

Abstract
In the heart, coupling between excitation of the surface membrane and activation of contractile apparatus is mediated by Ca released from the sarcoplasmic reticulum (SR). Several components of Ca machinery are perfectly arranged within the SR network and the T-tubular system to generate a regular Ca cycling and thereby rhythmic beating activity of the heart. Among these components, ryanodine receptor (RyR) and SR Ca ATPase (SERCA) complexes play a particularly important role and their dysfunction largely underlies abnormal Ca homeostasis in diseased hearts such as in heart failure. The abnormalities in Ca regulation occur at practically all main steps of Ca cycling in the failing heart, including activation and termination of SR Ca release, diastolic SR Ca leak, and SR Ca uptake. The contributions of these different mechanisms to depressed contractile function and enhanced arrhythmogenesis may vary in different HF models. This brief review will therefore focus on modifications in RyR and SERCA structure that occur in the failing heart and how these molecular modifications affect SR Ca regulation and excitation-contraction coupling.
Keywords: Sarcoplasmic reticulum, Ca-induced Ca release, Ryanodine Receptor, SR Ca ATPase
Sarcoplasmic reticulum Ca handling in the heart
In the heart, excitation-contraction coupling (ECC) relies on well-controlled intracellular Ca cycling. Playing a particularly important role in this cycle is the sarcoplasmic reticulum (SR). The cardiac SR is equipped with Ca handling machinery, which is perfectly designed to regularly repeat two main steps of the Ca cycle, Ca release and uptake, over billions of times during the entire lifespan.
Activation of CICR
The SR Ca release predominantly occurs at subcellular microdomains called dyads. In these domains, L-type Ca channels (LTCCs) in the membrane of the T-tubules come into close contact with a cluster of ~20-100 ryanodine receptors (RyR) at the junction of the SR. During ECC, these RyR clusters are activated by a relatively small Ca influx via L-type Ca channels (LTCCs) (Fig 1A). This process, first described by Fabiato in 1983, is known as Ca-induced Ca release (CICR) [23]. The activation of a single RyR cluster generates a local increase in cytosolic Ca ([Ca]i) or Ca spark [18]. The spatio-temporal summation of thousands of Ca sparks during an action potential (AP) produces the global Ca transient that initiates contraction. The dyadic organization of ventricular myocytes provides the necessary local control of SR Ca release by LTCCs [95]. As a result, the amplitude of the global Ca transient can be gradually adjusted by recruiting varying amounts of RyR clusters.
Figure 1. Alteration of SR Ca cycling in the failing heart.
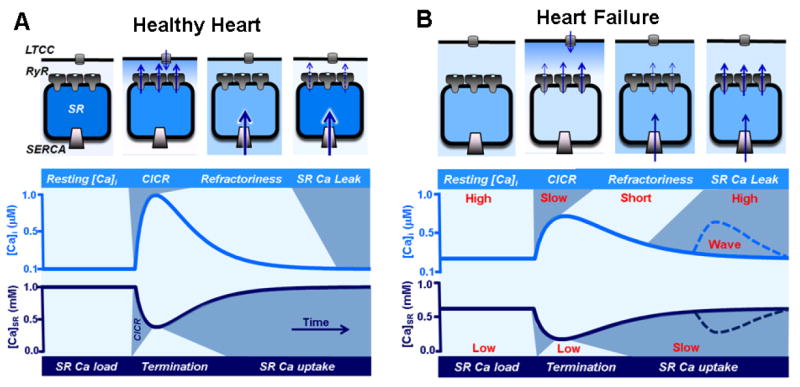
A, top: The diagram illustrates an SR Ca release unit composed by LTCCs in the T-tubule and RyR clusters in the junctional SR. SERCA pumps are predominantly located in the free SR. The activation of LTTCs during an AP increases [Ca] in the dyadic cleft. This local increase in [Ca] activates RyRs via the mechanism of CICR. Upon activation, RyRs transiently release a larger amount of Ca into the cytosol, causing to abruptly drop in [Ca]SR. The decrease in [Ca]SR deactivates RyRs, leading to termination of CICR. After SR Ca release termination, RyRs enter the refractory state which allows SERCA to replenish SR with Ca. During rest a few RyRs remain active, so the SR Ca uptake is balanced by Ca leak to finely tune the SR Ca content in preparation for the next Ca cycle. Bottom: The two traces illustrate dynamic changes of [Ca]i and [Ca]SR during ECC. All main steps of Ca cycling are marked by different colors (light and dark blue). B. In HF myocyte, the distance between LTCCs and RyRs is significantly increased. Such structural alteration decreases the fidelity of LTCC-RyR coupling causing less synchronized systolic SR Ca release. Due to the increased sensitivity of RyRs to luminal-SR [Ca], termination of systolic Ca release occurs later and at much lower [Ca]SR compared to healthy myocytes. Additionally, RyR refractoriness is shorter and RyR-mediated SR Ca leak is faster. These modifications of RyR activity lead to an enhanced propensity of pro-arrhythmic spontaneous Ca waves. Decreased SERCA activity and accelerated SR Ca leak both contribute to the depleted SR Ca content and elevated diastolic [Ca]i.
Termination of CICR
Since the discovery of CICR, the question of what causes SR Ca release to stop has attracted a great deal of attention. Several mechanisms have been proposed to explain CICR termination including cytosolic Ca-dependent inactivation, luminal SR Ca-dependent deactivation, adaptation and stochastic attrition [96]. Originally, it was thought that an increase in [Ca]i during Ca release inactivates RyRs via binding of Ca to cytosolic low affinity site(s) on the channel [24]. However, accumulated evidence indicates that inactivation of RyRs by [Ca]i plays very limited if any role in the termination process while the role of changes in [Ca]SR is critical. It was demonstrated that the RyR responds to changes in [Ca] at the luminal face of the channel [35;91]. As a result, intra-SR [Ca] ([Ca]SR)-dependent deactivation of the RyR has been proposed as a main underlying cause of termination of Ca sparks [100]. Indeed, it has been shown that Ca sparks cease when [Ca]SR falls to a certain critical level [16;99;115] supporting the functional link between partial [Ca]SR depletion and CICR termination. A fall in local [Ca]SR may terminate a spark by at least two different mechanisms. First, [Ca]SR depletion can drive unbinding of Ca from an intra-SR regulatory site on the RyR and thus promote the channel closing [43;99]. In addition to the direct regulation of the RyR, local [Ca]SR depletion could terminate Ca spark by reducing the unitary current via the RyR, decreasing local [Ca]i, and thus breaking the positive feedback of local CICR within a cluster [34;54;79]. Therefore, it seems that CICR termination is mediated by the interaction of Ca with several binding sites located on both sides of the RyR.
Refractoriness of CICR
For the maintenance of normal heart function, restraining mechanisms must exist to prevent the occurrence of spontaneous cell-wide SR Ca release events during diastole (i.e. Ca waves) and thereby after-contractions (Fig 1A). One possible explanation why spontaneous Ca waves do not normally happen during diastole involves regulation of the RyR activity by [Ca]SR. It has been suggested that [Ca]SR must reach a certain threshold level before spontaneous Ca waves can occur (the mechanism was called store-overload-induced Ca release or SOICR) [44;102]. However, recent direct measurements of [Ca]SR dynamics during global Ca transients suggested that this mechanism by itself is not sufficient to trigger Ca waves, even under conditions where the RyR sensitivity to Ca is increased [9]. It was shown that the refilling of the SR after Ca release occurs quickly, reaching the pre-release level long before the initiation of spontaneous Ca waves. Although exact molecular mechanisms of refractoriness of CICR remain undetermined, it is plausible that the initial drop in [Ca]SR causes allosteric changes in the RyR complex that evoke a protracted shift in the RyR sensitivity to luminal and cytosolic [Ca], making channels less available for activation.
SR Ca leak
Unlike inactivation of voltage-gated Ca and Na channels, RyRs are not completely closed during diastole and spontaneous openings of RyRs can generate a substantial SR Ca leak [88;114] (Fig 1A). SR Ca uptake (discussed below) and diastolic Ca leak together determine SR Ca load and, therefore, the amplitude of Ca transients that initiate contraction. At normal physiological conditions, the steep leak-load relationship [88;114] serves as an important protective mechanism against pro-arrhythmic SR Ca overload. When [Ca]SR approaches a dangerous level, increased SR Ca leak will partially discharge the SR, limiting SR Ca overload [14;21]. However, in pathological conditions associated with increased RyR activity, Ca leak can reach a critical point when the released Ca during spontaneous sparks diffuses to neighboring release clusters, triggers CICR, and generates diastolic Ca waves. By activating the electrogenic Na-Ca exchanger (NCX), spontaneous Ca waves can generate delayed afterdepolarizations (DADs), an effective trigger of cardiac arrhythmias [80].
RyR complex
The type-2 RyR is not only the major SR Ca release channel but also a giant scaffolding protein on which several regulatory proteins and enzymes are concentrated. On the cytosolic side, RyR interacts with calmodulin (CaM), FK-506 binding proteins (FKBP), and sorcin. It has been shown that FKBP12.6 can affect the RyR by stabilizing the interaction between the channel subunits [64], whereas CaM and sorcin affect the channel activity via Ca-dependent mechanisms [4;25]. The RyR also forms complex with two major protein kinases (protein kinase A (PKA) and Ca/Calmodulin-dependent kinase type II (CaMKII)) and three phosphatases (PP1, PP2A and PP2B) indicating the importance of RyR phosphorylation [12;63]. So far three functionally relevant RyR phosphorylation sites have been identified, including two PKA sites Ser-2808 and Ser-2030 and one (CaMKII) site Ser-2814. Two SR-membrane proteins, junctin and triadin, are believed to be crucial for the RyR’s ability to sense changes in [Ca]SR via interactions with calsequestrin (CASQ) [36] and histidine-rich Ca-binding proteins (HRC) [76]. In addition, RyR has multiple sites for regulation by Ca, Mg, ATP, and redox active compounds.
SR Ca uptake and SERCA complex
For relaxation to occur, not only CICR must terminate, but a main portion of Ca should be pumped from the cytosol back into the SR (Fig 1A). This function is achieved by the SR Ca-ATPase (SERCA). SERCA2a isoform is one of most abundant proteins in the cardiac SR, indicating a key role of this Ca transporter. Although SERCA interacts with a wide array of proteins (including HRC, PP1, calreticulin, S100A, and sarcolipin), phospholamban (PLB) is the most important regulator of the pump activity [50]. At the unphosphorylated state, PLB inhibits SERCA activity by lowering the apparent affinity of the pump for Ca. Phosphorylation of PLB at Ser-16 by PKA [90] relieves this inhibition, increasing the pumping rate by several fold. Stimulation of SR Ca uptake by PLB phosphorylation appears to be the major mechanism of acceleration of relaxation (lusitropic effect) during β-adrenergic receptor stimulation. SERCA activity can be also increased via PLB phosphorylation by CaMKII at Thr-17 [90]. This mechanism has been suggested to be responsible for a stimulation frequency-dependent acceleration of SR Ca uptake. In addition to the PLB regulation, SERCA is highly sensitive to changes in the metabolic status of the cytosol, including the ATP/ADP ratio, pH and the redox potential.
Although some mechanisms of SR Ca handling are not completely understood, it becomes increasingly clear that normal heart function highly relies on synchronized SR Ca release with robust termination, well-balanced diastolic SR Ca leak, and effective SR Ca uptake (Fig 1A). Just as the tightly controlled RyR and SERCA function play essential roles in Ca homeostasis of the healthy heart, defects in this control cause abnormality in contractile function and promote cardiac arrhythmia in the failing heart.
Alteration of sarcoplasmic reticulum Ca handling in HF
Although factors that cause HF may vary (myocardial infarction, changes in neurohumoral tone and pacing rate), decreased ability of the SR to retain Ca seems to be a consistent finding that can explain depressed contraction of the failing heart. This SR Ca mishandling occurs as a result of an alteration in expression and/or activity of several Ca transporters, including SERCA and RyR. Moreover, the alteration of cardiomyocyte architecture has been suggested playing an important role in the abnormality of SR Ca release in failing myocytes. The relative contributions of these different mechanisms may vary in different models of HF and with different degrees of severity of failure.
Diminished fidelity of CICR activation in HF
It appears that depressed AP-induced Ca transients in HF is a result of decreased fidelity of LTCC-RyR coupling, leading to less synchronized SR Ca release [31]. It has been suggested that on the subcellular level some RyR clusters have a delayed response to the stimulus due to downregulation of LTCC current [40;58]. HF myocytes is also characterized by substantial loss of T-tubules [3;19;40;60;61;93] possibly due to reduction in levels of junctophilins [104], proteins that tether junctional SR to T-tubules for precise positioning of LTCCs and RyR clusters within a 30-100 nm space [27] (Fig 1B). This could further decrease the ability of RyR clusters to properly and timely respond to activation by LTCCs, providing an additional basis for unsynchronized SR Ca release and depressed AP-induced Ca transients in HF (Fig 1B). Furthermore, numerous publications demonstrated a reduction of RyR expression at the mRNA and protein levels in HF [1;2;15;30;51;92;110] (but some publications demonstrate no change [3;31;45]). It is unclear however whether the reduction in RyR levels by itself can contribute to the diminished SR Ca release in HF. It seems that the exact contribution by each of these different mechanisms depends on specific causes and stages of HF. Nevertheless, the loss of strict dyadic organization resulting in diminished fidelity of LTCC-RyR coupling is considered to be one of the main reasons for depressed AP-induced Ca transients in HF. The diminished LTCC-RyR coupling may also promote Ca alternans [17]. These beat-to-beat variations of Ca transient amplitude evoke alternations in the AP duration at the cellular level, promoting electrical disbalance in the whole heart and thereby providing a substrate for cardiac arrhythmia.
Depressed SR Ca uptake in HF
Although several recent studies showed no change in SERCA function in intact as well as in permeabilized HF myocytes [7;10], a large number of publications indicated a reduction of SR Ca uptake in HF [20;45;74;81;85] (Fig 1B). Moreover, most studies of HF myocytes showed that the decrease in Ca transient amplitude was accompanied by diminished SR Ca content [41;45;57;70;73] (Fig 1B). Based on these results, it has been suggested that defective SR Ca uptake is one of the primary causes of depressed SR Ca release and contraction in HF. Decreased SR Ca uptake can also contribute to an increase in diastolic [Ca]i (Fig 1B). In addition, recent experimental and modeling studies also implicated a reduced SERCA-mediated SR Ca uptake in enhanced propensity for Ca-dependent alternans and thus cardiac arrhythmias [6;105]. Therefore, reduction in SERCA activity can cause the impairment of both systolic and diastolic function in the failing myocardium. As a result, overexpression of the SERCA pump or phospho-mimetic PLB can stabilize the SR Ca load and improve cardiac function in animal HF models (for review see [59]). Recent phase 2 clinical trials, conducted in patients with advanced HF, showed promising results when increasing SERCA expression using adeno-associated viral gene transfer [116]. Currently, a continuation of these trials is underway using a larger patient sample size. However, it should be noted that restoring the SERCA activity in HF is not always beneficial. For example, augmentation of SR Ca uptake due to PLB ablation or SERCA overexpression can potentially promote development of HF in mice with enhanced RyR function [46;113], suggesting that in certain conditions the benefit of accelerated SR Ca uptake can be limited by exacerbated RyR-mediated Ca leak.
Impaired CICR termination and refractoriness in HF
It has been suggested that depleted SR Ca load in HF myocytes would reduce Ca transient even more dramatically if it were not compensated for by an increase in RyR activity [22;32;51;98]. It appears that during the early stages of HF, sensitization of the RyR allows myocytes to maintain Ca transients of nearly normal amplitude by shifting threshold of SR Ca release termination to lower [Ca]SR levels (Fig. 1B), despite the diminished SR Ca content [10;22]. However, in later stages of HF, abnormally high activity of RyRs due to increased sensitivity to [Ca]SR may lead to shortened refractoriness of SR Ca release during diastole (Fig 1B). Premature reopening of RyRs can slow relaxation, exacerbate reduction of SR Ca content, and promote generation of spontaneous Ca sparks which can ignite arrhythmogenic Ca waves [9;10] contributing to diastolic and systolic dysfunction of HF. Indeed, a growing body of evidence indicates that RyR-mediated Ca leak due to shortened refractoriness is significantly increased in HF [1;7;66;89;98;110;114] (Fig 1B). Augmentation of SR Ca leak is one of the earliest alterations of Ca handling during the progression of HF that precedes changes in the amplitude of the Ca transient [10]. Increased RyR-mediated Ca leak, besides contributing to a reduction of SR Ca content, can promote Ca-dependent arrhythmias. This also implies that during diastole SERCA cannot achieve its maximal thermodynamic efficiency and more ATP must be consumed to balance the increased SR Ca leak. Thus, SR Ca leak is energetically costly, particularly in the metabolically compromised failing heart.
Molecular basis of RyR dysfunction in HF
The RyR in HF myocytes is characterized by defective interactions between regulatory domains and/or dissociation of several important proteins form the RyR complex. Moreover, the RyR becomes more phosphorylated and oxidized. These modifications of the RyR have been suggested contributing to SR Ca mishandling, contractile dysfunction, and arrhythmias of the failing heart.
Rearrangements of the RyR complex in HF
Increased activity of the RyR in HF has been attributed to increased sensitivity to [Ca]SR [51]. This defect can be caused by chronic dissociation of several auxiliary proteins (junctin, triadin and CASQ) that form the luminal Ca sensor of the RyR [37]. Although the total level of CASQ does not change in HF [32;51;85], it has been suggested that abnormal post-translational processing of CASQ can cause its defective trafficking [48]. This can potentially lead to a local depletion of CASQ in the junctional SR. Additionally, the levels of triadin and junctin were reported as being dramatically reduced in human end stage HF [29].
It has been proposed that FKBP12.6 binds to the cytosolic domain of RyR to stabilize the interaction among RyR subunits [15;65] or between neighboring tetramers [64;65]. It has been shown that during HF, chronic RyR phosphorylation by PKA causes dissociation of FKBP12.6 from the channel. This alteration in RyR structure leads to an increase in the channel activity [110] [66]. Moreover, it has been suggested that FKBP12.6 is also responsible for stabilizing intra-domain interactions within the RyR subunit. In healthy hearts, zipping between N-terminal and central domains keeps the RyR in the closed state [42]. During HF, dissociation of FKBP12.6 from the channel causes domain unzipping and SR Ca leak [71;109]. However, despite of many years of research, there is still controversy about the functional role of FKBP12.6 and PKA-mediated RyR phosphorylation [33;106].
Additionally, it has been shown that a defective inter-domain interaction in the RyR from the failing heart can lead to dissociation of CaM from the complex [1;72] which may affect RyR function [108].
RyR phosphorylation in HF
The phosphorylation state of the RyR is regulated by the balanced activities of kinases (PKA and CaMKII), phosphatases (PP1, PP2A and PP2B) and phosphodiesterase (4D3), all of which are bound to the channel [5;12;55;63]. In many animal HF models and human HF, activity and expression of CaMKII are increased [1;94]. However, this does not always translate into increased levels of CaMKII in the complex with the RyR [8;66]. On the other hand, several groups have suggested that an increased level of RyR phosphorylation in HF can be explained by decreased levels of phosphatases PP1 and PP2A [1;8;66] and phosphodiesterase 4D3 [55] associated with the RyR. Functional consequences of RyR phosphorylation have been recently summarized in several reviews [28;69]. Despite a major collective effort, the role of PKA-dependent phosphorylation in modulation of RyR function in normal and failing hearts remains a subject of heated debate [13;101]. Studies from different laboratories have yielded conflicting results regarding the role of RyR phosphorylation at Ser-2808 in the progression of HF, which ranged from having an essential role [66;71;87;103] to having only a very limited function [8;11;45;111]. In contrast, increased RyR activity due to enhanced phosphorylation of RyR at CaMKII site Ser-2814 appears to be a more consistent finding. Phosphorylation of RyR at the CaMKII site has been demonstrated in numerous (but not all [52]) HF models, and CaMKII inhibition was shown to attenuate abnormalities in Ca handling in myocytes from HF [1;10;77;94;112].
RyR oxidation in HF
Modulation of RyR activity by reactive oxygen species (ROS) in HF has been suggested to play an important role in the observed abnormality in Ca regulation [68;98]. In the healthy heart, the RyR subunit contains ~21 cysteines that are in the reduced state [107]. During development of HF, the number of free thiols is dramatically decreased. Incubation of HF myocytes with ROS scavengers and antioxidants was able, to a large degree, to restore Ca transient amplitude and reduce RyR-mediated SR Ca leak [10]. It has also been suggested that normalization of redox status of RyRs from failing hearts reverses inter-domain unzipping, thereby stabilizing RyR function [68]. Although HF is commonly seen as a state of oxidative stress, effective antioxidant strategies for treatment of HF symptoms have been so far a complete failure [62;78]. The major limitation to general antioxidant therapies is the inability to target ROS at distinct organelles. Thus, identifying sources of ROS production in HF myocytes will help to design a more beneficial approach that can prevent oxidative stress locally.
Mechanism of SERCA dysfunction in HF
Alteration of the SERCA/PLB complex in HF
Downregulation of SERCA2a at the mRNA and protein levels have been shown in numerous studies of HF [2;20;26;39;45;49;67;70;97]. All these studies highlight the critical role of SERCA in abnormal Ca homeostasis and contractile dysfunction of HF. However, there are several publications that did not report any alteration in the SERCA expression level in human HF [56;82-84] and animal HF models [51;75]. Moreover, it has been shown that HF is also associated with a decrease in the PLB expression level [2;45;70], but to a lesser degree than SERCA [38;49;67]. The decreased SERCA-PLB ratio can lead to inhibition of SR Ca uptake in HF, because an increased fraction of SERCA would be inhibited by PLB binding. On the contrary, the level of PLB remains unchanged in HF models characterized by an unchanged SERCA level [1;51;56;82;83].
In addition, the diminished SR Ca uptake in HF can be a result of post-translational modifications of the SERCA/PLB complex. It has been shown that the responsiveness of SERCA to activation by PKA was significantly decreased in the failing heart compared to non-failing myocardium [82]. As a result, the PLB phosphorylation level at the PKA-specific site was significantly decreased in HF. Several studies associated diminished SERCA activity in HF with decreased phosphorylation of PLB at both PKA and CaMKII sites [84;85]. Conversely, Currie and Smith demonstrated that PLB phosphorylation was increased in a rabbit HF model [20]. Decreased SR Ca uptake in the failing heart was explained by down-regulation of the SERCA expression level. Another study showed that the PLB phosphorylation level was increased at the CaMKII site but was decreased at the PKA site [1]. The authors suggested that the overall PLB phosphorylation level in HF would have only a minor effect on SERCA activity.
There is only very limited information regarding post-translational modifications of the SERCA protein in HF. It has been reported that the contractile dysfunction in Gαq-induced cardiomyopathy was mediated by inhibition of SERCA activity via oxidation of cysteine residue(s) on the pump [53]. Another study has shown that the depressed SR Ca uptake of failing myocytes is associated with dissociation of the small ubiquitin-related modifier 1 (SUMO1) from SERCA [47]. It has been suggested that restoring the sumoylation level of SERCA can stabilize the pump structure and improve SR Ca uptake in mouse and human failing hearts.
Conclusion and Perspective
Heart failure remains a major health problem of the western world. Essential to designing new therapeutic approaches for treating HF symptoms is a better understanding of the molecular mechanisms of this pathogenesis. Over the last two decades, significant progress has been made in this direction. Numerous studies demonstrate that abnormal SR Ca handling in HF myocytes is the primary defect that causes contractile dysfunction and promotes arrhythmogenesis. It becomes clear that functional impairment of two major Ca regulatory systems, the RyR and the SERCA complexes, contributes in great degree to aberrant SR Ca homeostasis. Although it has not yet been systematically explored, the degree of RyR or SERCA malfunctions in SR Ca mishandling has been shown to be dependent on the stage and etiology of HF. For example, study of myocardium from human patients with HF revealed distinct differences in SR Ca handling between dilated cardiomyopathy (DCM) and ischemic cardiomyopathy (ICM) [86]. The abnormal Ca homeostasis in ICM is primarily due to impaired SR Ca uptake, whereas in DCM dysfunctional SR Ca release is the major contributor in Ca mishandling. Therefore, the development of etiology or stage-specific therapeutic strategies that target the particular malfunctioning component of SR Ca homeostasis remains the highest priority.
Acknowledgments
This work was supported by the McCormick Foundation and The Schweppe Foundation (AVZ), Rhode Island Foundation (DT) and the National Institute of Health Grant T32-HL094300-03 (JAR and WL).
REFERENCE LIST
- 1.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 2.Armoundas AA, Rose J, Aggarwal R, Stuyvers BD, O’Rourke B, Kass DA, Marban E, Shorofsky SR, Tomaselli GF, William BC. Cellular and molecular determinants of altered Ca2+ handling in the failing rabbit heart: primary defects in SR Ca2+ uptake and release mechanisms. Am J Physiol Heart Circ Physiol. 2007;292:H1607–H1618. doi: 10.1152/ajpheart.00525.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balijepalli RC, Lokuta AJ, Maertz NA, Buck JM, Haworth RA, Valdivia HH, Kamp TJ. Depletion of T-tubules and specific subcellular changes in sarcolemmal proteins in tachycardia-induced heart failure. Cardiovasc Res. 2003;59:67–77. doi: 10.1016/s0008-6363(03)00325-0. [DOI] [PubMed] [Google Scholar]
- 4.Balshaw DM, Xu L, Yamaguchi N, Pasek DA, Meissner G. Calmodulin binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor) J Biol Chem. 2001;276:20144–20153. doi: 10.1074/jbc.M010771200. [DOI] [PubMed] [Google Scholar]
- 5.Bauman AL, Michel JJ, Henson E, Dodge-Kafka KL, Kapiloff MS. The mAKAP signalosome and cardiac myocyte hypertrophy. IUBMB Life. 2007;59:163–169. doi: 10.1080/15216540701358593. [DOI] [PubMed] [Google Scholar]
- 6.Bayer JD, Narayan SM, Lalani GG, Trayanova NA. Rate-dependent action potential alternans in human heart failure implicates abnormal intracellular calcium handling. Heart Rhythm. 2010;7:1093–1101. doi: 10.1016/j.hrthm.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belevych A, Kubalova Z, Terentyev D, Hamlin RL, Carnes CA, Gyorke S. Enhanced ryanodine receptor-mediated calcium leak determines reduced sarcoplasmic reticulum calcium content in chronic canine heart failure. Biophys J. 2007;93:4083–4092. doi: 10.1529/biophysj.107.114546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belevych AE, Sansom SE, Terentyeva R, Ho HT, Nishijima Y, Martin MM, Jindal HK, Rochira JA, Kunitomo Y, Abdellatif M, Carnes CA, Elton TS, Gyorke S, Terentyev D. MicroRNA-1 and -133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PLoS One. 2011;6:e28324. doi: 10.1371/journal.pone.0028324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belevych AE, Terentyev D, Terentyeva R, Ho HT, Gyorke I, Bonilla IM, Carnes CA, Billman GE, Gyorke S. Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ Res. 2012;110:569–577. doi: 10.1161/CIRCRESAHA.111.260455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belevych AE, Terentyev D, Terentyeva R, Nishijima Y, Sridhar A, Hamlin RL, Carnes CA, Gyorke S. The relationship between arrhythmogenesis and impaired contractility in heart failure: role of altered ryanodine receptor function. Cardiovasc Res. 2011;90:493–502. doi: 10.1093/cvr/cvr025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benkusky NA, Weber CS, Scherman JA, Farrell EF, Hacker TA, John MC, Powers PA, Valdivia HH. Intact beta-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase A phosphorylation site in the cardiac ryanodine receptor. Circ Res. 2007;101:819–829. doi: 10.1161/CIRCRESAHA.107.153007. [DOI] [PubMed] [Google Scholar]
- 12.Bers DM. Macromolecular complexes regulating cardiac ryanodine receptor function. J Mol Cell Cardiol. 2004;37:417–429. doi: 10.1016/j.yjmcc.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 13.Bers DM. Ryanodine receptor S2808 phosphorylation in heart failure: smoking gun or red herring. Circ Res. 2012;110:796–799. doi: 10.1161/CIRCRESAHA.112.265579. [DOI] [PubMed] [Google Scholar]
- 14.Bovo E, Mazurek SR, Blatter LA, Zima AV. Regulation of sarcoplasmic reticulum Ca2+ leak by cytosolic Ca2+ in rabbit ventricular myocytes. J Physiol. 2011;589:6039–6050. doi: 10.1113/jphysiol.2011.214171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brillantes AM, Allen P, Takahashi T, Izumo S, Marks AR. Differences in cardiac calcium release channel (ryanodine receptor) expression in myocardium from patients with end-stage heart failure caused by ischemic versus dilated cardiomyopathy. Circ Res. 1992;71:18–26. doi: 10.1161/01.res.71.1.18. [DOI] [PubMed] [Google Scholar]
- 16.Brochet DX, Yang D, Di Maio A, Lederer WJ, Franzini-Armstrong C, Cheng H. Ca2+ blinks: rapid nanoscopic store calcium signaling. Proc Natl Acad Sci U S A. 2005;102:3099–3104. doi: 10.1073/pnas.0500059102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen B, Guo A, Zhang C, Chen R, Zhu Y, Hong J, Kutschke W, Zimmerman K, Weiss RM, Zingman L, Anderson ME, Wehrens XH, Song LS. Critical roles of junctophilin-2 in T-tubule and excitation-contraction coupling maturation during postnatal development. Cardiovasc Res. 2013;100:54–62. doi: 10.1093/cvr/cvt180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 19.Crossman DJ, Ruygrok PN, Soeller C, Cannell MB. Changes in the organization of excitation-contraction coupling structures in failing human heart. PLoS One. 2011;6:e17901. doi: 10.1371/journal.pone.0017901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Currie S, Smith GL. Enhanced phosphorylation of phospholamban and downregulation of sarco/endoplasmic reticulum Ca2+ ATPase type 2 (SERCA 2) in cardiac sarcoplasmic reticulum from rabbits with heart failure. Cardiovasc Res. 1999;41:135–146. doi: 10.1016/s0008-6363(98)00241-7. [DOI] [PubMed] [Google Scholar]
- 21.Diaz ME, Trafford AW, O’Neill SC, Eisner DA. Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release. J Physiol. 1997;501(Pt 1):3–16. doi: 10.1111/j.1469-7793.1997.003bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Domeier TL, Blatter LA, Zima AV. Alteration of sarcoplasmic reticulum Ca2+ release termination by ryanodine receptor sensitization and in heart failure. J Physiol. 2009;587:5197–5209. doi: 10.1113/jphysiol.2009.177576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol. 1983;245:C1–14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- 24.Fabiato A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol. 1985;85:247–289. doi: 10.1085/jgp.85.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farrell EF, Antaramian A, Rueda A, Gomez AM, Valdivia HH. Sorcin inhibits calcium release and modulates excitation-contraction coupling in the heart. J Biol Chem. 2003;278:34660–34666. doi: 10.1074/jbc.M305931200. [DOI] [PubMed] [Google Scholar]
- 26.Feldman AM, Weinberg EO, Ray PE, Lorell BH. Selective changes in cardiac gene expression during compensated hypertrophy and the transition to cardiac decompensation in rats with chronic aortic banding. Circ Res. 1993;73:184–192. doi: 10.1161/01.res.73.1.184. [DOI] [PubMed] [Google Scholar]
- 27.Garbino A, van Oort RJ, Dixit SS, Landstrom AP, Ackerman MJ, Wehrens XH. Molecular evolution of the junctophilin gene family. Physiol Genomics. 2009;37:175–186. doi: 10.1152/physiolgenomics.00017.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.George CH. Sarcoplasmic reticulum Ca2+ leak in heart failure: mere observation or functional relevance? Cardiovasc Res. 2008;77:302–314. doi: 10.1093/cvr/cvm006. [DOI] [PubMed] [Google Scholar]
- 29.Gergs U, Berndt T, Buskase J, Jones LR, Kirchhefer U, Muller FU, Schluter KD, Schmitz W, Neumann J. On the role of junctin in cardiac Ca2+ handling, contractility, and heart failure. Am J Physiol Heart Circ Physiol. 2007;293:H728–H734. doi: 10.1152/ajpheart.01187.2006. [DOI] [PubMed] [Google Scholar]
- 30.Go LO, Moschella MC, Watras J, Handa KK, Fyfe BS, Marks AR. Differential regulation of two types of intracellular calcium release channels during end-stage heart failure. J Clin Invest. 1995;95:888–894. doi: 10.1172/JCI117739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA, Lederer WJ. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276:800–806. doi: 10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- 32.Guo T, Ai X, Shannon TR, Pogwizd SM, Bers DM. Intra-sarcoplasmic reticulum free [Ca2+] and buffering in arrhythmogenic failing rabbit heart. Circ Res. 2007;101:802–810. doi: 10.1161/CIRCRESAHA.107.152140. [DOI] [PubMed] [Google Scholar]
- 33.Guo T, Cornea RL, Huke S, Camors E, Yang Y, Picht E, Fruen BR, Bers DM. Kinetics of FKBP12.6 binding to ryanodine receptors in permeabilized cardiac myocytes and effects on Ca sparks. Circ Res. 2010;106:1743–1752. doi: 10.1161/CIRCRESAHA.110.219816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo T, Gillespie D, Fill M. Ryanodine receptor current amplitude controls Ca2+ sparks in cardiac muscle. Circ Res. 2012;111:28–36. doi: 10.1161/CIRCRESAHA.112.265652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gyorke I, Gyorke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J. 1998;75:2801–2810. doi: 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gyorke I, Hester N, Jones LR, Gyorke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004;86:2121–2128. doi: 10.1016/S0006-3495(04)74271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gyorke S, Terentyev D. Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovasc Res. 2008;77:245–255. doi: 10.1093/cvr/cvm038. [DOI] [PubMed] [Google Scholar]
- 38.Hasenfuss G, Meyer M, Schillinger W, Preuss M, Pieske B, Just H. Calcium handling proteins in the failing human heart. Basic Res Cardiol. 1997;92(Suppl 1):87–93. doi: 10.1007/BF00794072. [DOI] [PubMed] [Google Scholar]
- 39.Hasenfuss G, Reinecke H, Studer R, Meyer M, Pieske B, Holtz J, Holubarsch C, Posival H, Just H, Drexler H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2+)-ATPase in failing and nonfailing human myocardium. Circ Res. 1994;75:434–442. doi: 10.1161/01.res.75.3.434. [DOI] [PubMed] [Google Scholar]
- 40.He J, Conklin MW, Foell JD, Wolff MR, Haworth RA, Coronado R, Kamp TJ. Reduction in density of transverse tubules and L-type Ca(2+) channels in canine tachycardia-induced heart failure. Cardiovasc Res. 2001;49:298–307. doi: 10.1016/s0008-6363(00)00256-x. [DOI] [PubMed] [Google Scholar]
- 41.Hobai IA, O’Rourke B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation. 2001;103:1577–1584. doi: 10.1161/01.cir.103.11.1577. [DOI] [PubMed] [Google Scholar]
- 42.Ikemoto N, Yamamoto T. Regulation of calcium release by interdomain interaction within ryanodine receptors. Front Biosci. 2002;7:d671–d683. doi: 10.2741/A803. [DOI] [PubMed] [Google Scholar]
- 43.Jiang D, Chen W, Wang R, Zhang L, Chen SR. Loss of luminal Ca2+ activation in the cardiac ryanodine receptor is associated with ventricular fibrillation and sudden death. Proc Natl Acad Sci U S A. 2007;104:18309–18314. doi: 10.1073/pnas.0706573104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H, Chen SR. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR) Proc Natl Acad Sci U S A. 2004;101:13062–13067. doi: 10.1073/pnas.0402388101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang MT, Lokuta AJ, Farrell EF, Wolff MR, Haworth RA, Valdivia HH. Abnormal Ca2+ release, but normal ryanodine receptors, in canine and human heart failure. Circ Res. 2002;91:1015–1022. doi: 10.1161/01.res.0000043663.08689.05. [DOI] [PubMed] [Google Scholar]
- 46.Kalyanasundaram A, Lacombe VA, Belevych AE, Brunello L, Carnes CA, Janssen PM, Knollmann BC, Periasamy M, Gyorke S. Up-regulation of sarcoplasmic reticulum Ca(2+) uptake leads to cardiac hypertrophy, contractile dysfunction and early mortality in mice deficient in CASQ2. Cardiovasc Res. 2013;98:297–306. doi: 10.1093/cvr/cvs334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E, Park WJ, Hajjar RJ. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 2011;477:601–605. doi: 10.1038/nature10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kiarash A, Kelly CE, Phinney BS, Valdivia HH, Abrams J, Cala SE. Defective glycosylation of calsequestrin in heart failure. Cardiovasc Res. 2004;63:264–272. doi: 10.1016/j.cardiores.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 49.Kiss E, Ball NA, Kranias EG, Walsh RA. Differential changes in cardiac phospholamban and sarcoplasmic reticular Ca(2+)-ATPase protein levels. Effects on Ca2+ transport and mechanics in compensated pressure-overload hypertrophy and congestive heart failure. Circ Res. 1995;77:759–764. doi: 10.1161/01.res.77.4.759. [DOI] [PubMed] [Google Scholar]
- 50.Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res. 2012;110:1646–1660. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kubalova Z, Terentyev D, Viatchenko-Karpinski S, Nishijima Y, Gyorke I, Terentyeva R, da Cunha DN, Sridhar A, Feldman DS, Hamlin RL, Carnes CA, Gyorke S. Abnormal intrastore calcium signaling in chronic heart failure. Proc Natl Acad Sci U S A. 2005;102:14104–14109. doi: 10.1073/pnas.0504298102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kushnir A, Shan J, Betzenhauser MJ, Reiken S, Marks AR. Role of CaMKIIdelta phosphorylation of the cardiac ryanodine receptor in the force frequency relationship and heart failure. Proc Natl Acad Sci U S A. 2010;107:10274–10279. doi: 10.1073/pnas.1005843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lancel S, Qin F, Lennon SL, Zhang J, Tong X, Mazzini MJ, Kang YJ, Siwik DA, Cohen RA, Colucci WS. Oxidative posttranslational modifications mediate decreased SERCA activity and myocyte dysfunction in Galphaq-overexpressing mice. Circ Res. 2010;107:228–232. doi: 10.1161/CIRCRESAHA.110.217570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laver DR, Kong CH, Imtiaz MS, Cannell MB. Termination of calcium-induced calcium release by induction decay: an emergent property of stochastic channel gating and molecular scale architecture. J Mol Cell Cardiol. 2013;54:98–100. doi: 10.1016/j.yjmcc.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 55.Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SL, Conti M, Marks AR. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123:25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Linck B, Boknik P, Eschenhagen T, Muller FU, Neumann J, Nose M, Jones LR, Schmitz W, Scholz H. Messenger RNA expression and immunological quantification of phospholamban and SR-Ca(2+)-ATPase in failing and nonfailing human hearts. Cardiovasc Res. 1996;31:625–632. [PubMed] [Google Scholar]
- 57.Lindner M, Erdmann E, Beuckelmann DJ. Calcium content of the sarcoplasmic reticulum in isolated ventricular myocytes from patients with terminal heart failure. J Mol Cell Cardiol. 1998;30:743–749. doi: 10.1006/jmcc.1997.0626. [DOI] [PubMed] [Google Scholar]
- 58.Litwin SE, Zhang D, Bridge JH. Dyssynchronous Ca(2+) sparks in myocytes from infarcted hearts. Circ Res. 2000;87:1040–1047. doi: 10.1161/01.res.87.11.1040. [DOI] [PubMed] [Google Scholar]
- 59.Lompre AM, Hajjar RJ, Harding SE, Kranias EG, Lohse MJ, Marks AR. Ca2+ cycling and new therapeutic approaches for heart failure. Circulation. 2010;121:822–830. doi: 10.1161/CIRCULATIONAHA.109.890954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Louch WE, Bito V, Heinzel FR, Macianskiene R, Vanhaecke J, Flameng W, Mubagwa K, Sipido KR. Reduced synchrony of Ca2+ release with loss of T-tubules-a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc Res. 2004;62:63–73. doi: 10.1016/j.cardiores.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 61.Lyon AR, MacLeod KT, Zhang Y, Garcia E, Kanda GK, Lab MJ, Korchev YE, Harding SE, Gorelik J. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci U S A. 2009;106:6854–6859. doi: 10.1073/pnas.0809777106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mak S, Newton GE. The oxidative stress hypothesis of congestive heart failure: radical thoughts. Chest. 2001;120:2035–2046. doi: 10.1378/chest.120.6.2035. [DOI] [PubMed] [Google Scholar]
- 63.Marks AR. Cardiac intracellular calcium release channels: role in heart failure. Circ Res. 2000;87:8–11. doi: 10.1161/01.res.87.1.8. [DOI] [PubMed] [Google Scholar]
- 64.Marx SO, Gaburjakova J, Gaburjakova M, Henrikson C, Ondrias K, Marks AR. Coupled gating between cardiac calcium release channels (ryanodine receptors) Circ Res. 2001;88:1151–1158. doi: 10.1161/hh1101.091268. [DOI] [PubMed] [Google Scholar]
- 65.Marx SO, Ondrias K, Marks AR. Coupled gating between individual skeletal muscle Ca2+ release channels (ryanodine receptors) Science. 1998;281:818–821. doi: 10.1126/science.281.5378.818. [DOI] [PubMed] [Google Scholar]
- 66.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 67.Meyer M, Schillinger W, Pieske B, Holubarsch C, Heilmann C, Posival H, Kuwajima G, Mikoshiba K, Just H, Hasenfuss G. Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation. 1995;92:778–784. doi: 10.1161/01.cir.92.4.778. [DOI] [PubMed] [Google Scholar]
- 68.Mochizuki M, Yano M, Oda T, Tateishi H, Kobayashi S, Yamamoto T, Ikeda Y, Ohkusa T, Ikemoto N, Matsuzaki M. Scavenging free radicals by low-dose carvedilol prevents redox-dependent Ca2+ leak via stabilization of ryanodine receptor in heart failure. J Am Coll Cardiol. 2007;49:1722–1732. doi: 10.1016/j.jacc.2007.01.064. [DOI] [PubMed] [Google Scholar]
- 69.Niggli E, Ullrich ND, Gutierrez D, Kyrychenko S, Polakova E, Shirokova N. Posttranslational modifications of cardiac ryanodine receptors: Ca(2+) signaling and EC-coupling. Biochim Biophys Acta. 2012 doi: 10.1016/j.bbamcr.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.O’Rourke B, Kass DA, Tomaselli GF, Kaab S, Tunin R, Marban E. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I: experimental studies. Circ Res. 1999;84:562–570. doi: 10.1161/01.res.84.5.562. [DOI] [PubMed] [Google Scholar]
- 71.Oda T, Yano M, Yamamoto T, Tokuhisa T, Okuda S, Doi M, Ohkusa T, Ikeda Y, Kobayashi S, Ikemoto N, Matsuzaki M. Defective regulation of interdomain interactions within the ryanodine receptor plays a key role in the pathogenesis of heart failure. Circulation. 2005;111:3400–3410. doi: 10.1161/CIRCULATIONAHA.104.507921. [DOI] [PubMed] [Google Scholar]
- 72.Ono M, Yano M, Hino A, Suetomi T, Xu X, Susa T, Uchinoumi H, Tateishi H, Oda T, Okuda S, Doi M, Kobayashi S, Yamamoto T, Koseki N, Kyushiki H, Ikemoto N, Matsuzaki M. Dissociation of calmodulin from cardiac ryanodine receptor causes aberrant Ca(2+) release in heart failure. Cardiovasc Res. 2010;87:609–617. doi: 10.1093/cvr/cvq108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Piacentino V, III, Weber CR, Chen X, Weisser-Thomas J, Margulies KB, Bers DM, Houser SR. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res. 2003;92:651–658. doi: 10.1161/01.RES.0000062469.83985.9B. [DOI] [PubMed] [Google Scholar]
- 74.Pieske B, Kretschmann B, Meyer M, Holubarsch C, Weirich J, Posival H, Minami K, Just H, Hasenfuss G. Alterations in intracellular calcium handling associated with the inverse force-frequency relation in human dilated cardiomyopathy. Circulation. 1995;92:1169–1178. doi: 10.1161/01.cir.92.5.1169. [DOI] [PubMed] [Google Scholar]
- 75.Pogwizd SM, Qi M, Yuan W, Samarel AM, Bers DM. Upregulation of Na(+)/Ca(2+) exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ Res. 1999;85:1009–1019. doi: 10.1161/01.res.85.11.1009. [DOI] [PubMed] [Google Scholar]
- 76.Pritchard TJ, Kranias EG. Junctin and the histidine-rich Ca2+ binding protein: potential roles in heart failure and arrhythmogenesis. J Physiol. 2009;587:3125–3133. doi: 10.1113/jphysiol.2009.172171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Respress JL, van Oort RJ, Li N, Rolim N, Dixit SS, deAlmeida A, Voigt N, Lawrence WS, Skapura DG, Skardal K, Wisloff U, Wieland T, Ai X, Pogwizd SM, Dobrev D, Wehrens XH. Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ Res. 2012;110:1474–1483. doi: 10.1161/CIRCRESAHA.112.268094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Santos CX, Anilkumar N, Zhang M, Brewer AC, Shah AM. Redox signaling in cardiac myocytes. Free Radic Biol Med. 2011;50:777–793. doi: 10.1016/j.freeradbiomed.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sato D, Bers DM. How does stochastic ryanodine receptor-mediated Ca leak fail to initiate a Ca spark? Biophys J. 2011;101:2370–2379. doi: 10.1016/j.bpj.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schlotthauer K, Bers DM. Sarcoplasmic reticulum Ca(2+) release causes myocyte depolarization. Underlying mechanism and threshold for triggered action potentials. Circ Res. 2000;87:774–780. doi: 10.1161/01.res.87.9.774. [DOI] [PubMed] [Google Scholar]
- 81.Schmidt U, Hajjar RJ, Helm PA, Kim CS, Doye AA, Gwathmey JK. Contribution of abnormal sarcoplasmic reticulum ATPase activity to systolic and diastolic dysfunction in human heart failure. J Mol Cell Cardiol. 1998;30:1929–1937. doi: 10.1006/jmcc.1998.0748. [DOI] [PubMed] [Google Scholar]
- 82.Schmidt U, Hajjar RJ, Kim CS, Lebeche D, Doye AA, Gwathmey JK. Human heart failure: cAMP stimulation of SR Ca(2+)-ATPase activity and phosphorylation level of phospholamban. Am J Physiol. 1999;277:H474–H480. doi: 10.1152/ajpheart.1999.277.2.H474. [DOI] [PubMed] [Google Scholar]
- 83.Schwinger RH, Bohm M, Schmidt U, Karczewski P, Bavendiek U, Flesch M, Krause EG, Erdmann E. Unchanged protein levels of SERCA II and phospholamban but reduced Ca2+ uptake and Ca(2+)-ATPase activity of cardiac sarcoplasmic reticulum from dilated cardiomyopathy patients compared with patients with nonfailing hearts. Circulation. 1995;92:3220–3228. doi: 10.1161/01.cir.92.11.3220. [DOI] [PubMed] [Google Scholar]
- 84.Schwinger RH, Bolck B, Munch G, Brixius K, Muller-Ehmsen J, Erdmann E. cAMP-dependent protein kinase A-stimulated sarcoplasmic reticulum function in heart failure. Ann N Y Acad Sci. 1998;853:240–250. doi: 10.1111/j.1749-6632.1998.tb08272.x. [DOI] [PubMed] [Google Scholar]
- 85.Schwinger RH, Munch G, Bolck B, Karczewski P, Krause EG, Erdmann E. Reduced Ca(2+)-sensitivity of SERCA 2a in failing human myocardium due to reduced serin-16 phospholamban phosphorylation. J Mol Cell Cardiol. 1999;31:479–491. doi: 10.1006/jmcc.1998.0897. [DOI] [PubMed] [Google Scholar]
- 86.Sen L, Cui G, Fonarow GC, Laks H. Differences in mechanisms of SR dysfunction in ischemic vs. idiopathic dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. 2000;279:H709–H718. doi: 10.1152/ajpheart.2000.279.2.H709. [DOI] [PubMed] [Google Scholar]
- 87.Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A, Dura M, Chen BX, Marks AR. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J Clin Invest. 2010;120:4375–4387. doi: 10.1172/JCI37649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shannon TR, Ginsburg KS, Bers DM. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res. 2002;91:594–600. doi: 10.1161/01.res.0000036914.12686.28. [DOI] [PubMed] [Google Scholar]
- 89.Shannon TR, Pogwizd SM, Bers DM. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res. 2003;93:592–594. doi: 10.1161/01.RES.0000093399.11734.B3. [DOI] [PubMed] [Google Scholar]
- 90.Simmerman HK, Collins JH, Theibert JL, Wegener AD, Jones LR. Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. J Biol Chem. 1986;261:13333–13341. [PubMed] [Google Scholar]
- 91.Sitsapesan R, Williams AJ. Regulation of the gating of the sheep cardiac sarcoplasmic reticulum Ca(2+)-release channel by luminal Ca2+ J Membr Biol. 1994;137:215–226. doi: 10.1007/BF00232590. [DOI] [PubMed] [Google Scholar]
- 92.Song LS, Pi Y, Kim SJ, Yatani A, Guatimosim S, Kudej RK, Zhang Q, Cheng H, Hittinger L, Ghaleh B, Vatner DE, Lederer WJ, Vatner SF. Paradoxical cellular Ca2+ signaling in severe but compensated canine left ventricular hypertrophy. Circ Res. 2005;97:457–464. doi: 10.1161/01.RES.0000179722.79295.d4. [DOI] [PubMed] [Google Scholar]
- 93.Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci U S A. 2006;103:4305–4310. doi: 10.1073/pnas.0509324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sossalla S, Fluschnik N, Schotola H, Ort KR, Neef S, Schulte T, Wittkopper K, Renner A, Schmitto JD, Gummert J, El-Armouche A, Hasenfuss G, Maier LS. Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ Res. 2010;107:1150–1161. doi: 10.1161/CIRCRESAHA.110.220418. [DOI] [PubMed] [Google Scholar]
- 95.Stern MD. Theory of excitation-contraction coupling in cardiac muscle. Biophys J. 1992;63:497–517. doi: 10.1016/S0006-3495(92)81615-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stern MD, Cheng H. Putting out the fire: what terminates calcium-induced calcium release in cardiac muscle? Cell Calcium. 2004;35:591–601. doi: 10.1016/j.ceca.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 97.Studer R, Reinecke H, Bilger J, Eschenhagen T, Bohm M, Hasenfuss G, Just H, Holtz J, Drexler H. Gene expression of the cardiac Na(+)-Ca2+ exchanger in end-stage human heart failure. Circ Res. 1994;75:443–453. doi: 10.1161/01.res.75.3.443. [DOI] [PubMed] [Google Scholar]
- 98.Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, de Blanco EC, Khanna S, Sen CK, Cardounel AJ, Carnes CA, Gyorke S. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res. 2008;103:1466–1472. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Terentyev D, Kubalova Z, Valle G, Nori A, Vedamoorthyrao S, Terentyeva R, Viatchenko-Karpinski S, Bers DM, Williams SC, Volpe P, Gyorke S. Modulation of SR Ca release by luminal Ca and calsequestrin in cardiac myocytes: effects of CASQ2 mutations linked to sudden cardiac death. Biophys J. 2008;95:2037–2048. doi: 10.1529/biophysj.107.128249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Terentyev D, Viatchenko-Karpinski S, Valdivia HH, Escobar AL, Gyorke S. Luminal Ca2+ controls termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circ Res. 2002;91:414–420. doi: 10.1161/01.res.0000032490.04207.bd. [DOI] [PubMed] [Google Scholar]
- 101.Valdivia HH. Ryanodine receptor phosphorylation and heart failure: phasing out S2808 and “criminalizing” S2814. Circ Res. 2012;110:1398–1402. doi: 10.1161/CIRCRESAHA.112.270876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Venetucci LA, Trafford AW, O’Neill SC, Eisner DA. The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc Res. 2008;77:285–292. doi: 10.1093/cvr/cvm009. [DOI] [PubMed] [Google Scholar]
- 103.Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci U S A. 2006;103:511–518. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, Weiss RM, Anderson ME, Cheng H, Song LS. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107:520–531. doi: 10.1161/CIRCRESAHA.109.212324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wilson LD, Jeyaraj D, Wan X, Hoeker GS, Said TH, Gittinger M, Laurita KR, Rosenbaum DS. Heart failure enhances susceptibility to arrhythmogenic cardiac alternans. Heart Rhythm. 2009;6:251–259. doi: 10.1016/j.hrthm.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xiao B, Sutherland C, Walsh MP, Chen SR. Protein kinase A phosphorylation at serine-2808 of the cardiac Ca2+-release channel (ryanodine receptor) does not dissociate 12.6-kDa FK506-binding protein (FKBP12.6) Circ Res. 2004;94:487–495. doi: 10.1161/01.RES.0000115945.89741.22. [DOI] [PubMed] [Google Scholar]
- 107.Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 108.Yang Y, Guo T, Oda T, Chakraborty A, Chen L, Uchinoumi H, Knowlton AA, Fruen BR, Cornea RL, Meissner G, Bers DM. Cardiac Myocyte Z-line Calmodulin is Mainly RyR2-Bound and Reduction is Arrhythmogenic and Occurs in Heart Failure. Circ Res. 2013 doi: 10.1161/CIRCRESAHA.114.302857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yano M, Okuda S, Oda T, Tokuhisa T, Tateishi H, Mochizuki M, Noma T, Doi M, Kobayashi S, Yamamoto T, Ikeda Y, Ohkusa T, Ikemoto N, Matsuzaki M. Correction of defective interdomain interaction within ryanodine receptor by antioxidant is a new therapeutic strategy against heart failure. Circulation. 2005;112:3633–3643. doi: 10.1161/CIRCULATIONAHA.105.555623. [DOI] [PubMed] [Google Scholar]
- 110.Yano M, Ono K, Ohkusa T, Suetsugu M, Kohno M, Hisaoka T, Kobayashi S, Hisamatsu Y, Yamamoto T, Kohno M, Noguchi N, Takasawa S, Okamoto H, Matsuzaki M. Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca(2+) leak through ryanodine receptor in heart failure. Circulation. 2000;102:2131–2136. doi: 10.1161/01.cir.102.17.2131. [DOI] [PubMed] [Google Scholar]
- 111.Zhang H, Makarewich CA, Kubo H, Wang W, Duran JM, Li Y, Berretta RM, Koch WJ, Chen X, Gao E, Valdivia HH, Houser SR. Hyperphosphorylation of the cardiac ryanodine receptor at serine 2808 is not involved in cardiac dysfunction after myocardial infarction. Circ Res. 2012;110:831–840. doi: 10.1161/CIRCRESAHA.111.255158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr, Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–417. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 113.Zhang T, Guo T, Mishra S, Dalton ND, Kranias EG, Peterson KL, Bers DM, Brown JH. Phospholamban ablation rescues sarcoplasmic reticulum Ca(2+) handling but exacerbates cardiac dysfunction in CaMKIIdelta(C) transgenic mice. Circ Res. 2010;106:354–362. doi: 10.1161/CIRCRESAHA.109.207423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zima AV, Bovo E, Bers DM, Blatter LA. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J Physiol. 2010;588:4743–4757. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zima AV, Picht E, Bers DM, Blatter LA. Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ Res. 2008;103:e105–e115. doi: 10.1161/CIRCRESAHA.107.183236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zsebo K, Yaroshinsky A, Rudy JJ, Wagner K, Greenberg B, Jessup M, Hajjar RJ. Long-Term Effects of AAV1/SERCA2a Gene Transfer in Patients With Severe Heart Failure: Analysis of Recurrent Cardiovascular Events and Mortality. Circ Res. 2014;114:101–108. doi: 10.1161/CIRCRESAHA.113.302421. [DOI] [PubMed] [Google Scholar]