

Abstract
Cirrhosis of the liver is associated with increased fucosylation of proteins in the plasma. We describe a data independent (DIA) strategy for comparative analysis of the site-specific glycoforms of plasma glycoproteins. A library of 161 glycoforms of 25 N-glycopeptides was established by data dependent LC-MS/MS analysis of a tryptic digest of 14 human protein groups retained on a multiple affinity removal column. The collision induced dissociation conditions were adjusted to maximize the yield of selective Y-ions which were quantified by a data-independent mass spectrometry workflow using a 10 Da acquisition window. Using this workflow, we quantified 125 glycoforms of 25 glycopeptides, covering 10 of the 14 proteins, without any further glycopeptide enrichment. Comparison of the proteins in the plasma of healthy controls and cirrhotic patients shows an average 1.5 fold increase in the fucosylation of bi-antennary glycoforms and 3 fold increase in the fucosylation of tri- and tetra- antennary glycoforms. These results show that the adjusted glycopeptide DIA workflow using soft collision-induced fragmentation of glycopeptides is suitable for site specific analysis of protein glycosylation in complex mixtures of analytes without glycopeptide enrichment.
Keywords: Data independent analysis, N-glycopeptide, fucosylation, GP-SWATH
Introduction
Glycosylation of proteins is an integral part of life where diverse classes of glycans carry out distinct functions (1;2). Heterogeneity of this common modification makes its analysis challenging (3–5). Recent innovations facilitate glycoproteomic research (6;7) but, in spite of these advances, properties of glycopeptides complicate detection and quantification by MS (8;9). Glycoproteomic studies frequently benefit from independent analysis of detached glycans because full structural characterization of glycans is difficult to complete on glycopeptides (10–12). Similarly, proteomic analyses are complicated by the attached glycans and most studies of glycoproteins are carried out after de-glycosylation (13–15). We need to study site specific glycoforms and quantify their changes in the context of diseases to understand the structure-function relationships that tie glycoproteins to pathophysiological processes (16–19). However, methods for quantitative comparisons of site-specific protein glycoforms have only begun to emerge (9). While the comparative analysis of proteins by data independent acquisition (DIA) has improved the efficiency of quantitative proteomics analyses (20;21), applications of DIA methods to the analysis of posttranslational modifications are limited and virtually non-existent for glycoproteins (22). In this paper, we introduce a workflow for DIA analysis of N-glycopeptides under soft CID fragmentation conditions and document applicability of the glycopeptide DIA (Glycopeptide SWATH or GP-SWATH) workflow to the comparative analysis of fucosylated complex glycoforms of ten abundant plasma glycoprotein groups in the context of liver cirrhosis.
Patients, Materials, and Methods
Study population
Data dependent analysis of the N-glycosylation of major serum glycoproteins was carried out using pooled samples of cirrhotic patients and healthy individuals enrolled in collaboration with the Department of Hepatology and Liver Transplantation, Georgetown University Hospital, Washington, D.C. under protocols approved by the Institutional Review Board as described previously (23). All of the cirrhotic patients had chronic hepatitis C viral infection as the primary diagnosis. Blood samples were collected using EDTA Vacutainer tubes (BD Diagnostics, Franklin Lakes, NJ). Plasma was collected according to the manufacturer’s protocol within 6 hours of the blood draw and was stored at −80°C until use. Comparative quantification of the N-glycopeptides by the GP-SWATH DIA workflow was carried out on paired pools of samples (equal volume of plasma of five participants per pool) from the same patient population. Participants were split into groups of 5 and plasma samples for each group were pooled for analysis (2 pools per group, 5 participants per pool); each pool was analyzed in triplicate to document reproducibility of the workflow. All of the groups were matched on age, race (60% Caucasian, 40% African-American), and gender (80% males); HCC and cirrhosis groups were further matched on MELD score as described in (23).
MARS 14 affinity chromatography of plasma samples and sample preparation
Affinity chromatography was carried out on HP 1100 HPLC system (Agilent, Santa Clara, CA) and Multiple Affinity Removal Column Human 14 (MARS 14, Agilent). Chromatography conditions were set according to manufacturer’s protocol. Briefly, human plasma (20 μl) was mixed with buffer A (85 μl) and injected on the column. Affinity buffer A and B were used as mobile phases, 100% A at a flow rate 0.25 ml/min at 0–9 min, 100% B at a flow rate 1 ml/min at 9–15 min, and 100% A at a flow rate 1 ml/min at 15–20 min. The peak of the retained fraction was manually collected. Amicon microspin column with 3,000 Da cutoff and 0.5 ml volume was used to concentrate 1/4 of the retained fraction (cca 320 μg of protein) and to exchange Agilent buffer B to 50mM ammonium bicarbonate. Rapigest (Waters, Milford, MA) was added to a final concentration of 0.05% and incubated with the proteins at 37°C for 1 hour. Samples were reduced with 5 mM DTT, alkylated with 15 mM IAA, residual IAA was quenched with 5 mM DTT, and the samples were digested overnight using trypsin Gold (Promega, V5280) in a ratio 1/20 to the total protein.
Endoglycosidase and exoglycosidase treatment of the N-glycopeptides
Tryptic digests (50 μg) were de-sialylated with neuraminidase (New England BioLabs, Ipswich, MA) at 37°C using pressurized accelerated digestion (barocycler) technology (Pressure Biosciences, South Easton, MA). Completion of the de-sialylation reaction was confirmed by LC-MS/MS analysis. De-sialylated glycopeptides were evaporated in a vacuum concentrator (Labconco, Kansas City, MO) and dissolved in mobile phase A (2% ACN, 0.1% FA) for LC-MS/MS measurements. For the analysis of site occupancy, aliquots of the tryptic digests (50 μg) were heated at 99°C for 10 mins to deactivate trypsin. Five μl of deglycosylation buffer G7 (New England BioLabs, Ipswich, MA) were added to 50 μg of each tryptic digest and the samples were evaporated in a vacuum concentrator (Labconco) and dissolved in 50 μl of 18O water for deglycosylation with 0.5 μl PNGase F (New England BioLabs) using a 1 hour barocycler assisted protocol as described previously (24).
Study of glycopeptide fragmentation and identification of glycopeptides by LC-MS/MS
Tryptic digest of glycoprotein standards with and without neuraminidase treatment (Hemopexin, Haptoglobin, SHBG) were measured by data dependent acquisition (DDA) and parallel reaction monitoring (PRM) LC-MS/MS analysis. Glycopeptide separation was achieved by a 45 min gradient using the MS/MS conditions described below. Collision energy was altered between conditions optimal for the analysis of peptides and low energy CID used to determine the Y-ions optimal for detection of the glycopeptides. The optimized CID conditions (soft fragmentation) were used for further analysis of glycopeptides in the MARS 14 fractions. Glycopeptide separation was achieved on a Nanoacquity LC (Waters, Milford, MA) using capillary trap, 180 μm × 0.5 mm, and analytical 75 μm × 150 mm Atlantis DB C18, 3 μm, 300 Å columns (Water, Milford, MA) interfaced with 6600 TripleTOF (Sciex, Framingham, MA). A 4 min trapping step using 2% ACN, 0.1% formic acid at 15 μl/min was followed by chromatographic separation at 0.4 μl/min as follows: starting conditions were 5% ACN, 0.1% formic acid; 1–35 min, 5–50% ACN, 0.1% formic acid; 35–37 min, 50–95% ACN, 0.1% formic acid; 37–40 min 95% ACN, 0.1% formic acid followed by equilibration to starting conditions for additional 20 min. For all of the runs, we injected 0.5 μl (0.5 μg of human plasma proteins derived from 14.3 nl of plasma) of tryptic digest directly onto the column. We have used a DDA workflow with one MS1 full scan (400–1800 m/z) and 3 MS/MS fragmentations (100–1800). MS/MS spectra were recorded in the range 400–2000 m/z with resolution 30,000 and mass accuracy less than 15 ppm using the following experimental parameters: declustering potential 80 V, curtain gas 30, ion spray voltage 2,300 V, ion source gas1 11, interface heater 150°C, entrance potential 10 V, collision exit potential 11 V. Glycopeptide identities were assigned manually. Four parameters were used for positive glycopeptide identification (retention time, HR precursor mass, charge state, and MS/MS spectra). Identified glycopeptides were quantified using peak area of the extracted ion chromatograms (XIC) of the product (Y) ions. Peak integration was performed manually using MultiQuant 2.0 software (Sciex) using a full cluster method (window width of 1.2 Da).
DIA analysis of the glycopeptides
Samples were prepared as described above and measured under optimized conditions using rolling CE (CE3+=0.03*M-5) and a 10 Da SWATH window step. Tryptic digests of the MARS 14 fraction were separated using a 90 min gradient elution as described above. Y-ion isotope cluster chromatograms, based on an isolation window of 1.2 Da, were extracted from the SWATH MS/MS and used for analysis of the glycopeptide intensities. We have used a maximum of 1.0 min RT difference from the RT of the major glycoform as a qualitative parameter for positive identification of the glycoforms. Areas of the fucosylated glycopeptides were normalized to the areas of corresponding non-fucosylated analytes for the analysis of the degree (%) of fucosylation. All of the quantifiable glycoform ratios (see Electronic Supplementary Material (ESM) Table S1) were averaged across the bi- and tri/tetra-antennary glycoforms to derive summary changes between healthy controls and liver cirrhosis patient groups. The total areas of all tri/tetra-antennary glycopeptides were normalized to the total areas of bi-antennary glycopeptides for the analysis of branching.
Results and Discussion
Fragmentation of glycopeptides under soft CID conditions
Glycoprotein standards were measured using data dependent (DDA) and scheduled (PRM) MS/MS methods in order to optimize the fragmentation of glycopeptides and maximize the yield of informative Y-ions. Lower energy, corresponding to approximately 50% collision energy optimal for the fragmentation of peptides, was found sufficient for an efficient fragmentation of the complex N-glycopeptides. Characteristic Y-ions, corresponding to the loss of one N-glycan arm due to the weak Man-GlcNAc glycosidic bond were identified as the major fragmentation pathway (Fig 1). This characteristic loss is reproducible across all of the structures of the complex glycans examined (Fig 1A) as well as across various peptide backbones (Fig 1B; ESM Fig S1). Peptide fragmentation is minimized (virtually eliminated) by the soft fragmentation conditions, which means that interference from peptides in the MS/MS spectra of samples with complex background are minimized as well (ESM Fig S2); this contributes to the high selectivity of the Y-ions and high overall specificity of complex N-glycopeptide detection by the GP-SWATH workflow. Specificity of the Y-ions is substantially higher than the specificity of the corresponding B-ions which may have higher intensity at higher collision energies but cannot achieve specific glycopeptide detection in complex samples. Sensitivity of the quantification of the Y-ions is by definition higher than quantification of the precursor ions. We want to point out that we do not propose relative quantification of the distribution of glycoforms in each sample by the soft fragmentation GP-SWATH workflow; we propose comparative quantification of specific glycoforms between samples as documented below on the analysis of fucosylated glycoforms in cirrhotic patients and healthy controls.
Fig 1.
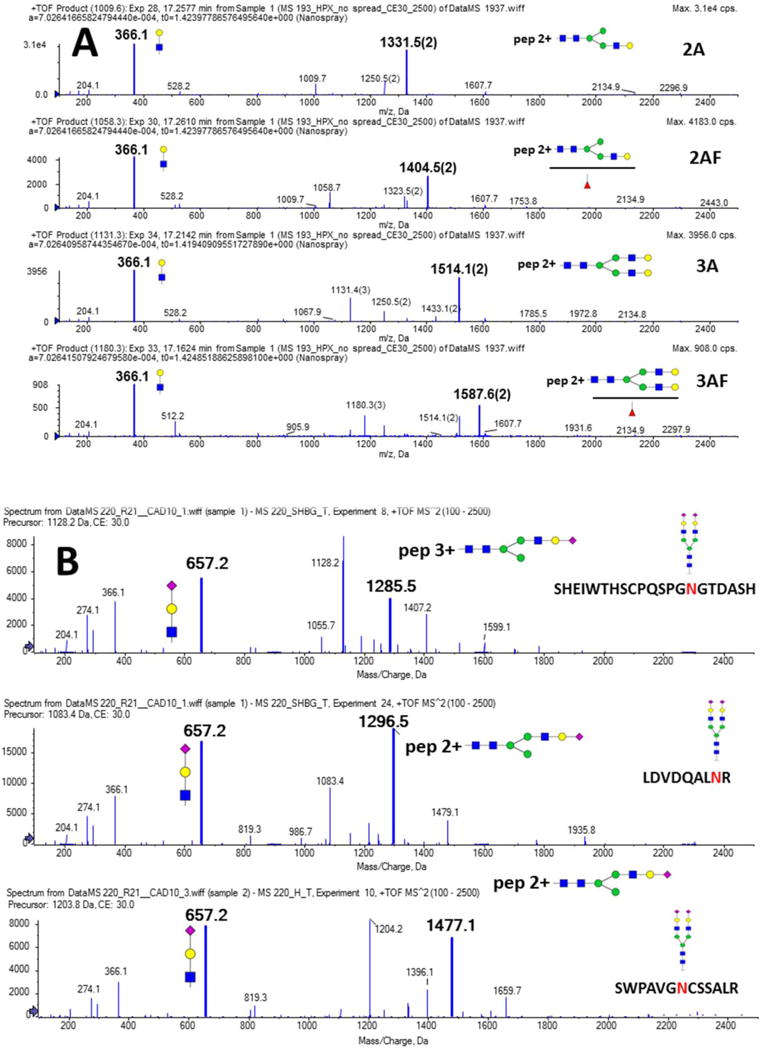
Soft fragmentation of the following analytes: A. SWPAVGNCSSALR glycopeptide of hemopexin with four different glycan structures attached (2A, 2AF, 3A, and 3AF); B. biantennary sialylated glycan (2A2SA) attached to the peptide backbones of SHBG (SHEIWTHSCPQSPGNGTDASH and LDVDQALNR) and hemopexin (SWPAVGNCSSALR). Structure, m/z, and charge state (M-1) of the major soft fragment (Y-ion) as well as the related oxonium ions (loss of glycan fragment, charge 1) are indicated in the figure.
Construction of a glycopeptide library
Tryptic digest of the MARS 14 retentate treated with neuraminidase was analyzed by DDA LC-MS/MS to construct a library of glycopeptides and their glycoforms. Retention characteristics under reversed phase chromatographic conditions and fragmentation characteristics under soft CID conditions were determined for the major glycoform of 18 glycopeptides derived from 10 glycoprotein groups retained on the MARS 14 column (Table 1). We selected the MARS 14 retentate because the majority of the glycoproteins and their glycoforms were previously examined by us and others (25;26) which serves as a reference for evaluation of this novel workflow.
Tab 1.
Database of major identified glycopeptide structures at each site of N-glycosylation with the retention time and dominant fragment selected for DIA quantification based on the DDA analysis of neuraminidase treated tryptic digest of the retained MARS 14 fraction.
| IgG Group | RT | m/z calculated | z | m/z observed | m/z fragment |
|---|---|---|---|---|---|
| EEQYNSTFR (A2G2F) | 18.5 | 986.7220 | 3 | 986.7270 | 1297.04 |
| EEQTNSTFR (A2G2F) | 20.5 | 976.0590 | 3 | 976.0620 | 1281.04 |
| EEQYNSYFR (A2F) | 19.2 | 873.3550 | 3 | 873.3610 | 1208.00 |
| Antitrypsin | |||||
| QLAHQSNSTNIFFSPVSIATAFAMLSLGTK (A2G2) | 68.5 | 961.6491 | 5 | 961.6491 | 1110.53 |
| ADTHDEILEGLNFNLTEIPEAQIHEGFQELLR (A2G2) | 52.8 | 1063.6870 | 5 | 1063.6936 | 1238.07 |
| YLGNATAIFFLPDEGK (A2G2) | 40.7 | 1126.8303 | 3 | 1126.8340 | 1507.18 |
| IgA Group | |||||
| PALEDLLLGSEANLTCTLTGLR (A2G2) | 52.7 | 995.7102 | 4 | 995.7145 | 1205.57 |
| TPLTANITK (A2G2F) | 19.8 | 909.7368 | 3 | 909.7404 | 1181.53 |
| Serotransferrin | |||||
| CGLVPVLAENYNK (A2G2) | 30.0 | 1033.7824 | 3 | 1033.7904 | 1367.60 |
| QQQHLFGSNVTDCSGNFCLFR (A2G2) | 36.1 | 1035.1818 | 4 | 1035.1876 | 1258.20 |
| Haptoglobin | |||||
| MVSHHNLTTGATLINEQWLLTTAK (A2G2) | 37.8 | 1076.2507 | 4 | 1076.2577 | 1312.95 |
| NLFLNHSENATAK (A2G2+A2G2) | 19.8 | 1176.7312 | 4 | 1176.7336 | 1446.93 |
| VVLHPNYSQVDIGLIK (A2G2) | 32.7 | 855.1535 | 4 | 855.1579 | 1018.16 |
| Fibrinogen Group | |||||
| DLQSLEDILHQVENK (A2G2) | 42.9 | 851.6276 | 4 | 851.6386 | 1013.46 |
| GTAGNALMDGASQLMGENR (A2G2) | 33.3 | 879.6155 | 4 | 879.6208 | 1050.77 |
| Alpha2-macroglobulin | |||||
| GCVLLSYLNETVTVSASLESVR (A2G2) | 49.9 | 1005.7090 | 4 | 1005.7109 | 1218.90 |
| IITILEEEMNVSVCGLYTYGK (A2G2) | 46.5 | 1014.4528 | 4 | 1014.4538 | 1230.56 |
| SINTTNVMGTSLTVR (A2G2) | 28.6 | 1072.8074 | 3 | 1072.8098 | 1426.14 |
| IYVLDYLNETQQLTPEIK (A2G2) | 42.5 | 951.4379 | 4 | 951.4357 | 1146.54 |
| VSNQTLSLF FTVLQDVPVR (A2G2) | 53.6 | 947.196 | 4 | 947.1987 | 1140.88 |
| Alpha1-acid glycoprotein | |||||
| QDQCIYNTTYLNVQR (A3G3) | 33.3 | 885.3749 | 4 | 885.3656 | 1058.45 |
| SVQEIQATFFYFTPNK (A3G3) | 26.7 | 977.6722 | 4 | 977.6689 | 1181.52 |
| IgM | |||||
| NNSDISSTR (A2G2) | 19.1 | 872.6851 | 3 | 872.6813 | 1125.96 |
| GLTFQQNASSMCVPDQDTAIR (A2G2F) | 29.8 | 1027.6842 | 4 | 1027.6914 | 1248.2 |
| complement C3 | |||||
| TVLTPATNHMGNVTFTIPANR (M5) | 31.8 | 1157.8657 | 3 | 1157.869 | 1229.62 |
As we discussed, the fragmentation of the complex N-glycopeptides is quite consistent and predictable. The Y-ions selected for quantification have high mass and predictable charge (typically one z less than the precursor) which increases reliability of their quantification. In addition, retention time alignment can be used as an important qualitative parameter for evaluation of the glycoforms of each glycopeptide as each additional neutral oligosaccharide decreases the retention time by a small amount under the reversed phase condition (27). Influence of the glycan structures on the retention time is predictable and we expect that formal models of retention behavior will be used in the future for automated data interpretation. We used a 1 minute retention window around the major glycoform as a criterion for identification of the peak as a related glycoform of the same peptide. For example, six glycoforms (2A, 2AF, 3A, 3AF, 4A, 4AF) of the VVLHPNYSQVDIGLIK tryptic peptide of haptoglobin align in retention time between 32.65 and 33.07 minutes (Fig 2) which further strengthens our confidence in the assignments. This figure also documents high selectivity of the Y-ions as seen with the minimal signal besides the peaks of the analytes of interest detected at the expected retention times.
Fig 2.

Extracted chromatograms of the Y-ions derived in the DIA LC-MS/MS workflow from the 2A, 2AF, 3A, 3AF, 4A, 4AF glycoforms of the tryptic peptide VVLHPNYSQVDIGLIK of haptoglobin.
We also carried out confirmatory analysis using PNGaseF de-glycosylation in the presence of 18O water as described previously (25) in order to verify the correct assignments of the glycopeptides by LC-MS/MS and to examine retention characteristics of the peptides. The final library consists of 161 glycoforms of 25 glycopeptides derived from 10 protein groups (ESM Table S1); all of the results were manually verified and 125 of these glycoforms were quantifiable in all of the samples examined. Comparative DIA quantification of these glycoforms in paired pools of plasma of the cirrhotic patients and healthy controls were examined in triplicate and summarized below. Our results suggest that the fragmentation and retention behavior of the glycopeptides is consistent enough to allow future expansion of the library to additional glycoforms and peptides that were not examined a priori by DDA analysis. At this point, we want to minimize false positive identifications and limit our analysis to the glycoforms observed by the DDA LC-MS/MS.
Analysis of fucosylation of the plasma proteins in the context of liver cirrhosis
The change of fucosylation in liver cirrhosis was determined on 18 glycopeptides covering 19 NxS/T sequons (Table 1 and ESM Table S1). We focus on the application of our DIA LC-MS/MS workflow to the comparative quantification of fucosylation because our prior work on isolated glycoproteins shows that fucosylation changes are an important characteristic of the development of liver cirrhosis (23;28). Our current results show that the quantitative changes of fucosylated glycoforms of peptides can be evaluated efficiently in a complex mixture (unfractionated digest of the MARS 14 retentate) in a multiplex format by the GP-SWATH workflow. We opted to use the MARS 14 retentate to minimize false positive identifications from the background of tryptic digest of the less abundant plasma proteins and to minimize potential impact of these peptides on ionization efficiency/accuracy of quantification. However, selectivity of the soft fragmentation of complex N-glycoforms suggests that the analysis can be done in samples of even higher complexity, including digest of unfractionated plasma. The specificity of detection of the N-glycoforms is demonstrated by the quality of the extracted Y-ion chromatograms of multiple glycoforms of the tryptic peptide VVLHPNYSQVDIGLIK of haptoglobin (Fig 2) where few other peaks were detected besides the expected analytes.
We have previously reported that the differences in fucosylation are site-specific, depend on the branching of the glycans, and differ between core and outer arm structures (23). In this paper we highlight the multiplex comparisons across different proteins and glycopeptides without glycopeptide enrichment. Our results confirm our previous observations (23;28) and expand the analysis to glycopeptides of additional proteins (ESM Table S1). Specific examples of the fucosylation changes (ratios of quantifiable pairs of glycoforms with/out fucose) are presented in Figs 3–5. Separately, we present data from two biological replicates (two pooled samples of cirrhosis and two control pooled samples, 5 individual plasma samples in each pool) analyzed each in triplicate, to demonstrate the reproducibility of the comparative quantification. The results shows that biological differences are typically higher than the technical variability of the measurements as seen, for example, on the A3G3F1 glycoforms of the VVLHPNYSQVDIGLIK peptide of haptoglobin (Fig 3A) or the A3G3F1 glycoform of QQQHLFGSNVTDCSGNFCLFRSETK peptide of serotransferin (Fig 3B). Average glycopeptide ratios of all the replicate measurements across all quantifiable pairs of glycoforms are summarized in the ESM Table S1.
Fig 3.
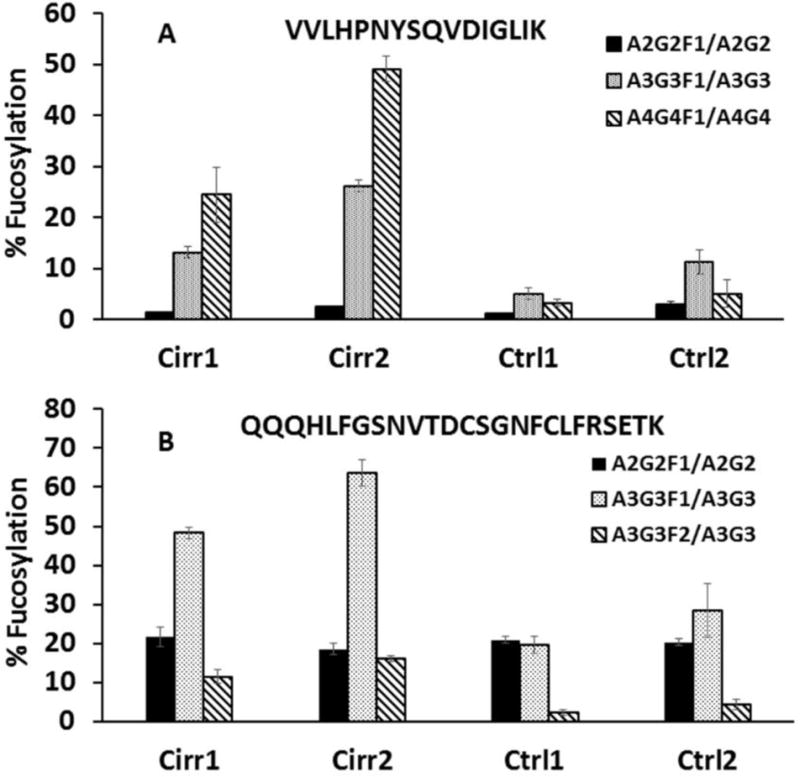
Increased ratio of fucosylated N-glycopeptides to their non-fucosylated counterparts as an example of increased fucosylation of liver secreted plasma proteins; triplicate (mean ± standard deviation) technical repeats of the two cirrhosis (Cirr) and control (Ctrl) samples each are presented. A. A2G2F1/A2G2, A3G3F1/A3G3, and A4G4F1/A4G4 glycoforms of the VVLHPNYSQVDIGLIK N-glycopeptide of haptoglobin; B. A2G2F1/A2G2, A3G3F1/A3G3, and A3G3F2/A3G3 glycoforms of the QQQHLFGSNVTDCSGNFCLFRSETK N-glycopeptide of serotransferrin.
Fig 5.
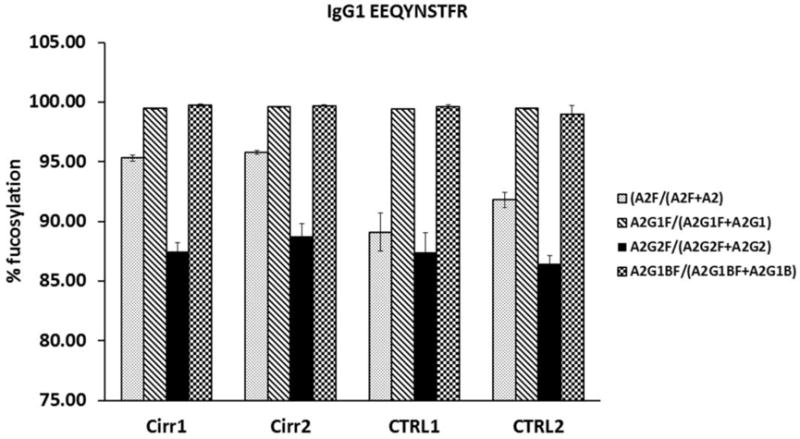
Analysis of the A2F/A2, A2G1F/A2G1, A2G2F/A2G2, and A2G1BF/A2G1B ratios of N-glycoforms of the tryptic peptide EEQYNSTFR of IgG1 documents constantly high fucosylation of this immune glycoprotein in control and cirrhosis samples. Triplicate (mean ± standard deviation) technical repeats of the two cirrhosis (Cirr) and control (Ctrl) samples each are presented.
Our results confirm that the changes in fucosylation are protein and site specific but we also observe some general trends. Fucosylation of the bi-antennary glycopeptides increases less than fucosylation of tri-/tetra-antennary glycoforms typically in cirrhosis. Only 4 of 12 bi-antennary glycopeptide-pairs, and 9 of 13 tri-/tetra-antennary glycopeptide-pairs show greater than 1.5 fold increase in cirrhosis (ESM Table S1). These differences are clearly protein and site specific as the increase of the A2G2F1/A2G2 ratio of the QQQHLFGSNVTDCSGNFCLFRSETK peptide of serotransferin in cirrhosis is 0.97 (Fig 3B), but the A2G2F1/A2G2 ratio of the CGLVPVLAENYNK peptide of the same protein increases 2.26 fold. The A2G2F1/A2G2 ratio of fibrinogen peptide GTAGNALMDGASQLMGENR increases 7.28 fold (ESM Table S1). A typical trend is observed in the case of the A2G2F1/A2G2 ratio of the VVLHPNYSQVDIGLIK peptide of haptoglobin (0.95 fold increase), A3G3F1/A3G3 ratio (2.41 fold increase), and the A4G4F1/A4G4 ratio (8.80 fold increase, Fig 3A). In addition, multiply fucosylated glycoforms were observed to increase more than singly fucosylated glycoforms, though only a few of these low intensity analytes could be quantified. For example, the ratio of A3G3F2/A3G3 glycoforms of serotransferin peptide QQQHLFGSNVTDCSGNFCLFRSETK increases 4.14 fold while the A3G3F1/A3G3 ratio on the same peptide increases 2.33 fold (Fig 3B) and the ratio of A3G3F2/A3G3 glycoforms of antitrypsin peptide ADTHDEILEGLNFNLTEIPEAQIHEGFQELLR increases 2.84 fold, while a 1.72 fold increase is observed in the A3G3F1/A3G3 ratio (ESM Table S1).
The increase in branching (tetra-antennary structures) reported previously (29) is also clearly detectable in our datasets. The ratio of the A4G4/A2G2 glycoforms of the VVLHPNYSQVDIGLIK peptide of haptoglobin increases 1.5 fold while the A3G3/A2G2 ratio increases 7.4 fold (Fig 4). It is interesting to note that, in general, immunoglobulins (IgG1–4, IgA, IgM) and immune system derived proteins like complement C3 show minimal increases in fucosylation (ESM Table S1). The example of IgG1 shows that the bi-antennary glycoforms typical of this immunoglobulin (A2F, A2G1F, A2G2F, A2G1BF) are fucosylated to a high degree (>85%) but the fucosylation does not further increase in cirrhosis, except for a 90.5→95.6 increase in the percentage of A2F (Fig 5). This could be related to the high degree of fucosylation, dominant bi-antennary structures of IgG, or even specific glycan structures or topologies (such as core vs. outer arm fucosylation). We do not fully explore these possibilities here but we do observe that IgA glycoforms which have a low percentage of fucosylation do not show an increase either and fucosylation of the tri-antennary glycoforms of complement C3 also do not increase in cirrhosis. It is therefore likely that in general the immune cells do not produce glycoproteins with increased fucosylation in the context of cirrhosis. Further evaluation will be needed to make this observation definitive.
Fig 4.
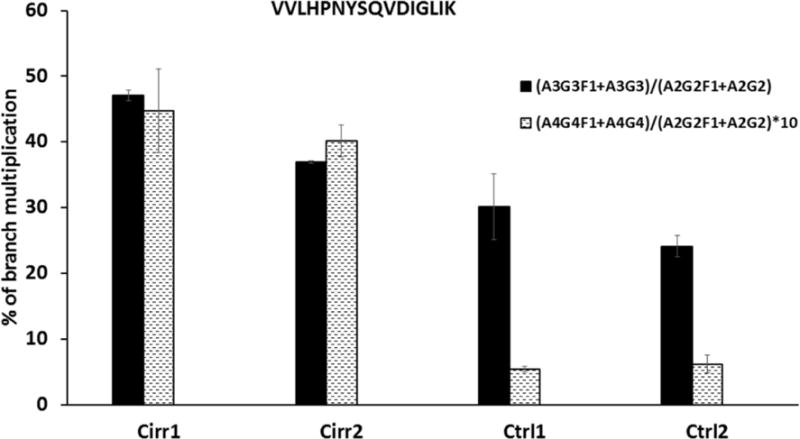
Increased branching of the VVLHPNYSQVDIGLIK N-glycopeptide of haptoglobin expressed as (A3G3F1+A3G3)/(A2G2F1+A2G2) and (A4G4F1+A4G4)/(A2G2F1+A2G2) ratios of its glycoforms. Data represent triplicate (mean ± standard deviation) technical repeats of the two cirrhosis (Cirr) and healthy control (Ctrl) samples. The (A4G4F1+A4G4)/(A2G2F1+A2G2) ratio is magnified 10-times for clarity of presentation.
In this paper, we do not pursue resolution of the outer arm and core fucosylation by exoglycosidase digests as described previously (23;25). We can, however, distinguish these types of glycoforms from the soft fragmentation data at least to some degree. Core fucosylated biantennary glycopeptides (like the glycopeptides of IgG1) (26;28) produce one dominant fucosylated Y-ion because the fucose of the glycan core remains intact under the soft CID conditions. Biantennary glycopeptides fucosylated on the outer arm produce two Y-ions of comparable intensity (with/without fucose) based on approximately equal fragmentation of both arms (like the glycopeptides of haptoglobin which carry negligible amount of core fucose) (30). This pair of ions is specific to the outer arm fucosylation under soft CID conditions. Assignment of the proportions in case of mixed outer arm/core fucosylated glycopeptides, which co-elute under the reversed phase chromatographic conditions, needs further study using synthetic glycopeptide standards. Assignments of the glycoforms are further validated by additional ions including the complementary B-ions (e.g. the 512 Da ion in case of outer arm fucosylated structures).
Conclusion
Fragmentation of complex N-glycopeptides under soft CID conditions (energy approximately half of the CE typically used for the fragmentation of peptides) yields predictable Y-ions with minimal fragmentation of non-glycosylated peptides. This selective fragmentation allows efficient extraction of the high mass Y-ions for quantitative DIA assessment of glycopeptides in complex samples (without glycopeptide enrichment). Selectivity of the Y-ions and retention time alignment of multiple glycoforms of the same peptide improve specificity of the DIA LC-MS/MS quantification. Our results show that the GP-SWATH workflow allows quantitative comparison of 130 glycoforms of 28 glycopeptides covering 10 protein groups in a complex background (without glycopeptide enrichment) of similar quality as our previous quantification of enriched glycopeptides from digests of simple glycoproteins mixtures (23;28). The results confirm that fucosylation of liver secreted glycoproteins increases in liver cirrhosis and shows that the increase in fucosylation depends on the degree of glycan branching and is protein- and site- specific. We expect that the soft fragmentation conditions will allow efficient and multiplex DIA comparisons of various glycoforms of proteins much needed for the quantitative assessment of protein glycosylation in various (patho)physiological processes.
Supplementary Material
Acknowledgments
Funding
This work was supported by National Institutes of Health Grants UO1 CA168926, UO1 CA171146, and RO1 CA135069 (to R.G.) and CCSG Grant P30 CA51008 (to Lombardi Comprehensive Cancer Center supporting the Proteomics and Metabolomics Shared Resource).
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethics approval and consent to participate
All the participants were enrolled and signed informed consent under protocols approved by the MedStar Health Research Institute – Georgetown University Oncology Institutional Review Board (IRB) under IRB # 2014-0804. All bio-specimens are de-identified and may not be linked to identifiable individual private information.
References
- 1.Haltiwanger RS, Lowe JB. Role of glycosylation in development. Annu Rev Biochem. 2004;73:491–537. doi: 10.1146/annurev.biochem.73.011303.074043. [DOI] [PubMed] [Google Scholar]
- 2.Freeze HH. Understanding human glycosylation disorders: biochemistry leads the charge. J Biol Chem. 2013 Mar 8;288(10):6936–45. doi: 10.1074/jbc.R112.429274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cummings RD. The repertoire of glycan determinants in the human glycome. Mol Biosyst. 2009 Oct;5(10):1087–104. doi: 10.1039/b907931a. [DOI] [PubMed] [Google Scholar]
- 4.Moremen KW, Tiemeyer M, Nairn AV. Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol. 2012 Jul;13(7):448–62. doi: 10.1038/nrm3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Canis K, McKinnon TA, Nowak A, Haslam SM, Panico M, Morris HR, et al. Mapping the N-glycome of human von Willebrand factor. Biochem J. 2012 Oct 15;447(2):217–28. doi: 10.1042/BJ20120810. [DOI] [PubMed] [Google Scholar]
- 6.Alley WR, Jr, Mann BF, Novotny MV. High-sensitivity analytical approaches for the structural characterization of glycoproteins. Chem Rev. 2013 Apr 10;113(4):2668–732. doi: 10.1021/cr3003714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thaysen-Andersen M, Packer NH, Schulz BL. Maturing Glycoproteomics Technologies Provide Unique Structural Insights into the N-glycoproteome and Its Regulation in Health and Disease. Mol Cell Proteomics. 2016 Jun;15(6):1773–90. doi: 10.1074/mcp.O115.057638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wuhrer M. Glycomics using mass spectrometry. Glycoconj J. 2013 Jan;30(1):11–22. doi: 10.1007/s10719-012-9376-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldman R, Sanda M. Targeted methods for quantitative analysis of protein glycosylation. Proteomics Clin Appl. 2015 Feb;9(1–2):17–32. doi: 10.1002/prca.201400152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harvey DJ. Derivatization of carbohydrates for analysis by chromatography; electrophoresis and mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2011 May 15;879(17–18):1196–225. doi: 10.1016/j.jchromb.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 11.Kailemia MJ, Ruhaak LR, Lebrilla CB, Amster IJ. Oligosaccharide analysis by mass spectrometry: a review of recent developments. Anal Chem. 2014 Jan 7;86(1):196–212. doi: 10.1021/ac403969n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ashline DJ, Hanneman AJ, Zhang H, Reinhold VN. Structural documentation of glycan epitopes: sequential mass spectrometry and spectral matching. J Am Soc Mass Spectrom. 2014 Mar;25(3):444–53. doi: 10.1007/s13361-013-0776-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu T, Qian WJ, Gritsenko MA, Camp DG, Monroe ME, Moore RJ, et al. Human plasma N–glycoproteome analysis by immunoaffinity subtraction, hydrazide chemistry, and mass spectrometry. J Proteome Res. 2005 Nov;4(6):2070–80. doi: 10.1021/pr0502065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stahl-Zeng J, Lange V, Ossola R, Aebersold R, Domon B. High sensitivity detection of plasma proteins by multiple reaction monitoring of N-glycosites. Mol Cell Proteomics. 2007 Aug;:28. doi: 10.1074/mcp.M700132-MCP200. [DOI] [PubMed] [Google Scholar]
- 15.Zielinska DF, Gnad F, Wisniewski JR, Mann M. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell. 2010 May 28;141(5):897–907. doi: 10.1016/j.cell.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 16.Rudd PM, Dwek RA. Glycosylation: heterogeneity and the 3D structure of proteins. Crit Rev Biochem Mol Biol. 1997;32(1):1–100. doi: 10.3109/10409239709085144. [DOI] [PubMed] [Google Scholar]
- 17.Thaysen-Andersen M, Packer NH. Site-specific glycoproteomics confirms that protein structure dictates formation of N-glycan type, core fucosylation and branching. Glycobiology. 2012 Jul;:13. doi: 10.1093/glycob/cws110. [DOI] [PubMed] [Google Scholar]
- 18.Chandler K, Goldman R. Glycoprotein disease markers and single protein-omics. Mol Cell Proteomics. 2013 Apr;12(4):836–45. doi: 10.1074/mcp.R112.026930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plomp R, Hensbergen PJ, Rombouts Y, Zauner G, Dragan I, Koeleman CA, et al. Site-specific N-glycosylation analysis of human immunoglobulin e. J Proteome Res. 2014 Feb 7;13(2):536–46. doi: 10.1021/pr400714w. [DOI] [PubMed] [Google Scholar]
- 20.Gillet LC, Navarro P, Tate S, Roest H, Selevsek N, Reiter L, et al. Targeted data extraction of the MS/MS spectra generated by data independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012 Jan;:18. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ting YS, Egertson JD, Payne SH, Kim S, MacLean B, Kall L, et al. Peptide-entric Proteome Analysis: An Alternative Strategy for the Analysis of Tandem Mass Spectrometry Data. Mol Cell Proteomics. 2015 Sep;14(9):2301–7. doi: 10.1074/mcp.O114.047035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keller A, Bader SL, Kusebauch U, Shteynberg D, Hood L, Moritz RL. Opening a SWATH Window on Posttranslational Modifications: Automated Pursuit of Modified Peptides. Mol Cell Proteomics. 2016 Mar;15(3):1151–63. doi: 10.1074/mcp.M115.054478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benicky J, Sanda M, Pompach P, Wu J, Goldman R. Quantification of fucosylated hemopexin and complement factor H in plasma of patients with liver disease. Anal Chem. 2014 Nov 4;86(21):10716–23. doi: 10.1021/ac502727s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szabo Z, Guttman A, Karger BL. Rapid release of N-inked glycans from glycoproteins by pressure-ycling technology. Anal Chem. 2010 Mar 15;82(6):2588–93. doi: 10.1021/ac100098e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pompach P, Brnakova Z, Sanda M, Wu J, Edwards N, Goldman R. Site-pecific glycoforms of haptoglobin in liver cirrhosis and hepatocellular carcinoma. Mol Cell Proteomics. 2013 May;12(5):1281–93. doi: 10.1074/mcp.M112.023259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Selman MH, Derks RJ, Bondt A, Palmblad M, Schoenmaker B, Koeleman CA, et al. Fc specific IgG glycosylation profiling by robust nano-everse phase HPLC-S using a sheath-low ESI sprayer interface. J Proteomics. 2012 Feb 2;75(4):1318–29. doi: 10.1016/j.jprot.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 27.Wang B, Tsybovsky Y, Palczewski K, Chance MR. Reliable determination of site-pecific in vivo protein N-glycosylation based on collision-nduced MS/MS and chromatographic retention time. J Am Soc Mass Spectrom. 2014 May;25(5):729–41. doi: 10.1007/s13361-013-0823-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan W, Sanda M, Wu J, Koomen J, Goldman R. Quantitative analysis of immunoglobulin subclasses and subclass specific glycosylation by LC-MS-MRM in liver disease. J Proteomics. 2015 Feb 26;116:24–33. doi: 10.1016/j.jprot.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mehta AS, Norton P, Liang H, Comunale MA, Wang M, Rodemich-Betesh L, et al. Increased levels of tetra-antennary N-linked glycan but not core fucosylation are associated with hepatocellular carcinoma tissue. Cancer Epidemiol Biomarkers Prev. 2012 Apr;:6. doi: 10.1158/1055-9965.EPI-11-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanda M, Pompach P, Brnakova Z, Wu J, Makambi K, Goldman R. Quantitative liquid chromatography-mass spectrometry-multiple reaction monitoring (LC-MS-MRM) analysis of site-specific glycoforms of haptoglobin in liver disease. Mol Cell Proteomics. 2013 May;12(5):1294–305. doi: 10.1074/mcp.M112.023325. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.