

Abstract
Objective
To describe in this review how research using mouse models developed to study the Fragile X premutation (PM) and Fragile X-associated tremor/ataxia syndrome (FXTAS) have contributed to understanding these disorders. PM carriers bear an expanded CGG trinucleotide repeat on the Fragile X Mental Retardation 1 (FMR1) gene, and are at risk for developing the late-onset neurodegenerative disorder FXTAS.
Conclusions
Much has been learned about these genetic disorders from the development and study of mouse models. This includes new insights into the early cellular and molecular events that occur in PM carriers and in FXTAS, the presence of multi-organ pathology beyond the CNS, immunological dysregulation, unexpected synthesis of a potentially toxic peptide in FXTAS (i.e., FMRpolyG), and evidence that the disease process may be halted or reversed by appropriate molecular therapies given early in the course of disease.
Keywords: Fragile X premutation (PM), FXTAS, intranuclear inclusions, Fragile X mental retardation gene (FMR1), Fragile X mental retardation protein (FMRP)
Introduction
The Fragile X 1 gene (FMR1) is located on the X chromosome (i.e., long arm at position 27.3q) and codes for the Fragile X mental retardation protein (FMRP), an important protein for normal brain development and synaptic plasticity (R. Hagerman & Hagerman, 2013; Willemsen, Levenga, & Oostra, 2011). The FMR1 gene contains a DNA segment called a CGG trinucleotide that is repeated in most people between 5–44 times. However, the length of the CGG repeat tract can expand over generations in some families. Individuals in which the CGG repeat tract has expanded to between 55–200 are called Fragile X premutation carriers (PM), and they are at risk for developing the late onset neurodegenerative disorder Fragile X-associated tremor/ataxia syndrome (FXTAS). In FXTAS there is tremor/ataxia, brain atrophy, cognitive impairment and dementia in some individuals. Individuals with larger CGG repeat numbers over 200 are called full mutation carriers and develop Fragile X syndrome (FXS). In FXS the FMR1 gene is epigenetically silenced, no FMRP is produced, dendritic spine morphology in the neocortex is abnormal, and there are learning disabilities and cognitive impairments. Additional neurological and physiological findings including facial dysmorphologies and macroorchidism (Hinton, Brown, Wisniewski, & Rudelli, 1991; Santoro, Bray, & Warren, 2012).
The laboratory mouse (Mus musculus) has been crucial for studying human genetic diseases for decades, ranging from simple Mendelian-inherited diseases such as FXS, to complex polygenic disorders including autism. In the case of PM and FXTAS, several mouse models have been developed to study the underlying neurobiology and pathogenesis of the complex neurodegenerative disease (Berman et al., 2014; Berman & Willemsen, 2009; Willemsen et al., 2003). This review will focus on these mouse models and what has been learned from them about the Fragile X premutation (PM) and Fragile X-associated tremor/ataxia syndrome (FXTAS).
How is FXTAS modeled in mice?
Modeling human diseases in animals, and rodents in particular, can be challenging but such models have yielded important information about many neurological disorders, including FXS, Parkinson’s, Huntington’s and Alzheimer’s diseases (Nestler & Hyman, 2010). Typically mouse models of genetic disorders are created by either direct genetic manipulation (insertion or deletion of a DNA segment) or through the use of mutagenic drugs that cause changes in the organism’s DNA. In order for mouse models to be clinically relevant they must have construct validity. This means that they must exhibit the key molecular and cellular pathologies and symptoms present in patients of a particular disorder. However, mouse models rarely, if ever, completely model all aspects of human disease, and this is true for the existing mouse models of the PM and FXTAS. Even with this caveat, mouse models can provide important information about the natural history of a disease process, from embryo to senescence, due to the relatively short 1–2 year life-span of mice compared to human’s 70-plus year life-span. This allows for longitudinal studies in mice, as well as molecular studies at various levels, from cellular to organ systems that are not feasible in humans. Finally, animal models provide a platform for evaluating therapeutic strategies, including development of new drugs to improve function in neurological disorders such as FXTAS.
In order to study FXS a mouse model was originally generated by replacing the native repeat tract of 14 CGGs in the mouse Fmr1 gene with a DNA segment of human origin containing 98 CGG repeats (Bontekoe et al., 2001). These mice are referred to as CGG knock-in mice (CGG KI). The repeat expansion showed modest instability increasing in length over generations until mice were obtained with more than 200 CGG repeats, well within the range of CGG expansions that causes FXS (Bontekoe et al., 2001). However, these mice did not show the expected silencing of the Fmr1 gene, FMRP levels were in the near-normal range, and the severe cognitive deficits seen in FXS were also absent (Brouwer et al., 2007; Entezam et al., 2007; Van Dam et al., 2005). Shortly after the development of this mouse model it was recognized that some carriers of the Fragile X premutation (i.e., CGG repeat expansions between 55–200), first thought to be without pathology, developed a late onset tremor/ataxia syndrome evidence of neurodegenerative disease (i.e., brain atrophy, white matter disease). This neurodegenerative disorder was labeled Fragile X-associated tremor/ataxia syndrome (FXTAS), and was found to occur in approximately 40% of male and 11–18% of female PM carriers over the age of 50 (P. J. Hagerman & Hagerman, 2004; R. J. Hagerman et al., 2001). It was subsequently recognized that the CGG KI mouse carrying between 70–200 CGG repeats actually provides a valid and useful model of several features seen in PM carriers and those that go on to develop FXTAS, including elevated levels of expression of FMR1, intranuclear protein inclusions, as well as motor and cognitive deficits (Table 1). Since that time additional models of the PM and FXTAS have been generated and are discussed within this review (Berman et al., 2014; Entezam et al., 2007).
Table 1.
FXTAS compared to the CGG knock-in (KI) mouse model
| Pathology | Human FXTAS | CGG KI mouse |
|---|---|---|
| CGG trinucleotide repeat Length |
55–199 CGG repeats Repeat instability |
70–300 CGG repeats Moderate repeat instability |
| Elevated FMR1 mRNA | Increased 2–8 fold | Increased 1.5–3 fold |
| FMRP levels | Reduced in several brain regions | Reduced in several brain regions |
| Intranuclear protein inclusions | In neurons and astrocytes, Correlated with CGG length Frequency increases with age | In neurons and astrocytes Correlated with CGG length Frequency increases with age |
| FMRpolyG peptide | Found in intranuclear inclusions | Found in intranuclear inclusions |
| Motor impairments | Tremor/ataxia, postural sway, Parkinsonism | Impaired on Rotarod & Ladder Rung tasks |
| Cognitive Deficits | Poor working memory, anxiety, depression, social phobia | Spatial memory deficits, increased anxiety |
Neuropathology
Similar to other neurodegenerative diseases, FXTAS is associated with presence of protein inclusions observed in neuronal nuclei of both neurons and astrocytes throughout the brain (Figure 1). These inclusions stain for ubiquitin, the FMR1 messenger RNA (mRNA) bearing the CGG repeat expansions, and several proteins associated with the mRNA involved in regulation of DNA repair processes and control of transcription (Greco et al., 2006; Greco et al., 2002; Iwahashi et al., 2006; Sellier et al., 2013; Sellier et al., 2010). Indeed, the presence of ubiquitin-positive inclusions has been considered a hallmark histological marker of FXTAS (Greco et al., 2006). The role of these intranuclear inclusions in pathology is still unresolved, and while they may be directly involved in the pathology, it is also possible that they are not in themselves cytotoxic or damaging per se to the nervous system and serve mainly as a marker of some underlying disease process. However, the number of intranuclear inclusions in neurons and astrocytes is positively correlated with the length of the CGG repeat expansions, and is inversely correlated to age of death in FXTAS (Greco et al., 2006). Several groups have confirmed the presence of these intranuclear inclusions in neurons of various brain regions in the CGG KI mice, including the cerebellum, amygdala, cerebral cortex, hippocampus and hypothalamus (Brouwer, Huizer, et al., 2008; Brouwer et al., 2007; Brouwer, Willemsen, & Oostra, 2009; Willemsen et al., 2003). In addition to neurons, inclusions were also found in the astrocytes and Bergmann glia of the brain (Wenzel, Hunsaker, Greco, Willemsen, & Berman, 2010). The presence of ubiquitin-positive inclusions in particular brain regions may be correlated with some of the clinical features of FXTAS in humans (Greco et al., 2006; Willemsen et al., 2003). For example, Purkinje cell loss was observed in a CGG KI mouse line that appears to be associated with motor deficits (Entezam et al., 2007). Research carried out in parallel with FXTAS patients and the CGG KI mice has also demonstrated that FXTAS is a multi-organ disorder, in which the intranuclear inclusions in both humans and mice were observed in a variety of organ systems besides the brain, including the heart, kidney, thyroid and pituitary glands, helping to explain the myriad of symptoms seen in PM carriers in FXTAS (Hunsaker, Greco, et al., 2011).
Fig. 1.
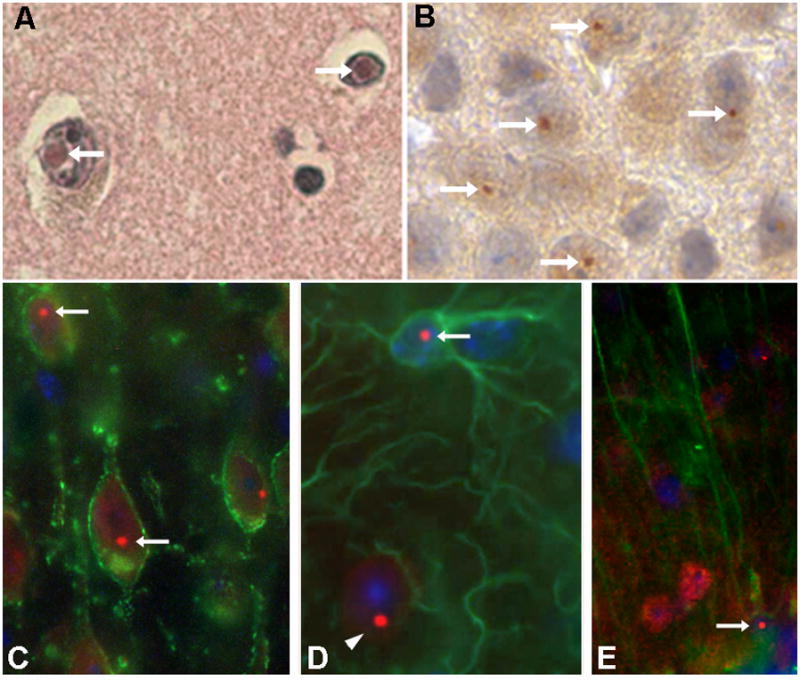
Intranuclear inclusions in neurons from human postmortem cortex (A) and CGG KI mouse cortex (B), stained with hematoxylin/eosin and diaminobenzidine, respectively. Immunofluorescent staining showing intranuclear inclusions (red fluorescent round structures) in CGG KI mouse neurons (C), astrocytes (D) and cerebellar Bergmann glia (E). Arrows point to inclusions, and arrow head in D shows an inclusions in an adjacent neuron. [Adapted from Wenzel, et al., 2010].
Molecular Findings in FXTAS and Mouse Models
Carriers of the PM and FXTAS patients also show a unique molecular pathology in blood, skin and brain. Specifically, they have a 3–8 fold increase in the levels of FMR1 mRNA suggesting increased transcription of the FMR1 gene due to some loss of regular transcriptional control (Tassone et al., 2007; Tassone et al., 2000). This is important because several “RNA binding” proteins have been found in the intranuclear inclusions apparently bound to the FMR1 mRNA. This sequestration of important proteins for DNA repair, transcriptional control and processing of microRNAs (miRNA) has been suggested to be the primary, or one of the primary molecular events underlying neurological impairments in PM carriers and the cell loss and brain atrophy in FXTAS (Iwahashi et al., 2006; Sellier et al., 2013; Sellier et al., 2010; Todd et al., 2013).
However, a competing hypothesis has recently emerged with the discovery that a potentially toxic peptide (FMRpolyG) may be produced in neurons by a process called repeat-associated non-AUG translation (RAN) translation (Todd et al., 2013). In RAN translation the expanded CGG trinucleotide repeat segment (i.e., …CGGCGGCGGCGG…), previously thought to be untranslated, may actually lead to the production of a potentially toxic polyglycine-containing peptide called FMRpolyG (Todd et al., 2013). Synthesis of this peptide occurs, in part, because the polyribosome uses an alternative start-site for translation on the FMR1 mRNA (i.e., non-AUG start site) resulting in recognition of an expanded GGC trinucleotide segment (i.e., …CGGCGGCGG… recognized as …GGCGGCGGC…). Because the trinucleotide GGC codes for glycine, the novel peptide contains a stretch of polyglycine along with additional flanking peptides. Interestingly, a second shift in translation also appears to occur in FXTAS, albeit at low levels, so that instead of GGC, a GCG segment (GCG codon for alanine) is translated producing a polyalanine-containing peptide called FMRpolyA. The FMRpolyG protein has been found to accumulate in the intranuclear inclusions in both FXTAS patients and CGG KI mice (Buijsen et al., 2014; Buijsen et al., 2016; Todd et al., 2013). The contributions that RAN translation and this unexpected polyglycine peptide make to disease pathology is under active investigation at the present time. If the polyglycine peptide plays an important role in FXTAS pathology it may be an important new target for the development of drug therapies.
In spite of an increase in levels of FMR1 mRNA, expression of FMRP is slightly to moderately decreased (i.e., 10–30%) from normal due to inefficient translation of the mRNA at the ribosome due to the present of the expanded CGG repeat in the FMR1 mRNA (Ludwig et al., 2014). Research from the CGG KI mouse models has made it clear that the greatest decrease in FMRP is seen with larger CGG repeat expansions. This provides support for the hypothesis of inefficient translation of the Fmr1 mRNA. Whether or not the decrease in FMRP contributes to pathology is currently unknown.
Mitochondrial dysfunction (MD) has also been found in FXTAS patients and to a lesser extent in PM carriers (Ross-Inta et al., 2010). Mitochondrial function is of particular interest because of the overlap in symptomatology between FXTAS and mitochondrial respiratory enzyme chain enzyme deficiencies (MRCDs), including gait ataxia, white matter disease, autonomic dysfunction, weakness and neuropsychiatric disorders. Mitochondrial studies carried out in cultured skin fibroblasts from FXTAS patients and PM carriers show decreased oxygen uptake rates, uncoupling between electron transport and ATP synthesis, and decreased levels of important mitochondrial proteins for ATP synthesis (ATPase β-subunit), electron transport (cytochrome c oxidase subunit IV) and antioxidant defense (MnSOD), but no change in the density of mitochondria. Similar results were found in postmortem cortical tissue from FXTAS patients (Ross-Inta et al., 2010). The severity of MD was directly related to the length of the CGG repeat tract, and the present of mitochondrial pathology in PM carriers suggests that MD may be an early event that precedes the appearance of FXTAS symptomatology. Interestingly, it has been suggested that MD in PM carriers may predispose them to other diseases involving mitochondria including Friedreich’s ataxia, Parkinson’s disease and Alzheimer’s disease (Napoli et al., 2010). Similar studies have not been reported in mouse fibroblasts from CGG KI mice. However, mitochondria in cultured hippocampus neurons from CGG KI mice have found increased oxygen consumption, and decreased density and movement of mitochondria along neurites (Kaplan et al., 2012). In this case, it is unclear why the results for MD differ between studies carried out in human versus CGG KI mouse tissues, but they point out the fact that results in mouse models do not always correspond directly or simply to results from human studies.
Translating Mouse Behavior to Human Symptoms
In addition to construct validity, a mouse model must also exhibit the key symptoms or behavioral changes associated with that disorder, this is referred to as face validity (Nestler & Hyman, 2010). By screening with a variety of behavioral assays, researchers are able to identify altered behaviors in mice which can then be correlated to the disease-related symptoms observed in patients. While not all PM carriers develop FXTAS, CGG KI mice have been shown to exhibit several behavioral symptoms of FXTAS including late-onset ataxia, memory impairments and sensorimotor gating deficits.
The CGG KI mice show deficits in motor learning and motor coordination based on performances in behavior tasks which are designed to assess the types of sensorimotor impairments observed in FXTAS patients (Diep et al., 2012; R. J. Hagerman et al., 2001; Hall, Hagerman, Hagerman, Jacquemont, & Leehey, 2006; von Leden et al., 2014). Moreover, reports suggest that these motor deficits may worsen with age and may also depend on the length of the CGG repeat expansion in the CGG KI mice (Diep et al., 2012; Hashem et al., 2009; Hunsaker, von Leden, et al., 2011; Van Dam et al., 2005). For example, CGG KI mice show poor foot placement (i.e., foot slips) on the “ladder rung” visuomotor test of ataxia in which they are required to cross a horizontal ladder (Hunsaker, von Leden, et al., 2011). Both young (< 6 months) and adult (> 6 months) CGG KI mice made significantly more foot placement faults than normal control mice, with larger repeat lengths (i.e., high versus low) associated with greater gait impairments in this test. FXTAS is associated with memory problems and executive function deficits, which may even progress to dementia in some patients (Cornish et al., 2008; Grigsby et al., 2008; P. J. Hagerman & Hagerman, 2004). The CGG KI mice also exhibit signs of cognitive impairments based on their performance in spatial learning and memory tasks, such as the Morris Water maze (Van Dam et al., 2005). Additional hippocampal-dependent cognitive deficits have also been reported including impairments in remembering the temporal order in which odors are presented, and enhanced sensitivity to interference in novelty detection tests (Borthwell, Hunsaker, Willemsen, & Berman, 2012). Consistent with FXTAS patients, the cognitive deficits present in the CGG KI mice were shown to also be age-dependent in a spatial processing task (Hunsaker, Wenzel, Willemsen, & Berman, 2009). Interestingly, this deficit in hippocampal-dependent learning and memory is also correlated with CGG repeat length (Borthwell et al., 2012; Hunsaker, Goodrich-Hunsaker, Willemsen, & Berman, 2010; Hunsaker, Kim, Willemsen, & Berman, 2012).
Sensorimotor gating is a neuronal process for filtering out unnecessary stimuli or information in the brain (Braff, Geyer, & Swerdlow, 2001). Deficits in sensorimotor gating measured by prepulse inhibition (PPI) of the acoustic startle response are impaired in male carriers of the PM and are more prominent in patients who have developed FXTAS (Schneider et al., 2012). Similarly, a recent study in CGG KI mice showed altered baseline acoustic startle responses and an age-dependent PPI deficits using a PPI procedure resembling that used in humans (Renoux et al., 2014).
Anxiety disorders are seen in a high proportion of individuals with FXS, and recent evidence shows that PM carriers are also at risk for anxiety disorders with a higher rate of anxiety disorder than expected in the general population (Bourgeois et al., 2007; Bourgeois et al., 2011; Cordeiro, Abucayan, Hagerman, Tassone, & Hessl, 2015). The development of anxiety-related behaviors has also been reported in CGG KI mice that show an age-dependent increase in anxiety in the open-field locomotor test. Specifically, at 72 weeks of age CGG KI mice spend less time exploring the center of an open compared to wildtype control mice, but do not differ from controls at 20 or 52 weeks of age, suggesting a late-onset increase in anxiety (Van Dam et al., 2005). It should be noted that evidence for decreased anxiety has been reported in the open-field test in a less frequently used CGG KI mouse model developed at the National Institutes of Health (NIH) (Qin et al., 2011). The discrepancy in anxiety behaviors seen between studies may be due to the very large decrease in brain levels of FMRP in the NIH mouse model (i.e., >70%) compared to that found in the CGG KI mouse model used by Van Dam, et al. (i.e., <50%). Serum corticosterone levels are also elevated in response to a mild stressor in 100 wk old CGG KI mice compared to control mice suggesting that sensitization of the HPA axis may contribute to the development of an anxiety disorder in response to stress (Brouwer, Severijnen, et al., 2008). It is interesting to note that intranuclear ubiquitin-positive inclusions are found in cells in both the pituitary and adrenal glands of PM carriers, and such pathology may contribute to sensitization of stress-related responses (Hunsaker, Greco, et al., 2011).
What have mouse models taught us about FXTAS?
As described above, the CGG KI mouse models much of the pathology seen in PM and FXTAS. In addition, several new findings have emerged from studies in the mouse models that have significantly altered current views on the development, etiology and reversibility of pathology as describe in this section.
Pathology is Not Limited to the Brain
Research using these established and validated mouse models of PM has shed light on our understanding of the underlying pathology and development of FXTAS. By immunostaining a variety of dissected tissues from CGG KI mice, Hunsaker et al. identified wide-spread cellular pathology that was not limited to the central nervous system (Hunsaker, Greco, et al., 2011). The hallmark FXTAS ubiquitin-positive intranuclear inclusions were observed in heart, pineal gland, colon, adrenal gland, thyroid, pancreas and pituitary tissue from dissected CGG KI mice. Postmortem tissue samples from FXTAS patients also showed the intranuclear inclusions in tissues from the heart, pineal, colon, kidney, thyroid, pancreas, adrenal gland, esophagus and testes. The presence of inclusions in different tissue types may play a role in the etiology of various co-morbidities associated with FXTAS, including gastrointestinal symptoms, peripheral neuropathy, neuroendocrine dysfunction, and cardiac arrhythmias.
The Immune System is Altered in the Fragile X Premutation
In addition to the numerous pathologies associated with FMR1 CGG-expansions, immunological issues have been reported in carriers as well (Careaga et al., 2014; Marek et al., 2012; Winarni et al., 2012). This has been particularly evident in female PM carriers, who were found to be at higher risk for developing autoimmune conditions when compared with controls (Winarni et al., 2012). Based on a survey study of 344 PM carriers and 72 controls, the incidence of immune-mediated disorders (IMD) was 44.8% in all carriers studied (n=344) versus 27.8% in controls (n=72). In older women with FXTAS the incidence was 72.7% versus 46. 5% in carriers without FXTAS and 31.58% in controls. The associated autoimmune conditions varied, but risk was mostly associated with systemic autoinflammatory conditions such as fibromyalgia and autoimmune thyroid disease (Winarni et al., 2012). No similar risk has been identified in male carriers, but this may be related to gender disparity seen in many autoimmune diseases, which are highly biased towards females (Ngo, Steyn, & McCombe, 2014). However, the discrepancy may also have resulted from insufficient power in the study by Winarni, et al (Winarni et al., 2012). A reduction in cytokine production has also been reported in female premutation carriers compared to controls. This included reductions in the cytokines IL-12(p40) and IL-1α, MCP-1 and INFγ, IL-1α and TNFα in unstimulated monocytes and peripheral blood lymphocytes, respectively. Subtle immune deficiencies were observed in both female and male CGG KI mice that were similar to those seen in human female PM carriers, suggesting that human male carriers may be immunologically affected as well (Careaga et al., 2014). This hypothesis is supported by observations by Marek et al. who found that male carriers diagnosed with FXTAS show elevated production of the anti-inflammatory cytokine IL-10 (Marek et al., 2012). Given the findings in both human and mice, PM carriers and FXTAS patients of both genders should be monitored with extra attention for the development or presence of autoimmune disorders.
Pathological Processes Begin Early in Development
In neurodegenerative disorders, such as Alzheimer’s disease, the cellular pathologies and patient-related symptoms are typically thought to manifest later in life. However, developmental studies of CGG KI mice have provided evidence for an earlier disease onset in FXTAS than was previously thought to occur based on molecular, histological and behavioral studies. First, elevations of Fmr1 mRNA levels seen in adult PM carriers and in FXTAS patients was detected throughout development from 1 to 72 weeks of age in the CGG KI mice (Willemsen et al., 2003). This early increase in Fmr1 mRNA may lead to developmental consequences in PM carriers which may, in turn, lead to the pathogenesis of FXTAS. For example, although gross brain morphology is relatively normal, there are fewer dendritic branches of reduced length and abnormal synaptic morphology in the cortex and hippocampus of CGG KI mice (Berman, Murray, Arque, Hunsaker, & Wenzel, 2012; Chen, 2010). Additionally, younger mice have smaller and fewer total numbers of intranuclear inclusions in neurons compared with older CGG KI mice. This gradual increase in size and density of inclusions over the lifetime of the mouse may parallel the progressive development of FXTAS in PM carriers (Jacquemont et al., 2004). Finally, embryonic development of the cortex in CGG KI mice is abnormal, with aberrant migration and fewer numbers of neuronal precursor cells within the developing neocortex (Cunningham et al., 2011). These observations are important because recognition that disease-related processes and cellular pathologies may occur before the clinical symptoms of FXTAS are seen, suggests that there may be critical time points earlier in development when drug treatments would be maximally effective in halting or preventing disease in PM carriers.
Reversibility
Exciting new evidence for reversibility of pathology in the PM and FXTAS has come from recent studies in a new “inducible” mouse model in which the disease process can be induced anytime during development under experimental control. Specifically, a CGG repeat expansion can be activated in these mice, and then turned off by adding or removing the drug doxycycline to their drinking water, respectively (i.e., dox-inducible mice). This new mouse model allows for studies to determine when during development activation of the CGG repeat expansions leads to full expression of disease, and whether the disease progression can be halted, or possibly reversed by stopping expression of the CGG repeat by removing doxycycline at various time points during the disease. In a recent study it was found that activating the expression of a CGG repeat expansion of 90 CGGs in the brains of dox-inducible mice resulted in the formation of the hallmark FXTAS ubiquitin-positive intranuclear inclusions in the brain within 8 weeks, with high expression in the hippocampus and cerebellum. Further study of the inclusion formation identified that expression of the expanded CGG repeat over a longer period of time led to an increase in the number as well as the size of the inclusions. Importantly, stopping expression of the expanded CGG90 repeat followed by a period without expression of 12 weeks, resulted in a significant decrease of both the number and size of the inclusions. However, if the expanded CGG repeat was expressed for a longer period of 12 or 16 weeks no reversibility was observed, but only a stop in further disease progression (Hukema et al., 2015). These neuropathological results were confirmed by testing the animals on the optokinetic reflex, a cerebellar-dependent movement of the eyes in response to head movement. This eye-reflex was impaired by expanded CGG repeat expression when animals were on doxycycline and the impairment worsened over time. However, further progression could be halted by taking the animals off doxycycline after 8 weeks, thereby halting expression of the expanded CGG repeat. These exciting results show that the appropriate pharmacological of gene-targeted therapy, if given early in development, could prevent or reverse some brain pathology associated with the PM (i.e., intranuclear inclusions), and may lessen or prevent some neurological deficits from developing.
Conclusions and Future Directions
Mouse models of the PM and FXTAS have contributed substantially to our understanding of the molecular mechanisms underlying the disease process in these disorders, and have provide new information about the accompanying neurobehavioral deficits. Mouse models have shown that the disease process has its beginning early in development, possibly in the embryo, and that the elevated Fmr1 mRNA and reduced FMRP can be seen early in development. Ongoing studies on the FMRpolyG peptide may determine that this molecular is a new potential target for new therapeutic drugs to improve neurological function in PM carriers and FXTAS patients. Finally, the exciting new evidence of reversibility of pathology opens the way for development of new gene-based therapeutic strategies. Mouse models are likely to play an important role in the development and preclinical screening of new therapeutics for FXTAS.
Acknowledgments
We wish to acknowledge the helpful comments and suggestions from Rob Willemsen and Renate Hukema during the preparation of this manuscript. This work was supported by NIH-NINDS grant number R01 NS079775.
Footnotes
Disclosures: None of the authors have any disclosures concerning this research or the writing of this manuscript.
Contributor Information
Molly M. Foote, Department of Neurological Surgery, University of California Davis, Davis, CA 95618 (239-699-4229)
Milo Careaga, Dept. Psychiatry and UC Davis M.I.N.D. Institute, University of California Davis, Davis, CA 95618 (916-703-0415).
Robert F. Berman, Dept. Neurological Surgery and the UC Davis M.I.N.D. Institute, University of California Davis, Davis, CA, 95618.
References
- Berman RF, Buijsen RA, Usdin K, Pintado E, Kooy F, Pretto D, … Hukema RK. Mouse models of the fragile X premutation and fragile X-associated tremor/ataxia syndrome. J Neurodev Disord. 2014;6(1):25. doi: 10.1186/1866-1955-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman RF, Murray KD, Arque G, Hunsaker MR, Wenzel HJ. Abnormal dendrite and spine morphology in primary visual cortex in the CGG knock-in mouse model of the fragile X premutation. Epilepsia. 2012;53(Suppl 1):150–160. doi: 10.1111/j.1528-1167.2012.03486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman RF, Willemsen R. Mouse models of fragile x-associated tremor ataxia. J Investig Med. 2009;57(8):837–841. doi: 10.231/JIM.0b013e3181af59d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bontekoe CJ, Bakker CE, Nieuwenhuizen IM, van der Linde H, Lans H, de Lange D, … Oostra BA. Instability of a (CGG)98 repeat in the Fmr1 promoter. Hum Mol Genet. 2001;10(16):1693–1699. doi: 10.1093/hmg/10.16.1693. [DOI] [PubMed] [Google Scholar]
- Borthwell RM, Hunsaker MR, Willemsen R, Berman RF. Spatiotemporal processing deficits in female CGG KI mice modeling the fragile X premutation. Behav Brain Res. 2012;233(1):29–34. doi: 10.1016/j.bbr.2012.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeois JA, Cogswell JB, Hessl D, Zhang L, Ono MY, Tassone F, … Hagerman RJ. Cognitive, anxiety and mood disorders in the fragile X-associated tremor/ataxia syndrome. Gen Hosp Psychiatry. 2007;29(4):349–356. doi: 10.1016/j.genhosppsych.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeois JA, Seritan AL, Casillas EM, Hessl D, Schneider A, Yang Y, … Hagerman RJ. Lifetime prevalence of mood and anxiety disorders in fragile X premutation carriers. J Clin Psychiatry. 2011;72(2):175–182. doi: 10.4088/JCP.09m05407blu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) 2001;156(2–3):234–258. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- Brouwer JR, Huizer K, Severijnen LA, Hukema RK, Berman RF, Oostra BA, Willemsen R. CGG-repeat length and neuropathological and molecular correlates in a mouse model for fragile X-associated tremor/ataxia syndrome. J Neurochem. 2008;107(6):1671–1682. doi: 10.1111/j.1471-4159.2008.05747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer JR, Mientjes EJ, Bakker CE, Nieuwenhuizen IM, Severijnen LA, Van der Linde HC, … Willemsen R. Elevated Fmr1 mRNA levels and reduced protein expression in a mouse model with an unmethylated Fragile X full mutation. Exp Cell Res. 2007;313(2):244–253. doi: 10.1016/j.yexcr.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer JR, Severijnen E, de Jong FH, Hessl D, Hagerman RJ, Oostra BA, Willemsen R. Altered hypothalamus-pituitary-adrenal gland axis regulation in the expanded CGG-repeat mouse model for fragile X-associated tremor/ataxia syndrome. Psychoneuroendocrinology. 2008;33(6):863–873. doi: 10.1016/j.psyneuen.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer JR, Willemsen R, Oostra BA. The FMR1 gene and fragile X-associated tremor/ataxia syndrome. Am J Med Genet B Neuropsychiatr Genet. 2009;150B(6):782–798. doi: 10.1002/ajmg.b.30910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buijsen RA, Sellier C, Severijnen LA, Oulad-Abdelghani M, Verhagen RF, Berman RF, … Hukema RK. FMRpolyG-positive inclusions in CNS and non-CNS organs of a fragile X premutation carrier with fragile X-associated tremor/ataxia syndrome. Acta Neuropathol Commun. 2014;2:162. doi: 10.1186/s40478-014-0162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buijsen RA, Visser JA, Kramer P, Severijnen EA, Gearing M, Charlet-Berguerand N, … Hukema RK. Presence of inclusions positive for polyglycine containing protein, FMRpolyG, indicates that repeat-associated non-AUG translation plays a role in fragile X-associated primary ovarian insufficiency. Hum Reprod. 2016;31(1):158–168. doi: 10.1093/humrep/dev280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Careaga M, Rose D, Tassone F, Berman RF, Hagerman R, Ashwood P. Immune dysregulation as a cause of autoinflammation in fragile X premutation carriers: link between FMRI CGG repeat number and decreased cytokine responses. PLoS One. 2014;9(4):e94475. doi: 10.1371/journal.pone.0094475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Tassone F, Berman RF, Hagerman PM, Hagerman RJ, Willemsen R, Pessah IN. Murine hippocampal neurons expressing Fmr1 gene premutation show early developmental deficits and late degeneration. Human Molecular Genetics. 2010;19(1):196–208. doi: 10.1093/hmg/ddp479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro L, Abucayan F, Hagerman R, Tassone F, Hessl D. Anxiety disorders in fragile X premutation carriers: Preliminary characterization of probands and non-probands. Intractable Rare Dis Res. 2015;4(3):123–130. doi: 10.5582/irdr.2015.01029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish KM, Li L, Kogan CS, Jacquemont S, Turk J, Dalton A, … Hagerman PJ. Age-dependent cognitive changes in carriers of the fragile X syndrome. Cortex. 2008;44(6):628–636. doi: 10.1016/j.cortex.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CL, Martinez Cerdeno V, Navarro Porras E, Prakash AN, Angelastro JM, Willemsen R, … Noctor SC. Premutation CGG-repeat expansion of the Fmr1 gene impairs mouse neocortical development. Hum Mol Genet. 2011;20(1):64–79. doi: 10.1093/hmg/ddq432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diep AA, Hunsaker MR, Kwock R, Kim K, Willemsen R, Berman RF. Female CGG knock-in mice modeling the fragile X premutation are impaired on a skilled forelimb reaching task. Neurobiol Learn Mem. 2012;97(2):229–234. doi: 10.1016/j.nlm.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entezam A, Biacsi R, Orrison B, Saha T, Hoffman GE, Grabczyk E, … Usdin K. Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene. 2007;395(1–2):125–134. doi: 10.1016/j.gene.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco CM, Berman RF, Martin RM, Tassone F, Schwartz PH, Chang A, … Hagerman PJ. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS) Brain. 2006;129(Pt 1):243–255. doi: 10.1093/brain/awh683. [DOI] [PubMed] [Google Scholar]
- Greco CM, Hagerman RJ, Tassone F, Chudley AE, Del Bigio MR, Jacquemont S, … Hagerman PJ. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain. 2002;125(Pt 8):1760–1771. doi: 10.1093/brain/awf184. [DOI] [PubMed] [Google Scholar]
- Grigsby J, Brega AG, Engle K, Leehey MA, Hagerman RJ, Tassone F, … Reynolds A. Cognitive profile of fragile X premutation carriers with and without fragile X-associated tremor/ataxia syndrome. Neuropsychology. 2008;22(1):48–60. doi: 10.1037/0894-4105.22.1.48. [DOI] [PubMed] [Google Scholar]
- Hagerman PJ, Hagerman RJ. Fragile X-associated tremor/ataxia syndrome (FXTAS) Ment Retard Dev Disabil Res Rev. 2004;10(1):25–30. doi: 10.1002/mrdd.20005. [DOI] [PubMed] [Google Scholar]
- Hagerman R, Hagerman P. Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol. 2013;12(8):786–798. doi: 10.1016/S1474-4422(13)70125-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, … Hagerman PJ. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57(1):127–130. doi: 10.1212/wnl.57.1.127. [DOI] [PubMed] [Google Scholar]
- Hall DA, Hagerman RJ, Hagerman PJ, Jacquemont S, Leehey MA. Prevalence of FMR1 repeat expansions in movement disorders. A systematic review. Neuroepidemiology. 2006;26(3):151–155. doi: 10.1159/000091656. [DOI] [PubMed] [Google Scholar]
- Hashem V, Galloway JN, Mori M, Willemsen R, Oostra BA, Paylor R, Nelson DL. Ectopic expression of CGG containing mRNA is neurotoxic in mammals. Hum Mol Genet. 2009;18(13):2443–2451. doi: 10.1093/hmg/ddp182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton VJ, Brown WT, Wisniewski K, Rudelli RD. Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet. 1991;41(3):289–294. doi: 10.1002/ajmg.1320410306. [DOI] [PubMed] [Google Scholar]
- Hukema RK, Buijsen RA, Schonewille M, Raske C, Severijnen LA, Nieuwenhuizen-Bakker I, … Willemsen R. Reversibility of neuropathology and motor deficits in an inducible mouse model for FXTAS. Hum Mol Genet. 2015;24(17):4948–4957. doi: 10.1093/hmg/ddv216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsaker MR, Goodrich-Hunsaker NJ, Willemsen R, Berman RF. Temporal ordering deficits in female CGG KI mice heterozygous for the fragile X premutation. Behav Brain Res. 2010;213(2):263–268. doi: 10.1016/j.bbr.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsaker MR, Greco CM, Spath MA, Smits AP, Navarro CS, Tassone F, … Hukema RK. Widespread non-central nervous system organ pathology in fragile X premutation carriers with fragile X-associated tremor/ataxia syndrome and CGG knock-in mice. Acta Neuropathol. 2011;122(4):467–479. doi: 10.1007/s00401-011-0860-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsaker MR, Kim K, Willemsen R, Berman RF. CGG trinucleotide repeat length modulates neural plasticity and spatiotemporal processing in a mouse model of the fragile X premutation. Hippocampus. 2012;22(12):2260–2275. doi: 10.1002/hipo.22043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsaker MR, von Leden RE, Ta BT, Goodrich-Hunsaker NJ, Arque G, Kim K, … Berman RF. Motor deficits on a ladder rung task in male and female adolescent and adult CGG knock-in mice. Behav Brain Res. 2011;222(1):117–121. doi: 10.1016/j.bbr.2011.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsaker MR, Wenzel HJ, Willemsen R, Berman RF. Progressive spatial processing deficits in a mouse model of the fragile X premutation. Behav Neurosci. 2009;123(6):1315–1324. doi: 10.1037/a0017616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahashi CK, Yasui DH, An HJ, Greco CM, Tassone F, Nannen K, … Hagerman PJ. Protein composition of the intranuclear inclusions of FXTAS. Brain. 2006;129(Pt 1):256–271. doi: 10.1093/brain/awh650. [DOI] [PubMed] [Google Scholar]
- Jacquemont S, Farzin F, Hall D, Leehey M, Tassone F, Gane L, … Hagerman RJ. Aging in individuals with the FMR1 mutation. Am J Ment Retard. 2004;109(2):154–164. doi: 10.1352/0895-8017(2004)109<154:AIIWTF>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan ES, Cao Z, Hulsizer S, Tassone F, Berman RF, Hagerman PJ, Pessah IN. Early mitochondrial abnormalities in hippocampal neurons cultured from Fmr1 pre-mutation mouse model. J Neurochem. 2012;123(4):613–621. doi: 10.1111/j.1471-4159.2012.07936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig AL, Espinal GM, Pretto DI, Jamal AL, Arque G, Tassone F, … Hagerman PJ. CNS expression of murine fragile X protein (FMRP) as a function of CGG-repeat size. Hum Mol Genet. 2014;23(12):3228–3238. doi: 10.1093/hmg/ddu032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek D, Papin S, Ellefsen K, Niederhauser J, Isidor N, Ransijn A, … Tanackovic G. Carriers of the fragile X mental retardation 1 (FMR1) premutation allele present with increased levels of cytokine IL-10. J Neuroinflammation. 2012;9:238. doi: 10.1186/1742-2094-9-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli E, Ross-Inta C, Wong S, Omanska-Klusek A, Barrow C, Iwahashi C, … Giulivi C. Altered zinc transport disrupts mitochondrial protein processing/import in fragile X-associated tremor/ataxia syndrome. Hum Mol Genet. 2010;20(15):3079–3092. doi: 10.1093/hmg/ddr211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci. 2010;13(10):1161–1169. doi: 10.1038/nn.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo ST, Steyn FJ, McCombe PA. Gender differences in autoimmune disease. Front Neuroendocrinol. 2014;35(3):347–369. doi: 10.1016/j.yfrne.2014.04.004. [DOI] [PubMed] [Google Scholar]
- Qin M, Entezam A, Usdin K, Huang T, Liu ZH, Hoffman GE, Smith CB. A mouse model of the fragile X premutation: effects on behavior, dendrite morphology, and regional rates of cerebral protein synthesis. Neurobiol Dis. 2011;42(1):85–98. doi: 10.1016/j.nbd.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renoux AJ, Sala-Hamrick KJ, Carducci NM, Frazer M, Halsey KE, Sutton MA, … Todd PK. Impaired sensorimotor gating in Fmr1 knock out and Fragile X premutation model mice. Behav Brain Res. 2014;267:42–45. doi: 10.1016/j.bbr.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Inta C, Omanska-Klusek A, Wong S, Barrow C, Garcia-Arocena D, Iwahashi C, … Giulivi C. Evidence of mitochondrial dysfunction in fragile X-associated tremor/ataxia syndrome. Biochem J. 2010;429(3):545–552. doi: 10.1042/BJ20091960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro MR, Bray SM, Warren ST. Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol. 2012;7:219–245. doi: 10.1146/annurev-pathol-011811-132457. [DOI] [PubMed] [Google Scholar]
- Schneider A, Ballinger E, Chavez A, Tassone F, Hagerman RJ, Hessl D. Prepulse inhibition in patients with fragile X-associated tremor ataxia syndrome. Neurobiol Aging. 2012;33(6):1045–1053. doi: 10.1016/j.neurobiolaging.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Freyermuth F, Tabet R, Tran T, He F, Ruffenach F, … Charlet-Berguerand N. Sequestration of DROSHA and DGCR8 by Expanded CGG RNA Repeats Alters MicroRNA Processing in Fragile X-Associated Tremor/Ataxia Syndrome. Cell Rep. 2013 doi: 10.1016/j.celrep.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, … Charlet-Berguerand N. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. Embo J. 2010;29(7):1248–1261. doi: 10.1038/emboj.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F, Beilina A, Carosi C, Albertosi S, Bagni C, Li L, … Hagerman PJ. Elevated FMR1 mRNA in premutation carriers is due to increased transcription. Rna. 2007;13(4):555–562. doi: 10.1261/rna.280807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, Hagerman PJ. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet. 2000;66(1):6–15. doi: 10.1086/302720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, … Paulson HL. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron. 2013;78(3):440–455. doi: 10.1016/j.neuron.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dam D, Errijgers V, Kooy RF, Willemsen R, Mientjes E, Oostra BA, De Deyn PP. Cognitive decline, neuromotor and behavioural disturbances in a mouse model for fragile-X-associated tremor/ataxia syndrome (FXTAS) Behav Brain Res. 2005;162(2):233–239. doi: 10.1016/j.bbr.2005.03.007. [DOI] [PubMed] [Google Scholar]
- von Leden RE, Curley LC, Greenberg GD, Hunsaker MR, Willemsen R, Berman RF. Reduced activity-dependent protein levels in a mouse model of the fragile X premutation. Neurobiol Learn Mem. 2014;109:160–168. doi: 10.1016/j.nlm.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel HJ, Hunsaker MR, Greco CM, Willemsen R, Berman RF. Ubiquitin-positive intranuclear inclusions in neuronal and glial cells in a mouse model of the fragile X premutation. Brain Res. 2010;1318:155–166. doi: 10.1016/j.brainres.2009.12.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen R, Hoogeveen-Westerveld M, Reis S, Holstege J, Severijnen LA, Nieuwenhuizen IM, … Oostra BA. The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum Mol Genet. 2003;12(9):949–959. doi: 10.1093/hmg/ddg114. [DOI] [PubMed] [Google Scholar]
- Willemsen R, Levenga J, Oostra B. CGG repeat in the FMR1 gene: size matters. Clin Genet. 2011;80(3):214–225. doi: 10.1111/j.1399-0004.2011.01723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winarni TI, Chonchaiya W, Sumekar TA, Ashwood P, Morales GM, Tassone F, … Hagerman RJ. Immune-mediated disorders among women carriers of fragile X premutation alleles. Am J Med Genet A. 2012;158A(10):2473–2481. doi: 10.1002/ajmg.a.35569. [DOI] [PMC free article] [PubMed] [Google Scholar]