

Abstract
Residues E402 and R404 of the E. coli serine chemoreceptor, Tsr, appear to form a salt bridge that spans the interfaces between neighboring dimers in the Tsr trimer of dimers, a key structural component of receptor core signaling complexes. To assess their functional roles we constructed full sets of single amino acid replacement mutants at E402 and R404 and characterized their signaling behaviors with a suite of in vivo assays. Our results indicate that the E402 and R404 residues of Tsr play their most critical signaling roles at their inner locations near the trimer axis where they likely participate in stabilizing trimer-of-dimer packing and the kinase-ON state of core signaling complexes. Mutant receptors with a variety of sidechain replacements still accessed both the ON and OFF signaling states, suggesting that core signaling complexes produce kinase activity over a range of receptor conformations and dynamic motions. Similarly, the kinase-OFF state may not be a discrete conformation, but rather a range of structures outside the range of those suitable for kinase activation. Consistent with this idea, some structural lesions at both E402 and R404 produced signaling behaviors that are not compatible with discrete two-state models of core complex signaling states. Those lesions might stabilize intermediate receptor conformations along the OFF-ON energy landscape. Amino acid replacements produced different constellations of signaling defects at each residue, indicating that they play distinct structure-function roles. R404, but not E402, was critical for high signal cooperativity in the receptor array.
Keywords: chemotaxis, signal transduction, kinase control, salt bridge, dynamic motions
Graphical abstract
INTRODUCTION
Bacteria sense and respond to their environment via transmembrane receptors. Sensitive detection and adaptive response to environmental stimuli are critical to bacterial survival and proliferation. Many motile bacteria use chemoreceptors known as methyl-accepting chemotaxis proteins (MCPs) to modulate their locomotor responses to environmental chemicals (see [1, 2] for reviews). Escherichia coli’s MCPs have been extensively studied and are good experimental models for elucidating molecular mechanisms of transmembrane and intracellular signaling.
E. coli has four canonical MCPs: Tar, Tap, Trg, and Tsr, which have similar functional architecture and signal transducing mechanisms [1, 2]. These receptor proteins detect various small molecules through a periplasmic sensing domain; their cytoplasmic domains modulate the activity of an associated histidine autokinase CheA, which is coupled to receptor control by a third protein, CheW (Fig. 1a). Following autophosphorylation, CheA donates the phosphoryl group to the response regulator CheY. Phosphorylated CheY (P-CheY) molecules bind at the bases of flagellar motors, raising their probability of clockwise (CW) rotation [3, 4]. Counter-clockwise (CCW) motor rotation produces forward swimming movements, whereas CW reversals cause tumbling episodes that randomly reorient the cell’s swimming direction. Cellular P-CheY is shortlived, owing to a specific phosphatase, CheZ [5, 6].
Fig. 1. Structural features of the Tsr trimer-of-dimers tip.
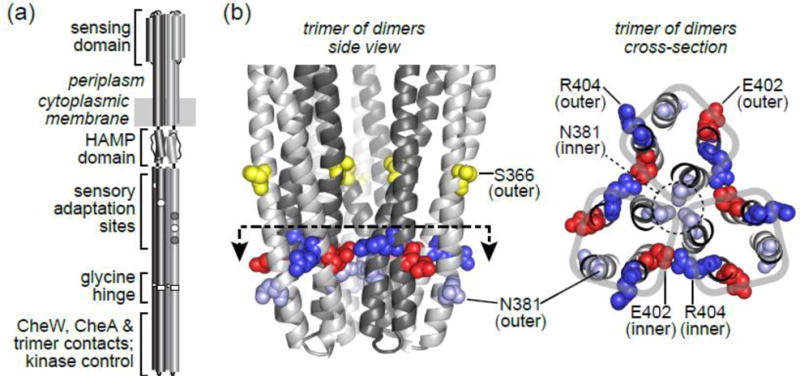
(a) Functional architecture of the Tsr homodimer. One subunit is shaded light gray, the other is dark gray. White circles represent adaptation sites with E residues; gray circles represent sites with Q residues (see text).
(b) The trimer-of-dimers tip. The dashed line with arrows in the side view indicates the plane and viewing direction of the cross-section image shown at right. The subunits of each dimer (light gray and dark gray) form a 4-helix bundle: N and N’ helices before the hairpin turns, C and C’ helices following the hairpin turns. The N helix (in the dark gray subunit) of each dimer lies at the trimer axis; the N, C, and C’ helices of each dimer lie at the trimer periphery. The N381 residues at the trimer axis interact to stabilize the trimer interface [11, 18]. Other trimer contact residues are not shown. The S366 residues in the N helices adopt a trigonal geometry just above the trimer interface; cysteine replacements at this position provide crosslinking reporter sites for the trifunctional thiol-reactive TMEA reagent [19] (see text). Residues E402 and R404 on the C and C’ helices occupy inner and outer locations with respect to the trimer axis. They may form a salt-bridge interaction between adjacent dimers at their inner location. Their counterparts in the outer helices are solvent-exposed at the trimer periphery.
Chemoreceptor/CheW/CheA signaling complexes generally conform to two-state behavior, having an “on” state with high kinase activity (ON) and an “off” state with very low kinase activity (OFF). Chemical stimuli elicit chemotactic responses by shifting MCP signaling complexes toward the ON or OFF state. Subsequent covalent modifications to the receptor signaling domain, catalyzed by a glutamyl methyl-transferase (CheR) and a deamidase and methyl-esterase (CheB), reverse the shift in ON/OFF equilibrium to terminate stimulus responses. This sensory adaptation system enables the swimming cell to detect and respond to temporal changes in chemoeffector levels over a wide concentration range.
Native MCP molecules are homodimers; protomers of Tsr, the serine chemoreceptor and subject of the current study, are 551 residues in length. Tsr subunits contain five sites for adaptational modifications, initially translated as glutamyl (E304, E392, E502) and glutaminyl (Q297, Q311) residues (Fig. 1a). CheB converts the Q sites to E sites through irreversible deamidation reactions; CheR converts E sites to glutamyl methyl-esters (Em) which can be reversibly hydrolyzed back to E sites by CheB. Q and Em sites shift receptor signaling complexes toward the ON state; E sites shift output toward the OFF state. Receptor modification state is under negative feedback control: Signaling complexes in the ON state are substrates for CheB action; complexes in the OFF state are substrates for CheR action [2, 7, 8]. Stimulusinduced shifts in the ON-OFF equilibrium trigger changes in the relative activities of CheB and CheR that drive the system back to its pre-stimulus set-point.
E. coli chemoreceptors operate in networked arrays that have highly cooperative signaling properties. The basic unit of receptor function, the core complex, contains six MCP molecules, organized as two trimers of dimers, two CheW molecules and one CheA homodimer [9]. The cytoplasmic domains of receptor molecules comprise antiparallel four-helix bundles with contact sites at their tips for trimer and core complex assembly (Fig. 1a). Receptor trimers-of-dimers and CheA/CheW binding interactions are critical for array formation [10] and kinase control [11–13]. Although the architecture of receptor arrays has been elucidated at the molecular level [14–16], the mechanism(s) of CheA control are still unclear, largely due to the many protein-protein interaction surfaces within the signaling arrays, which confound analysis of any single interaction. For example, residues at the receptor hairpin tip could play multiple roles by promoting interactions with several different partner proteins. Such structure-function complexity presents a difficult challenge to experimental identification of the component interactions and their signaling roles.
Residues E402 and R404 in the hairpin tip of Tsr are highly conserved across the MCP superfamily [17], implying important functional role(s). However, they occupy two different structural environments in the Tsr trimer of dimers (Fig. 1B&C). In each dimer, near the trimer axis, an inner E402 residue packs against an inner R404 residue from a neighboring dimer at the interdimer interface [18] (Fig. 1b&c). At the periphery of the trimer, the outer E402 and R404 residues are solvent exposed and their sidechains lie apart from one another (Fig. 1). The close proximity of two nearly invariant, oppositely-charged residues at inter-dimer interfaces of the trimer suggests that E402 and R404 sidechains might participate in an ionic interaction that contributes to trimer structure or stability. But could these residues also play important roles at the trimer periphery, for example, as binding determinants for CheA or CheW?
To investigate whether the E402 and/or R404 sidechains are critical for Tsr function, whether they interact structurally, and whether that interaction is functionally important, we constructed and characterized a complete set of single amino acid replacement mutants at each residue. Each mutant receptor was subjected to a panel of in vivo tests to assess: chemotaxis performance; trimer, core complex and array formation; kinase activation and stimulus control; and sensory adaptation. These studies revealed that E402 and R404 play functionally critical roles only at the inter-dimer interface of receptor trimers, not at the trimer periphery. The E402 and R404 sidechains may interact, for example, in a salt bridge, to promote trimer stability, but amino acid replacements that disrupt that interaction produce different signaling defects at the two positions, attesting to non-equivalent structural roles. These findings cast new light on the nature of CheA signaling states and their mechanism of control in chemoreceptor core complexes and arrays.
RESULTS
Mutational survey of Tsr residues E402 and R404
We constructed a complete set of amino acid replacement mutants at Tsr residues E402 and R404 in tsr expression plasmid pCS53 (see Methods). This plasmid encodes Tsr molecules with a cysteine replacement at residue S366, a crosslinking reporter site for trimer-of-dimer formation [19]. In receptor-less host strain UU2612, which contains both sensory adaptation enzymes, CheR and CheB (hereafter designated R+B+), pCS53-expressed Tsr-S366C promotes serine chemotaxis (in tryptone soft-agar plates) comparable to that promoted by wild-type Tsr, expressed from plasmid pRR53 [19]. Unless otherwise indicated, the wild-type Tsr phenotypes mentioned below refer to Tsr-S366C. In strain UU2612 (R+B+) some mutant receptors (designated E402* or R404*) supported chemotaxis towards serine in tryptone soft-agar plates, but many of those did so at reduced efficiencies (Fig. 2). Seven E402* mutants (V, L, I, G, P, K, R) and 12 R404* mutants (C, L, M, K, Y, T, N, E, D, W, F, P) could not support any serine chemotaxis in soft agar assays (Fig. 2). To determine whether any of these phenotypes were due to altered expression or stability of the mutant receptor, we measured the steady-state levels of the mutant proteins in receptor-less strain UU2610 (R−B−), which lacks the MCP-modifying enzymes CheR and CheB (see Methods). The expression levels of the mutant receptors ranged from 0.5 to 1.1 of the wild-type level (Fig. 2). We conclude that the E402* and R404* receptor proteins do not have impaired expression or stability.
Fig. 2. Chemotaxis functions and expression levels of Tsr-E402* and Tsr-R404* mutants.
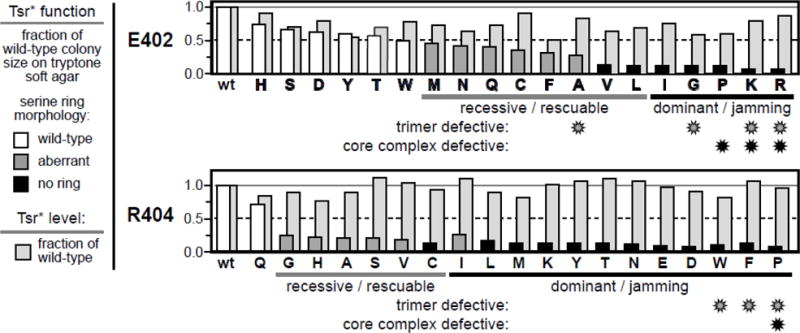
Properties of pCS53-E402* derivatives (upper panel) and pCS53-R404* derivatives (lower panel) are indicated by histogram bars that show performance relative to wild-type Tsr from pCS53. Amino acid replacements are denoted in single-letter notation; wt = wildtype. Bars in front indicate the chemotactic ability, assessed as colony size on tryptone soft agar plates (see Methods), of strain UU2612 (R+B+) carrying each mutant plasmid. Colony morphologies were classified based on the presence or absence of a chemotactic ring of cells at the colony border: wild-type (white), aberrant (dark gray), or no ring (black). The light gray histogram bars in back indicate the steady-state intracellular levels of the mutant proteins relative to the wild-type in strain UU2610 (R−B−) (see Methods). The functional defects of null and aberrant mutants (dominant or recessive in complementation tests; rescuable or jamming in epistasis tests; see Methods) are indicated below the oneletter codes for the mutant amino acid replacements. Mutant receptors that exihibited substantial defects in trimer-of-dimer formation (in TMEA crosslinking tests; see Methods) and/or in core complex formation (fluorescence light microscopy assays; see Methods) are identified with gray or black asterisks, respectively (see Fig. S1 and Tables S1 & S2). To facilitate comparisons of mutant behaviors each panel has a solid gray line at 1.0 and a dashed black line at 0.5 of the wild-type values.
Dominance and jamming properties of E402* and R404* receptors
To assess the severity of their functional alterations, we expressed the E402* and R404* mutant receptors in strain UU1623 (R+B+), which has a wild-type aspartate receptor gene (tar), but lacks the tsr, tap, trg and aer receptor genes. By forming mixed trimers of dimers with Tar receptors [19], some Tsr mutants can block Tar function, an epistasis effect known as jamming [10]. Other Tsr defects regain function in combination with wild-type Tar, an effect known as functional rescue [10]. We found that nearly all loss-of-function E402* or R404* receptors jammed Tar function, indicating formation of mixed, but defective, trimer-of-dimer products (Fig. 2). Most of the partial loss-of-function Tsr* mutants regained full function in UU1623 (R+B+), indicating formation of mixed trimers of dimers that rescued Tsr* function (Fig. 2).
To assess the severity of the structural alterations in E402* and R404* mutant receptors, we asked whether their functional defects prevailed upon co-expression at 1:1 stoichiometries with Tsr subunits that have a known recessive lesion (R69E or T156K) at the serine-binding site [20, 21]. All jamming defects proved dominant in these tests; all rescuable defects proved recessive (Fig. 2). We conclude that the recessive lesions, when present in one subunit, do not greatly perturb the structure of Tsr dimers. In contrast, the receptors that jammed Tar function might have structural lesions that spoil function when present in only one subunit of (E402* or R404*) / (R69E or T154) heterodimers, although we cannot exclude the possibility that a minority of mutant homodimers (E402*/E402* or R404*/R404*) might be sufficient to jam function in Tsr trimers of dimers.
Trimer and ternary complex formation by E402* and R404* receptors
We used an in vivo crosslinking assay to directly assess trimer formation by the mutant receptors in a strain (UU1581) that lacks all receptors and core complex components. The S366C reporter sites in Tsr adopt a trigonal geometry at the trimer interface (see Fig. 1) and can be efficiently cross-linked with the trifunctional thiol-reactive reagent TMEA [19]. In S366C/S366C homodimers, the outer receptor subunits of trimers of dimers are not cross-linked by TMEA, thus producing about 50% unlinked one-subunit products and roughly equivalent numbers of twosubunit and three-subunit forms [11, 12, 19, 22]. Many E402* and R404* receptors formed twoand three-subunit TMEA products less efficiently than did the wild-type control, implying defects in trimer formation or stability (Fig. S1; Table S1; Table S2). Four E402* receptors (A, G, K, R) and three R404* receptors (W, F, P) showed essentially no three-subunit products (gray asterisks in Fig. 2). These findings are consistent with the idea that the E402 and R404 sidechains contribute to the packing stability of Tsr trimers of dimers (see Fig. 1).
Receptor trimers of dimers are obligate assembly intermediates for receptor/CheW/CheA ternary complexes and core signaling units [9, 11, 12, 19, 23]. Accordingly, we evaluated E402* and R404* receptors expressed in receptor-less strain UU2610 (R−B−) for ternary complex formation by fluorescence microscopy, using YFP-CheZ as a clustering reporter. YFP-CheZ binds to CheAS [24], an alternative CheA translation product [25] present in ternary signaling complexes [26]. In this test, some mutant receptors that exhibited substantial reductions in trimer-related TMEA products (E402A, E402G; R404W, R404F) formed receptor clusters at normal levels (Fig. 2; Fig. S1; Table S1; Table S2), which suggests that binding of CheA and CheW to those trimer-compromised receptors enhanced their ability to form trimers of dimers and core signaling complexes. In contrast, the charge-reversal mutants E402K and E402R and the proline replacement mutants E402P and R404P produced no clusters, consistent with a defective binding interaction with CheA and/or CheW.
Adaptational modifications of E402* and R404* receptors
In hosts lacking both sensory adaptation enzymes, Tsr molecules have a homogeneous [QEQEE] modification state. In hosts containing both CheR and CheB, wild-type Tsr molecules have heterogeneous modification states that average about one Em or Q site per subunit [7, 27]. To determine whether E402* and R404* receptors undergo adaptational modifications, we expressed mutant Tsr* plasmids in three receptor-less host strains, UU2611 (R−B+), UU2632 (R+B−), and UU2612 (R+B+), and analyzed their Tsr molecules by electrophoresis in denaturing polyacrylamide gels in which relative band positions reflect subunit modification state [28, 29]. We also expressed mutant receptor molecules in host UU2610 (R−B−) to determine whether in the [QEQEE] state any of them had intrinsic mobility shifts; none did (see Fig. S2 & S3). Modification patterns of the E402* receptors are shown in Fig. S2; those of the R404* receptors are shown in Fig. S3.
We found that most E402* and R404* receptors were modified to approximately wild-type extent in both UU2611 (R−B+) and UU2632 (R+B−). In UU2612 (R+B+), many mutant receptors were driven to more highly modified states by a saturating serine stimulus, as was wild-type Tsr. A few mutant receptors (E402G, R404T, R404N, R404E, R404D) exhibited modification patterns consistent with OFF-shifted outputs: reduced deamidation by CheB and excessive methylation by CheR. The E402P and R404P receptors were very poor substrates for either type of modification, suggesting that those mutant receptors have substantially perturbed structures. Examples of normal and mutant modification patterns are shown in Fig. S4 to define the symbols used in subsequent summary figures of E402* and R404* functional properties.
Assessing the signaling properties of E402* and R404* receptors
We measured the signaling properties of mutant receptors with an in vivo FRET-based kinase assay [30, 31]. This assay measures receptor-controlled CheA autophosphorylation activity by monitoring the interaction between CheY-YFP (FRET acceptor) and CheZ-CFP (FRET donor) molecules. Only the phosphorylated form of CheY interacts appreciably with CheZ, so at steady-state, the FRET signal reflects the autophosphorylation activity of the CheA kinase. In analyzing the assay results, we assume that CheA control by receptors approximates two-state behavior [32]: In the absence of attractant ligands most wild-type Tsr signaling complexes have a kinase-on state; attractant binding shifts the signaling equilibrium toward a kinase-off state. Modification of receptors by the CheR and/or CheB enzymes can also shift the OFF-ON equlibrium: CheR-mediated methylation shifts output toward a kinase-on state; deamidation or demethylation by CheB shifts output toward a kinase-off state.
We first assayed plasmid-encoded E402* and R404* receptors in two receptor-less host strains: UU2567 (R−B−), which lacks the CheR and CheB adaptation enzymes; and UU2700 (R+B+), which has both CheR and CheB [27]. Wild-type Tsr molecules in the [QEQEE] state, i.e., in strain UU2567 (R−B−), produced serine responses of moderate sensitivity (K1/2 ~30 μM) and high cooperativity (Hill coefficient ~14) (Fig. 3; Table S3; Table S4). In UU2700 (R+B+), the lower modification state of wild-type Tsr confers enhanced detection sensitivity (K1/2 ~0.2 μM), but reduced cooperativity (Hill coefficient ~2.5) (Fig. 3; Table S3; Table S4). Like wild-type Tsr, many of the mutant receptors showed enhanced response sensitivities and lower cooperativities in the adaptation-proficient UU2700 (R+B+) host (Fig. 3; Table S3; Table S4), indicating that their signaling properties were subject to adaptational modification control. FRET kinase assays of mutant receptors in UU2697 (R+B−), a CheR-only host, and in UU2699 (R−B+), a CheB-only host, exhibited similar signs of adaptational modification control (Fig. S5; Table S5; Table S6). Response thresholds were generally higher in UU2697 (R+B−) and generally lower in UU2699 (R−B+) relative to the corresponding behaviors in the adaptation-deficient strain UU2567 (R−B−) (Fig. 3; Fig. S5; Table S3; Table S4; Table S5; Table S6).
Fig. 3. Signaling properties of Tsr-E402* and Tsr-R404* mutants.
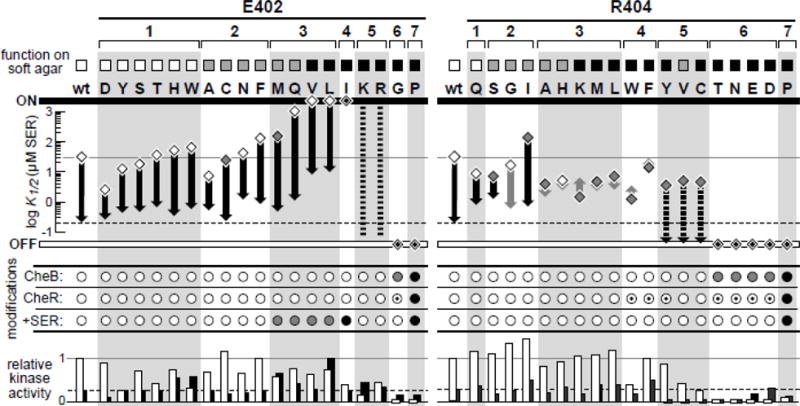
The figure summarizes K1/2 values and relative kinase activities from in vivo FRET kinase assays (see Methods; Table S3 & Table S4) and propensities of the mutant receptors for adaptational modifications in various hosts (see Methods, Fig. S2; Fig. S3; Fig. S4). Squares at the top correspond to the chemotaxis assignments in Fig. 2: wild-type serine ring (white), aberrant serine ring (gray), and no serine ring (black).
upper: Serine responses of mutant receptors in strain UU2567 (R−B−; diamonds) and strain UU2700 (R+B+; vertical arrowheads) are indicated on the K1/2 scale. The solid gray and dashed black lines indicate the respective wild-type values in those two hosts. Gray diamonds indicate low cooperativity responses (Hill coefficient <5) in UU2567 (R−B−); gray vertical arrows indicate low cooperativity responses (Hill coefficient <1.4) in UU2700 (R+B+). White diamonds containing a smaller black diamond denote mutant receptors locked in a kinase-on or kinase-off state that failed to respond in either host. Dashed vertical lines indicate mutant receptors (E402K, E402R) with decaying stimulus responses that were not amenable to quantitative analysis. Dashed vertical arrows indicate mutant receptors (R404Y, R404V, R404C) that lost all kinase activity in the UU2700 (R+B+) host.
middle: Summary of the modification properties of mutant receptors in three host strains: CheB = strain UU2611 (R−B+); CheR = strain UU2632 (R+B−); +SER = strain UU2612 (R+B+) subjected to a saturating serine stimulus (see Methods). Experimental data are given in Figures S2 & S3; modification symbols are defined in Fig. S4: white = wild-type extent of modification; dark gray = less than wild-type extent of modification; black = little or no modification; white with black center = more than wild-type extent of modification. bottom: Histogram bars indicate the maximum kinase activity, derived from in vivo FRET assays, of mutant receptors in the two host strains relative to that of wild-type Tsr signaling complexes in UU2567 (R−B−). White bars = UU2567 (R−B−); black bars = UU2700 (R+B+). Horizontal lines indicate the wild-type values in UU2567 (R−B−; solid gray) and UU2700 (R+B+; dashed black) for comparison purposes. Small bars straddling the zero line indicate no measurable activity. Each set of receptor mutants defines seven phenotypically distinct functional classes that are discussed in the text.
Sensory adaptation behaviors of E402* and R404* receptors
We determined the maximal kinase activity produced by mutant receptors in these various host strains from the magnitude of the FRET change elicited by a saturating serine stimulus [27, 31]. In cases where the receptor failed to respond to serine, we estimated CheA kinase activity from the FRET change elicited by treatment of the cells with 3 mM KCN [27], which depletes intracellular ATP [33], the phospho-donor for the CheA autophosphorylation reaction [5, 34, 35]. In general, the kinase activities of wild-type and many mutant receptor signaling complexes were higher in the UU2567 (R−B−) and UU2697 (R+B−) hosts than in the UU2700 (R+B+) and UU2699 (R−B+) hosts, consistent with modification state control of their signaling properties (Fig. 3; Fig, S5).
To assess the sensory adaptation properties of mutant receptors that were capable of responding to a serine stimulus in the adaptation-proficient host UU2700 (R+B+), we followed the timecourse of their response to a K1/2 concentration of serine until adaptation was complete or the kinase signal was no longer changing (Fig. 4). Overall, the ability of the mutant receptors to promote serine chemotaxis in host UU2612 (R+B+) in soft agar assays strongly correlated with their sensory adaptation properties in UU2700 (R+B+) (Fig. 4).
Fig. 4. Adaptation behaviors of Tsr-E402* and Tsr-R404* mutants.
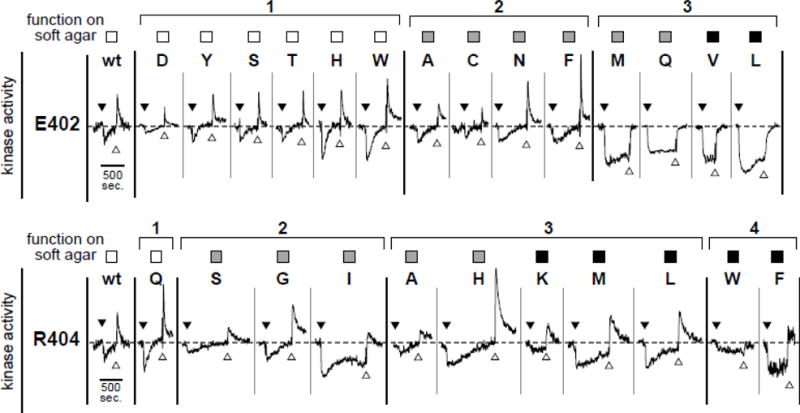
Squares at the top of each panel denote the chemotaxis proficiency of strain UU2612 (R+B+) carrying that mutant receptor, as summarized in Fig. 2: wild-type serine ring (white), aberrant serine ring (gray), and no serine ring (black). Data are the YFP/CFP ratio values from in vivo FRET kinase assays in strain UU2700 (R+B+) plotted at the same vertical and horizontal scales for each mutant receptor. Black triangles indicate addition of a K1/2 concentration serine stimulus. White triangles indicate serine removal. The horizonal dashed line indicates the pre-stimulus baseline level of kinase activity to which the system returns upon full adaptation and/or stimulus removal. Transient kinase activity spikes upon stimulus removal occur because of methylation increases that took place during the adaptation phase of the response.
Signaling phenotypes of E402* and R404* receptors
The FRET responses of E402* and R404* receptors in the UU2567 (R−B−) and UU2700 (R+B+) hosts, in combination with their chemotaxis, trimer, and clustering properties (Fig. 2) and their adaptational modification patterns and behaviors (Fig. 4; Fig. S2; Fig. S3; Fig. S4; Fig. S5), enabled us to assign the mutant receptors to phenotypically distinct classes with different extents and severities of functional defects (Fig. 3). Class 1 mutants at both residues exhibited approximately wild-type functionality in all assays; class 2 mutants were similar, but partly compromised in chemotaxis performance on soft agar (Fig. 3). Notably, ten E402* receptors fell into classes 1 and 2, whereas only four R404* receptors did, indicating that sidechain character at residue 404 is more critical to receptor function than it is at residue 402. Class 3 mutants at both residues had moderate to severe chemotaxis defects, but differed in their sensory adaptation behaviors. Following a serine stimulus, class 3 E402* receptors were poorly modified by CheR (Fig. 3) and fully defective in adaption (Fig. 4), whereas class 3 R404* receptors were modified well by CheR (Fig. 3) and underwent sensory adaptation, albeit more slowly than wild-type Tsr (Fig. 4). The class 4 R404* mutants exhibited adaptation defects comparable to those of the class 3 E402* mutants (Fig. 4). The class 4 E402* mutants and class 5 mutants at both residues exhibited little or no response to serine in the UU2700 host (R+B+) (Fig. 3). Class 6 mutants at both residues had essentially no kinase activity in either FRET host strain (Fig. 3). Finally, receptors with a proline replacement at either residue (E402P, R404P) were quite defective in essentially all functional measures (Fig. 2; Fig. 3). The mechanistic implications of these results are considered further in the Discussion.
DISCUSSION
Structural environments and functional roles of Tsr residues E402 and R404
The trimer-of-dimers arrangement creates different structural environments for corresponding residues in each protomer of receptor dimers: E402 and R404 sidechains in their outer locations are solvent-exposed; at their inner locations they pack against one another and trimer contact residues L378 and/or L380 near the trimer axis (Fig. 5a; Fig. S6). E402 and R404 residues at the trimer periphery could conceivably serve as CheA or CheW binding determinants to promote assembly or control of core signaling complexes. However, the functional properties of E402* and R404* mutant receptors indicate that these residues do not play such roles. If either residue resided on a critical docking surface, large sidechain replacements should sterically hinder the docking interaction. If either residue was an important binding determinant, nearly all sidechain replacements at the critical position would be expected to disrupt that interaction. We found that both small and large sidechain replacements at E402 and R404 preserved core unit assembly and functionality. For example, mutant receptors with an alanine or a phenylalanine replacement at either position formed kinase-active core complexes that responded to attractant stimuli and to adaptational modifications (Fig. 3).
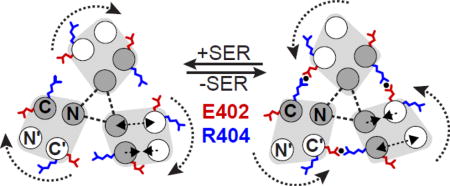
Fig. 5. Mechanistic model of the E402–R404 role in receptor signal state control.
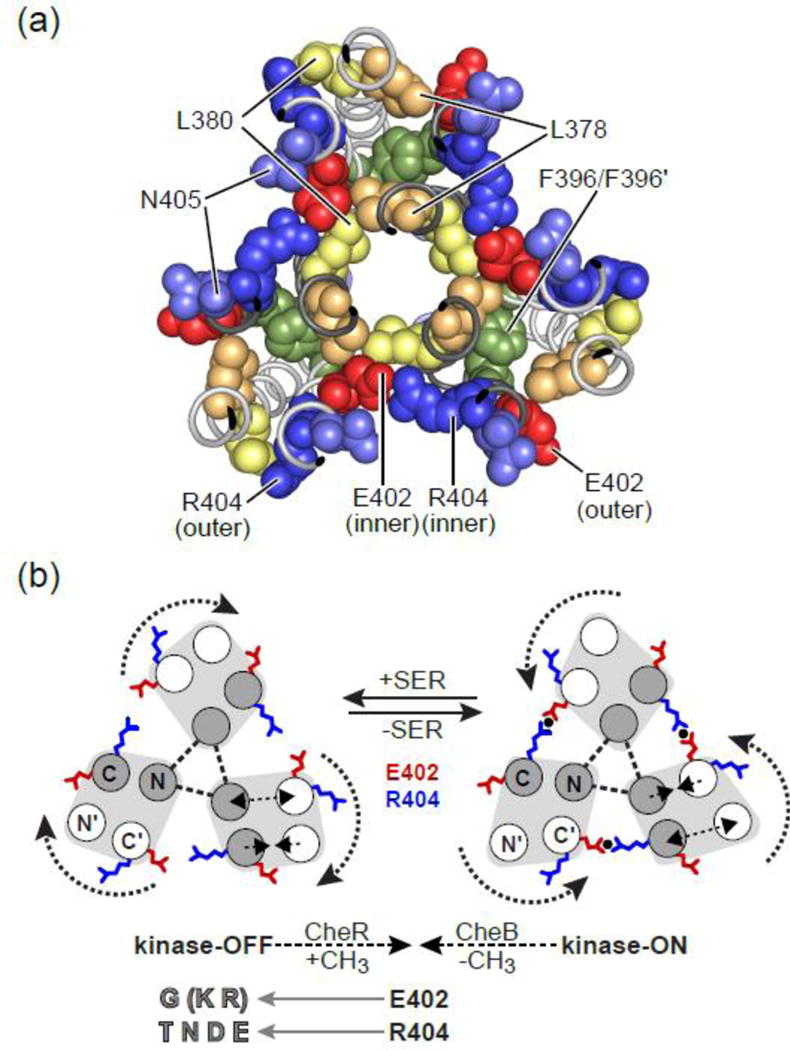
(a) Structural environment of the inner and outer E402–R404 residues at the trimer tip; a cross-section slab viewed from the membrane-proximal side. Subunit shading conventions follow those in Fig. 1: The three receptor dimer subunits that contribute the trimer contact residues at the trimer axis have dark gray backbone ribbons; the three more peripheral dimer subunits have light gray backbone ribbons. One inner and one outer location of residues L378, L380, E402, R404, N405 is labeled; their counterparts in other dimers are related by the three-fold symmetry of the trimer. The F396 and F396′ residues interact at the subunit interfaces of each dimer.
(b) Mechanistic model of the E402–R404 interaction. The cartoons depict cross-section views of the trimer of dimers like that shown in Fig. 1b and in panel (a) above. Gray circles represent the dimer subunits whose N helices interact at the trimer axis; white circles represent the other dimer subunits in the trimer. In the proposed kinase-OFF signaling conformation [40], one subunit (white helices) in each dimer rotates away from the neighboring dimer, increasing the distance between E402 and R404 side chains in adjacent dimers. In the kinase-ON conformation, those receptor subunits rotate toward their dimer neighbors, bringing the inner E402 and R404 residues into close proximity. The present study indicates that an ionic or H-bonded interaction (black circle) between the E402 and R404 sidechains at their inner location contributes to stability of the ON-state trimer conformation. The three N helices at the trimer axis (gray) interact stably (dashed triangle) and serve as a fulcrum for these signaling conformational changes. E402 and R404 amino acid replacements that effectively lock signal output are listed and discussed in the text.
These results indicate that sidechain character at the outer 402 and 404 positions is not critical to core complex assembly or operation. We cannot, however, exclude the possibility that one or both residues play a structural role of secondary importance at the trimer periphery. For example, in their outer locations, E402 packs against the L378 and N405 sidechains and R404 impinges on the L380 sidechain (Fig. 5a). These packing interactions could serve to stabilize the tips of receptor dimers and to constrain their motions within the trimer of dimers. Although some of the aberrant signaling properties of E402* and R404* receptors might arise from perturbation of an outer structural role, several lines of evidence, detailed below, suggest that the functional consequences of E402 and R404 amino acid replacements arise predominantly from the structural changes they create at their inner locations in the trimer of dimers (Fig. 5a)
Evidence for a trimer-stabilizing E402–R404 interaction at their inner location
At their inner location, the E402 sidechain from one dimer abuts the R404 sidechain of a neighboring dimer in the trimer (Fig. 5a). The residues meet in a crevice at the dimer-dimer boundary with the E402 sidechain partially buried by surrounding hydrophobic (L378, L380) and hydrophilic (N405) residues (Fig. 5a; Fig. S6). The R404 sidechain is more solvent-exposed, but its guanidinium group lies close to the sidechain carboxyl group of E402 in a bidentate arrangement characteristic of a salt-bridge interaction (Fig. 5a) [36]. Cassidy et al., also identified a probable salt bridge interaction between the E402–R404 counterparts in a molecular dynamics study of a Thermotoga maritima receptor modeled in a trimer of dimers arrangement [16]. Internal salt bridges can appreciably enhance the structural stability of a protein, whereas salt bridge interactions between surface residues generally do not [37, 38]. The partly buried inner environment of residues 402 and 404 suggests that an ionic interaction between their sidechains might make a detectable contribution to the structural stablity of the Tsr trimer of dimers.
The high-resolution structure of the Tsr trimer-of-dimers came from an X-ray study of a soluble Tsr fragment containing Q residues (Em mimics) at modification sites 1–4 in each subunit [18]. Those fragments assemble signaling complexes, both in vivo and in vitro, with high CheA kinase activity, suggesting that the crystal structure depicts a kinase-ON conformation of the Tsr trimer tip [10, 39]. It follows that E402* and R404* alterations that weaken trimer packing or distort trimer structure should shift receptor signaling complexes toward a kinase-OFF state.
Most amino acid replacements at residues 402 and 404 had no substantive effects on trimer-of-dimer formation in TMEA crosslinking tests (Fig. 2; Fig. S1). Only small (G, A) or charge-reversal replacements (K, R) at E402 and helix-distorting (P) or large (F, W) sidechain replacements at R404 eliminated 3-subunit TMEA crosslinking products (Fig. 2; Fig. S1). Even so, in the presence of CheA and CheW the E402G, E402A, R404F, and R404W receptors efficiently formed ternary signaling complexes, implying relatively modest changes in trimer conformation or dynamic properties. The E402G core complexes produced no kinase activity, were poor substrates for CheB, which acts on ON-state receptors, and were over-zealous substrates for CheR, which acts on OFF-state receptors (Fig. 3). Although the R404F and R404W core complexes exhibited kinase activity, those receptors were also over-modified by CheR, implying OFF-shifted signaling properties (Fig. 3). We conclude that structural changes at the inner 402–404 interface can weaken or distort receptor trimers and influence their signaling properties.
Charge-reversal replacements at each residue produced similar, but more drastic trimer stability and signaling defects. The E402K and E402R receptors inefficiently assembled core complexes (Fig. 2) that irreversibly lost kinase activity following a serine stimulus (Fig. S7), implying substantial structural instability (Fig. 3). Similarly, R404D and R404E receptors made reduced levels of 3-subunit TMEA crosslinking products (Fig. 2). Their core signaling complexes produced no kinase activity, were poor substrates for CheB, and were over-modified by CheR (Fig. 3), all hallmarks of a locked-OFF output state. Thus, charge reversals at either residue destabilized trimers and/or core complexes and impaired the kinase-active output state. In contrast, doubly mutant receptors with charge-reversal replacements at both residues regained considerable functionality, including augmented trimer and core complex formation, enhanced kinase activity, and the ability to down-regulate kinase output in response to serine stimuli (Fig. S8). Taken together, these results suggest that destabilizing interactions between the inner 402 and 404 sidechains weaken Tsr trimers of dimers, impair core complex assembly, reduce kinase activity, and impact stimulus and adaptational control over signal output.
A two-state working model of receptor signaling conformations at the hairpin tip
The two-state model presented by Ortega, et al. [40] of receptor tip conformations in the trimer of dimers provides a useful conceptual framework for considering the signaling roles of Tsr residues E402 and R404 (Fig. 5b). Derived from molecular dynamics simulations of the Tsr cytoplasmic domain, the model proposes that conformational switches between kinase-on and kinase-off conformations occur concomitantly with a flip in the aromatic stacking of a phenylalanine residue (F396/F3962′) at the subunit interface in the tip of each receptor dimer (Fig. 5a). The proposed conformational changes within each dimer involve concerted increases and decreases in inter-subunit helix distances (double-headed arrows in Fig. 5b. In one F396 stacking arrangement, each dimer in the trimer resembles the putative ON-state crystal structure, characterized by the E402–R404 salt bridge interaction at the inter-dimer boundaries (Fig. 5b). In the other stacking arrangement, one subunit in each dimer (white helices in Fig. 5b) rotates away from the neighboring dimer, breaking the salt bridge between residues E402 and R404. The dimer subunits at the trimer axis (gray helices in Fig. 5b) remain stationary.
This working model accounts for the locked-OFF output properties of the E402G receptor and those with charge-reversals at either residue. At residue 404, N and T replacements also produced locked-OFF signaling properties comparable to those of the E402G, R404D, and R404E receptors (Fig. 3). However, the structural basis for the R404N and R404T output shifts is less apparent. It may be that these sidechain replacements not only lower ON-state stability by eliminating salt bridge formation with E402, but also enhance OFF-state stability through interactions of their polar sidechains with the aqueous environment at the outer 404 location. (Fig. 5b).
The R404G receptor exhibited very different functional properties than its E402G counterpart, attesting to distinct structural contributions from the salt bridge partners. Although the R404G mutant failed to support serine chemotaxis, its signaling behavior was near normal in other respects (Fig. 3; Fig. 4). This finding implies that the absence of sidechain structural contributions from residue 404 in the inner and outer environments has little effect on the relative stabilities of the ON and OFF states. Perhaps reduced stability of the ON state in the R404G receptor due to loss of the salt bridge interaction is offset by a loss of R404 sidechain structural contributions in the outer location that stabilize the OFF state.
Nature of the ON and OFF signaling states and inadequacies of two-state models
The signaling behaviors of most E402* and R404* receptors do not readily conform to discrete two-state models of receptor output control in which a core complex ON conformation activates CheA and an OFF conformation deactivates CheA. For example, class 1 and class 2 E402* and R404* receptors retained near-normal kinase activity (Fig. 3), stimulus-response (Fig. 3) and sensory adaptation behavior (Fig. 4) with a wide variety of sidechain replacements. Such disparate sidechain volumes and chemical characters are unlikely to produce precisely the same conformations and dynamic motions of the receptor trimers. Conceivably, the structural changes in the mutant receptors, in addition to altering the stabilities of the native output states, populate additional low-energy conformations along the OFF-ON energy landscape. Thus, the kinaseactive state of core complexes may not require a specific, stable receptor tip conformation, but rather a range of conformations involving close association of E402/R404 sidechains at their inner location or dynamic motions over that conformational range. In this way, many amino acid replacements at these two residues could still produce some kinase activity by preserving a portion of the wild-type dynamic and conformational range.
The phosphorylation site P1 domain of CheA also appears to be in dynamic motion in the kinase-active state of core complexes [41]. P1 movements might reflect underlying motions of the receptor tips in the trimers. In receptor signaling complexes locked in a kinase-OFF state, the CheA-P1 domains are more static. Conceivably, receptor tips and trimers are also more static in the OFF state, but none of the E402* or R404* mutant receptors make a compelling case for that idea.
Source(s) of cooperativity in receptor arrays
Attractant-induced down-regulation of CheA in core signaling units in an in vitro nanodisc system exhibits little cooperativity (Hill coefficients less than 2), suggesting that there is limited allosteric interaction among the members of individual receptor trimers [42]. In contrast, Tsr arrays in membrane vesicles [43] or cells [27, 44] can exhibit Hill coefficients as high as 10–20, implying that cooperative response behavior arises mainly from allosteric interactions between core signaling units. Disruption of those array connections by lesions at CheA-P5/CheW interface 2 leads to loss of core unit clustering and to low cooperativity response behavior comparable to that of solitary core units in nanodiscs [45].
A variety of amino acid replacements at Tsr residue 404 produced receptors that responded to serine with low Hill coefficients (Table S4; Fig. S9). Unlike interface 2 array mutants, they do not seem to have impaired array connections between core signaling units (Fig. S2). Perhaps these mutant receptors have lost allosteric interactions within core units that couple their signaling behaviors to neighboring core units. In this scenario, the R404 sidechain is an active participant in cooperative signaling transitions, whereas the E402 sidechain evidently is not (Fig. 3). Conceivably, many amino acid replacements at residue 404 reduce the structural stability of both signaling states, decoupling the conformational transitions of receptors in a trimer. The lowered serine response thresholds of these R404* mutant receptors resemble those of interface 2 array mutants [45].
Emerging evidence suggests that the signaling states of core units might influence the extent of their array connections [45, 46]. At high modification states and activity levels, wild-type Tsr produces high-threshold, high-cooperativity serine responses; receptors at low modification states and low kinase activities produce low-threshold, low-cooperativity responses. Over half of the low-cooperativity R404* receptors responded anomalously to adaptational modifications (R404K/A/L/M/W/F in Fig. 3) and could be defective in the mechanism that matches array connectivity to core complex kinase activity. In an adaptation-proficient host, these mutant receptors underwent adaptational modifications that reduced the activity of their associated CheA kinase, but the activity change did not alter their serine response sensitivity (Fig. 3). Further study of these R404* mutants may provide new mechanistic insights into how signaling state influences array connectivity.
Structural interplay between adaptational modifications and the receptor tip
A flexible region, bisected by a glycine hinge, mediates the conformational interplay between the methylation helix (MH) bundle and the receptor tip [17, 47] (Fig. 1a). Although the communication mechanism is still unclear, the structural interaction is presumably bidirectional, in which case conformational or dynamic changes at the receptor tip should influence the CheR/CheB substrate properties of the MH bundle. The E402P and R404P receptors provide the most dramatic example of structural interaction between the tip and MH bundle. Their proline replacements most likely destabilize helix packing at the tip. That structural disturbance propagates to the MH bundle, effectively preventing access to either the CheR or CheB substrate state (Fig. 3). The receptor mutants with locked-OFF outputs (E402G, R404T/N/E/D) also offer examples of tip-MH bundle structural interplay. Their OFF conformation at the tip makes them excellent substrates for CheR and poor substrates for CheB (Fig. 3).
Structural communication between the tip and MH bundles appeared to be aberrant in some mutant receptors. The class 3 E402* receptors (M/Q/V/L) exhibited elevated response thresholds in the QEQEE modification state, but enhanced serine sensitivities in adaptation-competent cells (Fig. 3), consistent with modification state control over their tip response thresholds. However, the mutant receptors failed to undergo net modification changes (Fig. 3) oi sensory adaptation in response to serine stimuli (Fig. 4), suggesting that the attractant-induced OFF conformation at their tip was structurally incompatible with the substrate properties of their MH bundle. In adaptation-competent cells, the class 3 R404* (A/H/K/M/L) receptors underwent net modification changes (Fig. 3) and sensory adaptation (Fig. 4) in response to a serine stimuli but their response sensitivities were comparable to those in adaptation-deficient cells (Fig. 3). Evidently, the modification states of their MH bundles did not affect the response thresholds at their tips.
Summary of key findings
Our results indicate that the E402 and R404 residues of Tsr play their most critical signaling roles at their inner locations near the trimer axis where they likely participate in a salt bridge interaction that stabilizes trimer-of-dimer packing and the kinase-ON state of core signalin complexes. Many mutant receptors with an amino acid replacement at either residue can achiev both ON and OFF signaling states, but seem unlikely to adopt the precise native signaling conformation(s). Instead, we suggest that kinase activity is produced over a range of receptor conformations and that the level of activity may reflect the amplitude or frequency of dynamic motions over that structural range. Similarly, the kinase-OFF state may not be a discrete conformation, but rather a range of dynamic motions outside the range of those suitable for kinase activation. Consistent with this idea, some structural lesions at both E402 and R404 produced signaling behaviors that are not compatible with discrete two-state models of core complex signaling states. Those lesions might stabilize intermediate receptor conformations along the OFF-ON energy landscape.
Amino acid replacements produced different constellations of signaling defects at each residue, indicating that they play distinct structure-function roles. R404, but not E402, may participate in coupling receptor structural changes in core complexes that lead to cooperative allosteric transitions between core complexes in the receptor array. Whether all members of a receptor trimer switch signaling states in concert remains an open question [42]. R404* lesions that reduce signal cooperativity also prevent activity-dependent changes in the size of allosteric signaling units in the receptor array and their study could provide valuable new insights into this array-remodeling mechanism.
MATERIALS AND METHODS
Bacterial strains
All strains used in this study were derivatives of E. coli K12 strain RP437 [48]. Their relevant genotypes are as follows:
UU1581 [Δ(flhD-flhA)4 Δ(tsr)7028 Δ(trg)100] [19];
UU1623 [Δ(tsr)7028 Δ(tap)3654 Δ(trg)100] [21];
UU2377 [tsr-R69E Δ(aer)1 Δ(tar-tap)5201 Δ(trg)4543 Δ(recA) [21];
UU2378 [tsr-T156K Δ(tar-tap)5201 Δ(trg)4543 Δ(recA) [21];
UU2567 [Δ(tar-cheZ)4211 Δ(tsr)5547 Δ(trg)4543 Δ(aer)1] [27];
UU2610 [Δ(aer)1 Δ(tar-cheB)4346 Δ(tsr)5547 Δ(trg)4543] [49];
UU2611 [Δ(aer)1 Δ(tar-cheR)4283 Δ(tsr)5547 Δ(trg)4543] [49];
UU2612 [Δ(aer)1 Δ(tar-tap)4530 Δ(tsr)5547 Δ(trg)4543] [49];
UU2632 [Δ(aer)1 Δ(tar-tap)4530 Δ(cheB)4345 Δ(tsr)5547 Δ(trg)4543] [49];
UU2697 [Δ(cheY-cheZ) 1215 Δ(aer)1 Δ(tar-tap)4530 Δ(cheB)4345 Δ(tsr)5547 Δ(trg)4543] [27];
UU2699 [Δ(cheY-cheZ) 1215 Δ(aer)1 Δ(tar-cheR)4283 Δ(tsr)5547 Δ(trg)4543] [27];
UU2700 [Δ(cheY-cheZ) 1215 Δ(aer)1 Δ(tar-tap)4530 Δ(tsr)5547 Δ(trg)4543] [27].
Plasmids
Plasmids used in this study were as follows: pRR48, a derivative of pBR322 that confers ampicillin resistance and has a tac promoter and an “ideal” lac operator under the control of a plasmid-encoded lacI repressor, which is inducible by isopropyl β-D-thiogalactopyranoside (IPTG) [22]; pCS53, a derivative of pRR48 that encodes Tsr-S366C under the control of IPTG [22]; pKG116, a derivative of pACYC184 that confers chloramphenicol resistance and has a sodium salicylate-inducible expression/cloning site [50]; pRZ30, a derivative of pKG116 that expresses cheY-yfp and cheZ-cfp gene fusions under salicylate control [7]; and pVS49, a derivative of pACYC184 that encodes a functional YFP-CheZ fusion protein under arabinose-inducible control [26].
Site-directed mutagenesis
Mutations in plasmid pCS53 were generated by QuikChange™ PCR mutagenesis and verified by sequencing the entire tsr coding region as previously described [10].
Expression levels of mutant Tsr proteins
Tsr expression from pCS53 derivatives was analyzed in strain UU2610 which lacks the CheR and CheB receptor modification enzymes. Protein expression samples were prepared and analyzed by SDS-PAGE and immunoblotting as described previously [12].
Chemotaxis assays on semi-solid agar media
Strains carrying tsr expression plasmids were assessed for chemotactic ability on tryptone semi-solid agar plates containing 50 μg/ml ampicillin and 100 μM IPTG as previously described [51]. Plates were incubated at 30–32.5°C for 6–8 h. Serine chemotaxis was evaluated in strain UU2612; dominance of tsr functional defects was measured in strains UU2377 and UU2378; ability of the mutant defect to jam operation of wild-type aspartate (Tar) receptors was evaluated in strain UU1623.
Dominance, rescue, and jamming tests
For dominance tests, mutant receptor plasmids were transferred into strains UU2377 and UU2378, which encode Tsr molecules with recessive lesions at the serine-binding site [21]. For rescue and jamming tests, mutant plasmids were transferred into UU1623, which encodes wild-type Tar as its only receptor [21]. Plasmid-containing cells were tested for Tsr and/or Tar function on tryptone soft agar plates, as described above.
Receptor clustering assays
Mutant pCS53 derivatives were introduced into UU2610 cells harboring plasmid pVS49. Cells containing these two compatible plasmids were grown to mid-exponential phase at 30°C in tryptone broth containing antibiotics (50 μg/ml ampicillin and 12.5 μg/ml chloramphenicol) and inducers (100 μM IPTG for Tsr expression and 0.005% arabinose for YFP-CheZ expression) and analyzed by fluorescence microscopy as previously described [10, 12].
Crosslinking assays for receptor trimers of dimers
UU1581 cells containing mutant pCS53 derivatives were grown to late-exponential phase (OD600 ~ 0.7) at 30°C in tryptone broth containing 50 μg/ml ampicillin and 100 μM IPTG, washed twice with KEP (10 mM potassium phosphate buffer pH 7.0, 0.1 mM potassium EDTA), and resuspended in KEP at (OD600 = 2). Cells were treated with 50 μM TMEA (tris-2-maleimidoethylamide; Pierce Chemical Co.), a trifunctional thiol reagent, for 30 s, quenched with 10 mM N-ethyl maleimide (NEM), and lysed with SDS sample buffer [11, 19]. Crosslinking products were analyzed by SDS-PAGE and immunoblotting, as described previously [19, 22].
Assessing receptor modification patterns
Strains carrying Tsr expression plasmids were prepared as described above for crosslinking assays. Cell lysates were analyzed by SDS-PAGE and Tsr protomers in different modifications states were visualized by immunoblotting as previously described [52].
FRET-based measurement of in vivo kinase activity
The assay protocol and data analysis followed the procedures previously described in detail [27]. Briefly, FRET signals were collected from cells expressing the reporter pair CheY-YFP and CheZ-CFP from plasmid pRZ30 and Tsr receptors from plasmid pCS53. FRET data were processed and fitted to a multisite Hill equation using KaleidaGraph 4.5 software (Synergy Software) to obtain K1/2 and Hill coefficient values. Absolute kinase activities were calculated from the FRET change to a saturating serine stimulus and/or to 3 mM KCN [27].
Protein structural display
Structure images were prepared with PyMOL (Mac) software (http://www.pymol.org).
Supplementary Material
Highlights.
Receptor trimers are key structural components of highly cooperative signaling arrays.
Tsr residues E402 and R404 form salt bridges in trimers that promote kinase-on output.
Receptor complexes produce kinase activity over a range of trimer stability states.
R404 is critical for sensory adaptation and high response cooperativity; E402 is not.
Allosteric interactions in core signaling units may contribute to array cooperativity.
Acknowledgments
We thank Davi Ortega (Caltech) and David Goldenberg (U. Utah) for helpful discussions. This work was supported by research grant GM19559 from the National Institute of General Medical Sciences. DNA sequencing and primer synthesis were carried out by the Protein-DNA Core Facility at the University of Utah, which receives support from National Cancer Institute grant CA42014 to the Huntsman Cancer Institute.
Abbreviations
- MCP
methyl-accepting chemotaxis protein
- FRET
Förster resonance energy transfer
- IPTG
isopropyl-β-D-thiogalactopyranoside
- P-CheY
phospho-CheY
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hazelbauer GL, Falke JJ, Parkinson JS. Bacterial chemoreceptors: high-performance signaling in networked arrays. Trends in biochemical sciences. 2008;33:9–19. doi: 10.1016/j.tibs.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parkinson JS, Hazelbauer GL, Falke JJ. Signaling and sensory adaptation in Escherichia coli chemoreceptors: 2015 update. Trends in microbiology. 2015;23:257–66. doi: 10.1016/j.tim.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ravid S, Matsumura P, Eisenbach M. Restoration of flagellar clockwise rotation in bacterial envelopes by insertion of the chemotaxis protein CheY. Proc Natl Acad Sci USA. 1986;83:7157–61. doi: 10.1073/pnas.83.19.7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Welch M, Oosawa K, Aizawa S-I, Eisenbach M. Phosphorylation-dependent binding of a signal molecule to the flagellar switch of bacteria. Proc Natl Acad Sci USA. 1993;90:8787–91. doi: 10.1073/pnas.90.19.8787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hess JF, Oosawa K, Kaplan N, Simon MI. Phosphorylation of three proteins in the signaling pathway of bacterial chemotaxis. Cell. 1988;53:79–87. doi: 10.1016/0092-8674(88)90489-8. [DOI] [PubMed] [Google Scholar]
- 6.Cluzel P, Surette M, Leibler S. An ultrasensitive bacterial motor revealed by monitoring signaling proteins in single cells. Science. 2000;287:1652–5. doi: 10.1126/science.287.5458.1652. [DOI] [PubMed] [Google Scholar]
- 7.Han XS, Parkinson JS. An unorthodox sensory adaptation site in the Escherichia coli serine chemoreceptor. J Bacteriol. 2014;196:641–9. doi: 10.1128/JB.01164-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitanovic S, Ames P, Parkinson JS. A Trigger Residue for Transmembrane Signaling in the Escherichia coli Serine Chemoreceptor. J Bacteriol. 2015;197:2568–79. doi: 10.1128/JB.00274-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li M, Hazelbauer GL. Core unit of chemotaxis signaling complexes. Proc Natl Acad Sci U S A. 2011;108:9390–5. doi: 10.1073/pnas.1104824108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ames P, Studdert CA, Reiser RH, Parkinson JS. Collaborative signaling by mixed chemoreceptor teams in Escherichia coli. Proc Natl Acad Sci U S A. 2002;99:7060–5. doi: 10.1073/pnas.092071899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gosink KK, Zhao Y, Parkinson JS. Mutational analysis of N381, a key trimer contact residue in Tsr, the Escherichia coli serine chemoreceptor. J Bacteriol. 2011;193:6452–60. doi: 10.1128/JB.05887-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mowery P, Ostler JB, Parkinson JS. Different signaling roles of two conserved residues in the cytoplasmic hairpin tip of Tsr, the Escherichia coli serine chemoreceptor. J Bacteriol. 2008;190:8065–74. doi: 10.1128/JB.01121-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swain KE, Gonzalez MA, Falke JJ. Engineered socket study of signaling through a four-helix bundle: evidence for a yin-yang mechanism in the kinase control module of the aspartate receptor. Biochemistry. 2009;48:9266–77. doi: 10.1021/bi901020d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Briegel A, Li X, Bilwes AM, Hughes KT, Jensen GJ, Crane BR. Bacterial chemoreceptor arrays are hexagonally packed trimers of receptor dimers networked by rings of kinase and coupling proteins. Proc Natl Acad Sci U S A. 2012;109:3766–71. doi: 10.1073/pnas.1115719109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu J, Hu B, Morado DR, Jani S, Manson MD, Margolin W. Molecular architecture of chemoreceptor arrays revealed by cryoelectron tomography of Escherichia coli minicells. Proc Natl Acad Sci U S A. 2012;109:E1481–8. doi: 10.1073/pnas.1200781109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cassidy CK, Himes BA, Alvarez FJ, Ma J, Zhao G, Perilla JR, et al. CryoEM and computer simulations reveal a novel kinase conformational switch in bacterial chemotaxis signaling. eLife. 2015;4 doi: 10.7554/eLife.08419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexander RP, Zhulin IB. Evolutionary genomics reveals conserved structural determinants of signaling and adaptation in microbial chemoreceptors. Proc Natl Acad Sci U S A. 2007;104:2885–90. doi: 10.1073/pnas.0609359104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim KK, Yokota H, Kim SH. Four-helical-bundle structure of the cytoplasmic domain of a serine chemotaxis receptor. Nature. 1999;400:787–92. doi: 10.1038/23512. [DOI] [PubMed] [Google Scholar]
- 19.Studdert CA, Parkinson JS. Crosslinking snapshots of bacterial chemoreceptor squads. Proc Natl Acad Sci U S A. 2004;101:2117–22. doi: 10.1073/pnas.0308622100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ames P, Zhou Q, Parkinson JS. Mutational analysis of the connector segment in the HAMP domain of Tsr, the Escherichia coli serine chemoreceptor. J Bacteriol. 2008;190:6676–85. doi: 10.1128/JB.00750-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou Q, Ames P, Parkinson JS. Mutational analyses of HAMP helices suggest a dynamic bundle model of input-output signalling in chemoreceptors. Mol Microbiol. 2009;73:801–14. doi: 10.1111/j.1365-2958.2009.06819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Studdert CA, Parkinson JS. Insights into the organization and dynamics of bacterial chemoreceptor clusters through in vivo crosslinking studies. Proc Natl Acad Sci U S A. 2005;102:15623–8. doi: 10.1073/pnas.0506040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Briegel A, Wong ML, Hodges HL, Oikonomou CM, Piasta KN, Harris MJ, et al. New insights into bacterial chemoreceptor array structure and assembly from electron cryotomography. Biochemistry. 2014;53:1575–85. doi: 10.1021/bi5000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cantwell BJ, Manson MD. Protein domains and residues involved in the CheZ/CheAS interaction. J Bacteriol. 2009;191:5838–41. doi: 10.1128/JB.00280-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith RA, Parkinson JS. Overlapping genes at the cheA locus of Escherichia coli. Proc Natl Acad Sci U S A. 1980;77:5370–4. doi: 10.1073/pnas.77.9.5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sourjik V, Berg HC. Localization of components of the chemotaxis machinery of Escherichia coli using fluorescent protein fusions. Mol Microbiol. 2000;37:740–51. doi: 10.1046/j.1365-2958.2000.02044.x. [DOI] [PubMed] [Google Scholar]
- 27.Lai RZ, Parkinson JS. Functional suppression of HAMP domain signaling defects in the E. coli serine chemoreceptor. Journal of molecular biology. 2014;426:3642–55. doi: 10.1016/j.jmb.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engstrom P, Hazelbauer GL. Multiple methylation of methyl-accepting chemotaxis proteins during adaptation of E. coli to chemical stimuli. Cell. 1980;20:165–71. doi: 10.1016/0092-8674(80)90244-5. [DOI] [PubMed] [Google Scholar]
- 29.Sherris D, Parkinson JS. Posttranslational processing of methyl-accepting chemotaxis proteins in Escherichia coli. Proc Natl Acad Sci U S A. 1981;78:6051–5. doi: 10.1073/pnas.78.10.6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sourjik V, Berg HC. Receptor sensitivity in bacterial chemotaxis. Proc Natl Acad Sci U S A. 2002;99:123–7. doi: 10.1073/pnas.011589998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sourjik V, Vaknin A, Shimizu TS, Berg HC. In vivo measurement by FRET of pathway activity in bacterial chemotaxis. Methods Enzymol. 2007;423:363–91. doi: 10.1016/S0076-6879(07)23017-4. [DOI] [PubMed] [Google Scholar]
- 32.Asakura S, Honda H. Two-state model for bacterial chemoreceptor proteins. The role of multiple methylation. Journal of molecular biology. 1984;176:349–67. doi: 10.1016/0022-2836(84)90494-7. [DOI] [PubMed] [Google Scholar]
- 33.Taylor BL, Miller JB, Warrick HM, Koshland DE., Jr Electron acceptor taxis and blue light effect on bacterial chemotaxis. J Bacteriol. 1979;140:567–73. doi: 10.1128/jb.140.2.567-573.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hess JF, Oosawa K, Matsumura P, Simon MI. Protein phosphorylation is involved in bacterial chemotaxis. Proc Natl Acad Sci U S A. 1987;84:7609–13. doi: 10.1073/pnas.84.21.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wylie D, Stock A, Wong CY, Stock J. Sensory transduction in bacterial chemotaxis involves phosphotransfer between Che proteins. Biochemical & Biophysical Research Communications. 1988;151:891–6. doi: 10.1016/s0006-291x(88)80365-6. [DOI] [PubMed] [Google Scholar]
- 36.Donald JE, Kulp DW, DeGrado WF. Salt bridges: geometrically specific, designable interactions. Proteins. 2011;79:898–915. doi: 10.1002/prot.22927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Makhatadze GI, Loladze VV, Ermolenko DN, Chen X, Thomas ST. Contribution of surface salt bridges to protein stability: guidelines for protein engineering. Journal of molecular biology. 2003;327:1135–48. doi: 10.1016/s0022-2836(03)00233-x. [DOI] [PubMed] [Google Scholar]
- 38.Strop P, Mayo SL. Contribution of surface salt bridges to protein stability. Biochemistry. 2000;39:1251–5. doi: 10.1021/bi992257j. [DOI] [PubMed] [Google Scholar]
- 39.Kim SH, Wang W, Kim KK. Dynamic and clustering model of bacterial chemotaxis receptors: Structural basis for signaling and high sensitivity. Proc Natl Acad Sci U S A. 2002;99:11611–5. doi: 10.1073/pnas.132376499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ortega DR, Yang C, Ames P, Baudry J, Parkinson JS, Zhulin IB. A phenylalanine rotameric switch for signal-state control in bacterial chemoreceptors. Nature communications. 2013;4:2881. doi: 10.1038/ncomms3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Briegel A, Ames P, Gumbart JC, Oikonomou CM, Parkinson JS, Jensen GJ. The mobility of two kinase domains in the Escherichia coli chemoreceptor array varies with signalling state. Mol Microbiol. 2013;89:831–41. doi: 10.1111/mmi.12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li M, Hazelbauer GL. Selective allosteric coupling in core chemotaxis signaling complexes. Proc Natl Acad Sci U S A. 2014;111:15940–5. doi: 10.1073/pnas.1415184111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li G, Weis RM. Covalent modification regulates ligand binding to receptor complexes in the chemosensory system of Escherichia coli. Cell. 2000;100:357–65. doi: 10.1016/s0092-8674(00)80671-6. [DOI] [PubMed] [Google Scholar]
- 44.Sourjik V, Berg HC. Functional interactions between receptors in bacterial chemotaxis. Nature. 2004;428:437–41. doi: 10.1038/nature02406. [DOI] [PubMed] [Google Scholar]
- 45.Pinas GE, Frank V, Vaknin A, Parkinson JS. The source of high signal cooperativity in bacterial chemosensory arrays. Proceedings of the National Academy of Sciences of the United States of America. 2016 doi: 10.1073/pnas.1600216113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frank V, Vaknin A. Prolonged stimuli alter the bacterial chemosensory clusters. Mol Microbiol. 2013;88:634–44. doi: 10.1111/mmi.12215. [DOI] [PubMed] [Google Scholar]
- 47.Coleman MD, Bass RB, Mehan RS, Falke JJ. Conserved glycine residues in the cytoplasmic domain of the aspartate receptor play essential roles in kinase coupling and on-off switching. Biochemistry. 2005;44:7687–95. doi: 10.1021/bi0501479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parkinson JS, Houts SE. Isolation and behavior of Escherichia coli deletion mutants lacking chemotaxis functions. J Bacteriol. 1982;151:106–13. doi: 10.1128/jb.151.1.106-113.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou Q, Ames P, Parkinson JS. Biphasic control logic of HAMP domain signalling in the Escherichia coli serine chemoreceptor. Mol Microbiol. 2011;80:596–611. doi: 10.1111/j.1365-2958.2011.07577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buron-Barral M, Gosink KK, Parkinson JS. Loss- and gain-of-function mutations in the F1-HAMP region of the Escherichia coli aerotaxis transducer Aer. J Bacteriol. 2006;188:3477–86. doi: 10.1128/JB.188.10.3477-3486.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parkinson JS. cheA, cheB, and cheC genes of Escherichia coli and their role in chemotaxis. J Bacteriol. 1976;126:758–70. doi: 10.1128/jb.126.2.758-770.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Slocum MK, Parkinson JS. Genetics of methyl-accepting chemotaxis proteins in Escherichia coli: null phenotypes of the tar and tap genes. Journal of Bacteriology. 1985;163:586–94. doi: 10.1128/jb.163.2.586-594.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.