, Yeates TO. 2015. Exploring bacterial organelle interactomes: a model of the protein-protein interaction network in the Pdu microcompartment. PLoS Comput Biol 11:e1004067. doi: 10.1371/journal.pcbi.1004067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bonacci W, Teng PK, Afonso B, Niederholtmeyer H, Grob P, Silver PA, Savage DF. 2012. Modularity of a carbon-fixing protein organelle. Proc Natl Acad Sci U S A 109:478–483. doi: 10.1073/pnas.1108557109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Held M, Kolb A, Perdue S, Hsu SY, Bloch SE, Quin MB, Schmidt-Dannert C. 2016. Engineering formation of multiple recombinant Eut protein nanocompartments in E. coli. Sci Rep 6:24359. doi: 10.1038/srep24359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jakobson CM, Chen Y, Slininger MF, Valdivia E, Kim EY, Tullman-Ercek D. 2016. Tuning the catalytic activity of subcellular nanoreactors. J Mol Biol 428:2989–2996. doi: 10.1016/j.jmb.2016.07.006. [DOI] [PubMed] [Google Scholar]
- 67.Menon BB, Dou Z, Heinhorst S, Shively JM, Cannon GC. 2008. Halothiobacillus neapolitanus carboxysomes sequester heterologous and chimeric RubisCO species. PLoS One 3:e3570. doi: 10.1371/journal.pone.0003570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Parsons JB, Frank S, Bhella D, Liang M, Prentice MB, Mulvihill DP, Warren MJ. 2010. Synthesis of empty bacterial microcompartments, directed organelle protein incorporation, and evidence of filament-associated organelle movement. Mol Cell 38:305–315. doi: 10.1016/j.molcel.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 69.Iancu CV, Morris DM, Dou Z, Heinhorst S, Cannon GC, Jensen GJ. 2010. Organization, structure, and assembly of alpha-carboxysomes determined by electron cryotomography of intact cells. J Mol Biol 396:105–117. doi: 10.1016/j.jmb.2009.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cameron JC, Wilson SC, Bernstein SL, Kerfeld CA. 2013. Biogenesis of a bacterial organelle: the carboxysome assembly pathway. Cell 155:1131–1140. doi: 10.1016/j.cell.2013.10.044. [DOI] [PubMed] [Google Scholar]
- 71.Chen AH, Robinson-Mosher A, Savage DF, Silver PA, Polka JK. 2013. The bacterial carbon-fixing organelle is formed by shell envelopment of preassembled cargo. PLoS One 8:e76127. doi: 10.1371/journal.pone.0076127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tanaka S, Kerfeld CA, Sawaya MR, Cai F, Heinhorst S, Cannon GC, Yeates TO. 2008. Atomic-level models of the bacterial carboxysome shell. Science 319:1083–1086. doi: 10.1126/science.1151458. [DOI] [PubMed] [Google Scholar]
- 73.Wheatley NM, Gidaniyan SD, Liu Y, Cascio D, Yeates TO. 2013. Bacterial microcompartment shells of diverse functional types possess pentameric vertex proteins. Protein Sci 22:660–665. doi: 10.1002/pro.2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bertani G. 1951. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol 62:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Berkowitz D, Hushon JM, Whitfield HJ Jr, Roth J, Ames BN. 1968. Procedure for identifying nonsense mutations. J Bacteriol 96:215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]