

Abstract
Although the presence of antineutrophil cytoplasmic antibodies (ANCA) has been reported in patients with systemic sclerosis (SSc), the association of SSc and systemic vasculitis has rarely been described. We obtained information on cases of systemic vasculitis associated with SSc in France from the French Vasculitis Study Group and all members of the French Research Group on Systemic Sclerosis.
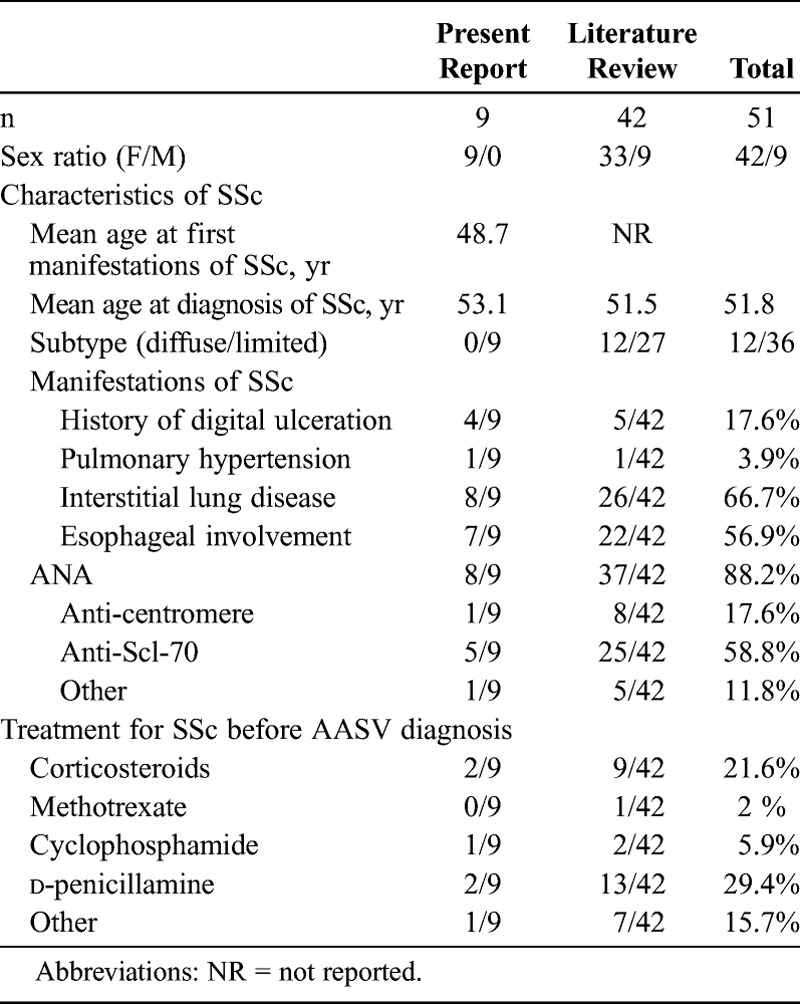
We identified 12 patients with systemic vasculitis associated with SSc: 9 with ANCA-associated systemic vasculitis (AASV) and 3 with mixed cryoglobulinemia vasculitis (MCV). In all AASV patients, SSc was of the limited type. The main complication of SSc was pulmonary fibrosis. Only 2 patients underwent a D-penicillamine regimen before the occurrence of AASV. The characteristics of AASV were microscopic polyangiitis (n = 7) and renal limited vasculitis (n = 2). Anti-myeloperoxidase antibodies were found in 8 of the 9 patients. The Five Factor Score was above 1 in 3 of the 9 patients. Of the 3 patients with MCV, Sjögren syndrome was confirmed in 2. We compared our findings with the results of a literature review (42 previously reported cases of AASV with SSc).
Although rare, vasculitis is a complication of SSc. AASV is the most frequent type, and its diagnosis can be challenging when the kidney is injured. Better awareness of this rare association could facilitate earlier diagnosis and appropriate management to reduce damage.
INTRODUCTION
Systemic sclerosis (SSc) is a chronic systemic fibrosing disease associated with autoimmune abnormalities such as antinuclear antibodies (ANA). The prevalence ranges from 4 to 489 cases per million individuals.13 The main manifestations are attributed to 3 features: tissue fibrosis, autoimmune disorder, and microvascular injury.
Fibrosis is responsible for the involvement of skin, lung, and gastrointestinal tract. Tissue biopsies reveal accumulation of extracellular matrix. Skin is almost always affected, except in the so-called sine scleroderma SSc. Scleroderma is usually classified into 2 subsets, according to the extent of skin involvement. In patients with limited cutaneous SSc (lcSSc), skin thickening is limited to the face, hands, and forearms, whereas in patients with diffuse cutaneous SSc (dcSSc), skin thickening affects the chest, abdomen, and/or upper arms.29 Interstitial lung disease, mainly nonspecific interstitial pneumonitis and less frequently usual interstitial pneumonitis, is found in up to 75% of SSc patients and still represents a therapeutic challenge, being a leading cause of mortality.44
ANA are detected in up to 95% of patients. The 2 main and specific subtypes are found to be exclusive; anti-Scl-70 (also called anti-topoisomerase I) antibodies seem to be more frequent in patients with dcSSc, interstitial lung disease, or scleroderma renal crisis. Anticentromere antibodies are more frequent in patients with lcSSc.39 More recently antibodies to anti-RNA-polymerase III have been found in patients with dcSSc, and are associated with increased risk for scleroderma renal crisis. Many other ANA can be found: anti-U3-RNP, anti-U1-RNP, and anti-Pm/Scl.
Small vessel vasculopathy is one of the first manifestations of the disease, preceding skin thickening and other symptoms. Raynaud phenomenon is associated with capillaroscopic changes, especially the presence of megacapillaries. Digital ulcers are usual during the course of disease. Two less frequent but potentially life-threatening features are related to the vascular component of SSc: pulmonary arterial hypertension, found in more than 10% of patients, and scleroderma renal crisis. Scleroderma renal crisis is usually revealed by hypertension, rapid progressive renal failure, and non-autoimmune hemolytic anemia with thrombocytopenia. Kidney biopsy reveals thrombotic microangiopathy with special features such as concentric edematous intimal proliferation and thickening of interlobular arteries leading to ischemic glomeruli. The treatment for scleroderma renal crisis is based on angiotensin-converting enzyme inhibitors.
Antineutrophil cytoplasmic antibodies (ANCA)-associated systemic vasculitis (AASV) is systemic necrotizing vasculitis of unknown etiology, including granulomatosis with polyangiitis (Wegener granulomatosis), microscopic polyangiitis, renal limited vasculitis, and Churg-Strauss syndrome. By indirect immunofluorescence, 2 major patterns of ANCA can be distinguished: a diffuse cytoplasmic staining (c-ANCA) mainly associated with anti-proteinase 3 (anti-PR3) antibodies, and a perinuclear pattern (p-ANCA) mainly associated with anti-myeloperoxidase (anti-MPO) antibodies. Drugs are a potential inductor of ANCA and AASV. Pauci-immune necrotizing glomerulonephritis is a frequent feature of AASV. The treatment of AASV is based on immunosuppressive drugs.
Vasculitis is not a typical finding in SSc. Nevertheless, the presence of ANCA has been observed in up to 11.7% of patients with SSc, and a few reports describe SSc patients with AASV. The association of these unrelated diseases has sometimes been attributed to a side effect of D-penicillamine treatment.21 Most cases are described as normotensive renal failure related to anti-MPO crescentic glomerulonephritis.5 Such cases of systemic vasculitis associated with SSc may be wrongly diagnosed as scleroderma renal crisis, resulting in inappropriate treatment.
We conducted the current study to assess the clinical features and prognosis of systemic vasculitis associated with SSc and to compare our cases with those reported in the literature.
METHODS
We obtained information on cases of systemic vasculitis and SSc in France from the French Vasculitis Study Group and members of the French Research Group on SSc. Data were collected by reviewing medical records in reference centers and departments with well-known experience in the field of SSc and systemic vasculitis.
We included only cases with a diagnosis of systemic vasculitis proven by biopsy. Clinical data on systemic vasculitis included general symptoms; cutaneous, neurologic, gastrointestinal, renal, or pulmonary involvement; delay between SSc and systemic vasculitis diagnosis; and delay between D-penicillamine regimen and occurrence of systemic vasculitis. The following laboratory data were recorded: C-reactive protein levels, creatininemia, proteinuria, urinary sediment, presence of ANCA, type of immunofluorescence, presence of anti-MPO and/or anti-PR3 antibodies, rheumatoid factor, cryoglobulinemia, and hepatitis B and hepatitis C virus serology.
All patients fulfilled the American Rheumatism Association classification criteria for SSc. SSc was classified according to the LeRoy classification criteria.29 Data on clinical (cutaneous, gastrointestinal, pulmonary, and cardiac involvement), laboratory, and immunologic manifestations were collected for each patient. All patients underwent transthoracic echocardiography, thoracic tomodensitometry during follow-up for SSc. Interstitial lung disease was defined as the presence of nonspecific interstitial pneumonia (NSIP) or usual interstitial pneumonia (UIP) findings on thoracic tomodensitometry. The following gastrointestinal manifestations were searched for: dysphagia, heartburn, alternating constipation and diarrhea, and pseudo-obstruction; the findings of endoscopic examinations were studied when the exams were performed.
The Five Factor Score was determined for each patient with ANCA-associated vasculitis.16
We performed a MEDLINE (National Library of Medicine, Bethesda, MD) literature review using the following key words: “systemic vasculitis,” “cryoglobulinemia,” “anti-neutrophil cytoplasmic antibody-associated vasculitis,” “Wegener’s granulomatosis,” “microscopic polyangiitis” and “systemic scleroderma.”
We compiled the results in tables describing the clinical and laboratory manifestations of SSc and systemic vasculitis in our cases, in the literature cases, and in the total 51 patients.
RESULTS
We identified 12 patients with systemic vasculitis associated with SSc: 9 patients with AASV and 3 with mixed cryoglobulinemia vasculitis (MCV). We previously reported 1 of the patients in the French literature.28 The cohort of patients with SSc is well known in 3 of the 5 centers participating in this study. These units are considered tertiary centers for SSc and systemic vasculitis. Six patients with AASV and 2 patients with MCV were identified in a cohort of 1120 patients with SSc.
The MEDLINE search yielded 51 cases of AASV associated with SSc. We analyzed 42 cases in the English or French literature.2–5,10–12,14,19–21,24,25,27,28,30–33,35,38,40,43,46,48–52 Seven additional patients had giant cell arteritis (n = 4) or Takayasu arteritis (n = 3) associated with SSc and were not included in this study.
Mixed Cryoglobulinemia
Three female patients (aged 60, 30, and 31 yr, respectively) presented with MCV (1 with type 2 cryoglobulinemia and 2 with type 3 cryoglobulinemia) and limited SSc (Table 1). No hepatitis C virus infection was detected. Associated Sjögren syndrome was diagnosed in 2 patients based on the American-European Consensus Group criteria. Vasculitis was diagnosed 6, 30, and 4 years, respectively, after SSc diagnosis. A diagnosis of cryoglobulinemia was proposed in view of the cutaneous and neurologic manifestations (multiple mononeuropathy). No other organ involvement was observed. Cutaneous biopsy confirmed the vasculitis, but immunofluorescence examination of tissue biopsy was not available. Low C4 values were observed in each case (respectively 3, 10, and 4 mg/dL). So the diagnosis of MCV was sustained on the basis of histopathologic findings (leukocytoclastic vasculitis), circulating cryoglobulins, and low C4 values. Treatment was based on corticosteroids, cyclophosphamide, and plasmapheresis. None of the patients developed scleroderma renal crisis after receiving high doses of corticosteroids. All patients had remission of MCV without relapse. One patient received mycophenolate mofetil as a maintenance regimen.
TABLE 1.
Patients With Mixed Cryoglobulinemia and Systemic Sclerosis, Present Report
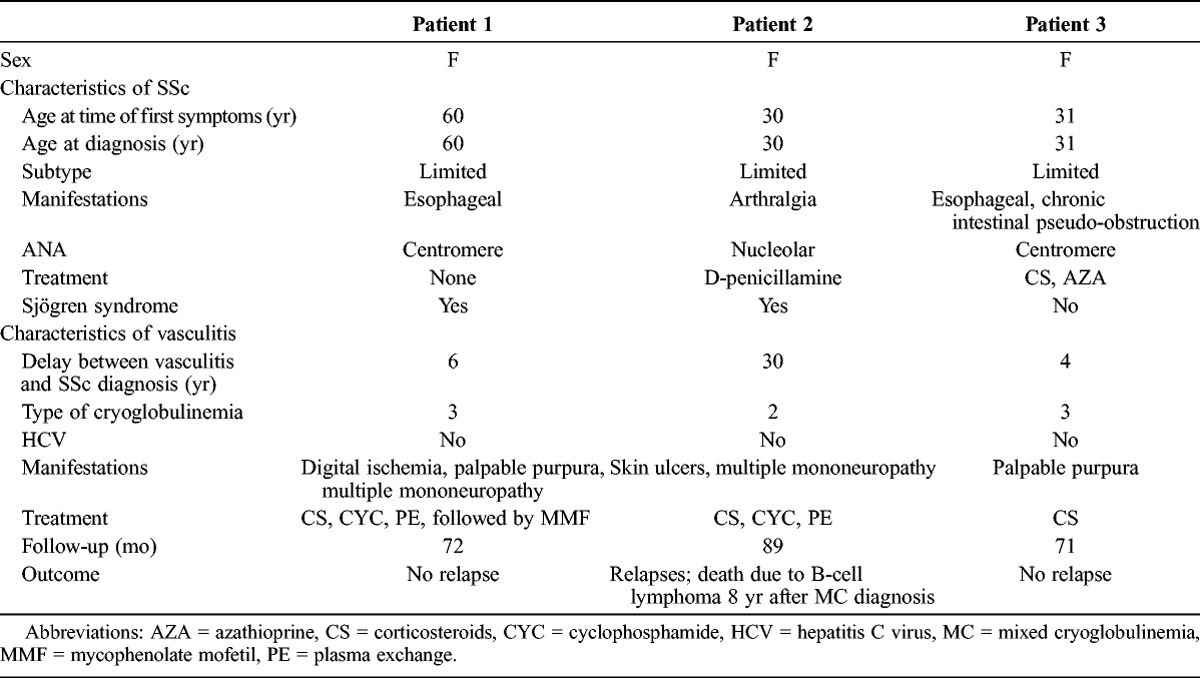
One of the 3 patients died as a result of B-cell lymphoma, diagnosed 8 years after the occurrence of type 2 cryoglobulinemia vasculitis.
ANCA-Associated Systemic Vasculitis
SSc Characteristics
Nine patients (all women; mean age of 53.1 yr at the time of SSc diagnosis) had limited SSc (Table 2). None presented with diffuse SSc. The main visceral complications were interstitial lung disease and esophageal involvement. ANA were found in 8 of the 9 patients, with anti-Scl-70 in 5 of 9 and anticentromere antibodies in 1 of 9 patients.
TABLE 2.
SSc Features, Present Report
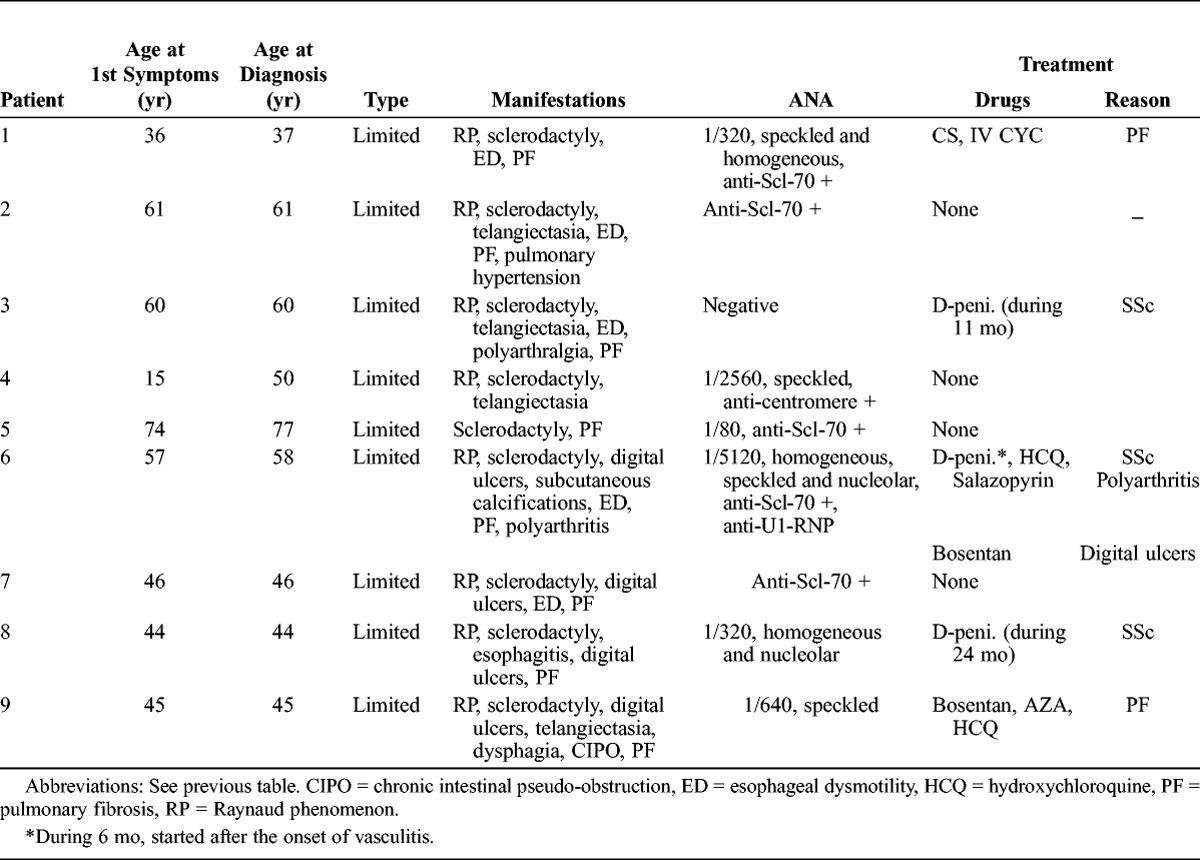
Previous treatment for SSc consisted of corticosteroids (2 patients), cyclophosphamide (1 patient), and D-penicillamine (2 patients received D-penicillamine at the time of AASV diagnosis for 11 and 24 mo, respectively).
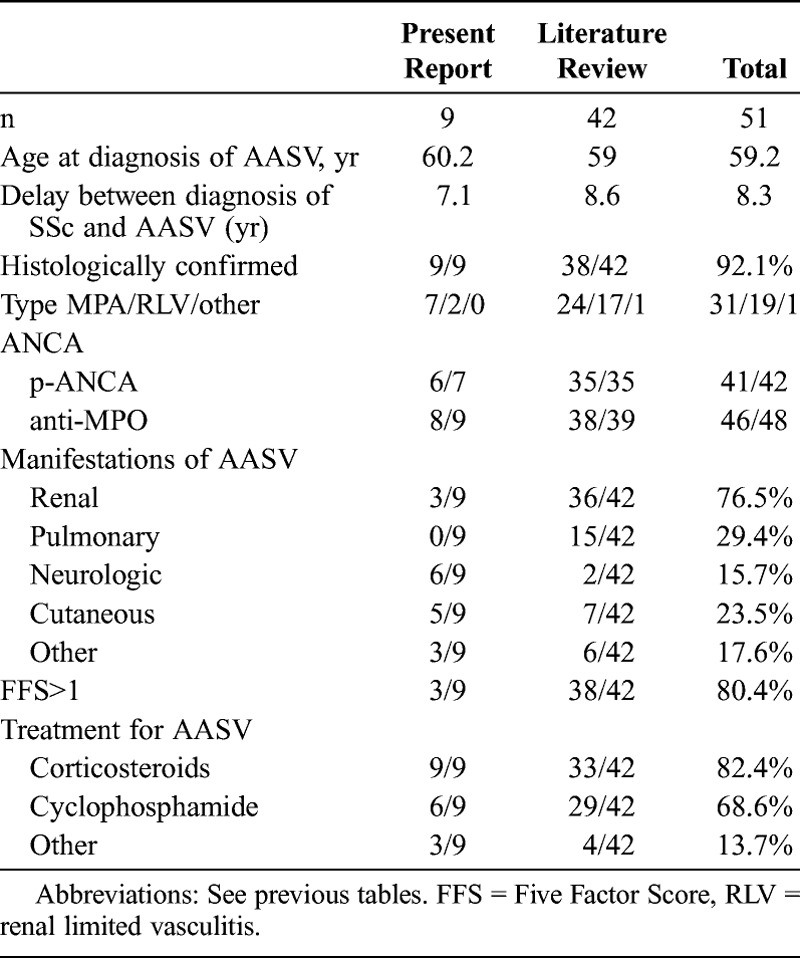
Characteristics of the 42 cases from the literature review are described in Table 3, along with characteristics of the 9 patients from the current study. Features of the patients from the literature and patients from the present series are quite similar, with a predominance of female patients, lcSSc, and interstitial lung disease. Anti-Scl-70 antibodies were the most prevalent autoimmune abnormality.
TABLE 3.
Characteristics of SSc Before Diagnosis of AASV, Present and Previous Reports
AASV Characteristics
Vasculitis was confirmed by histologic data in every case (cutaneous n = 4, renal n = 3, neurologic n = 1, muscular n = 1) (Table 4). AASV always occurred after SSc diagnosis, with a mean delay of 7.1 years (median, 0.5 yr; range, 0–24 yr). The final diagnoses were microscopic polyangiitis n = 7, renal limited vasculitis n = 2.
TABLE 4.
AASV Features, Present Report
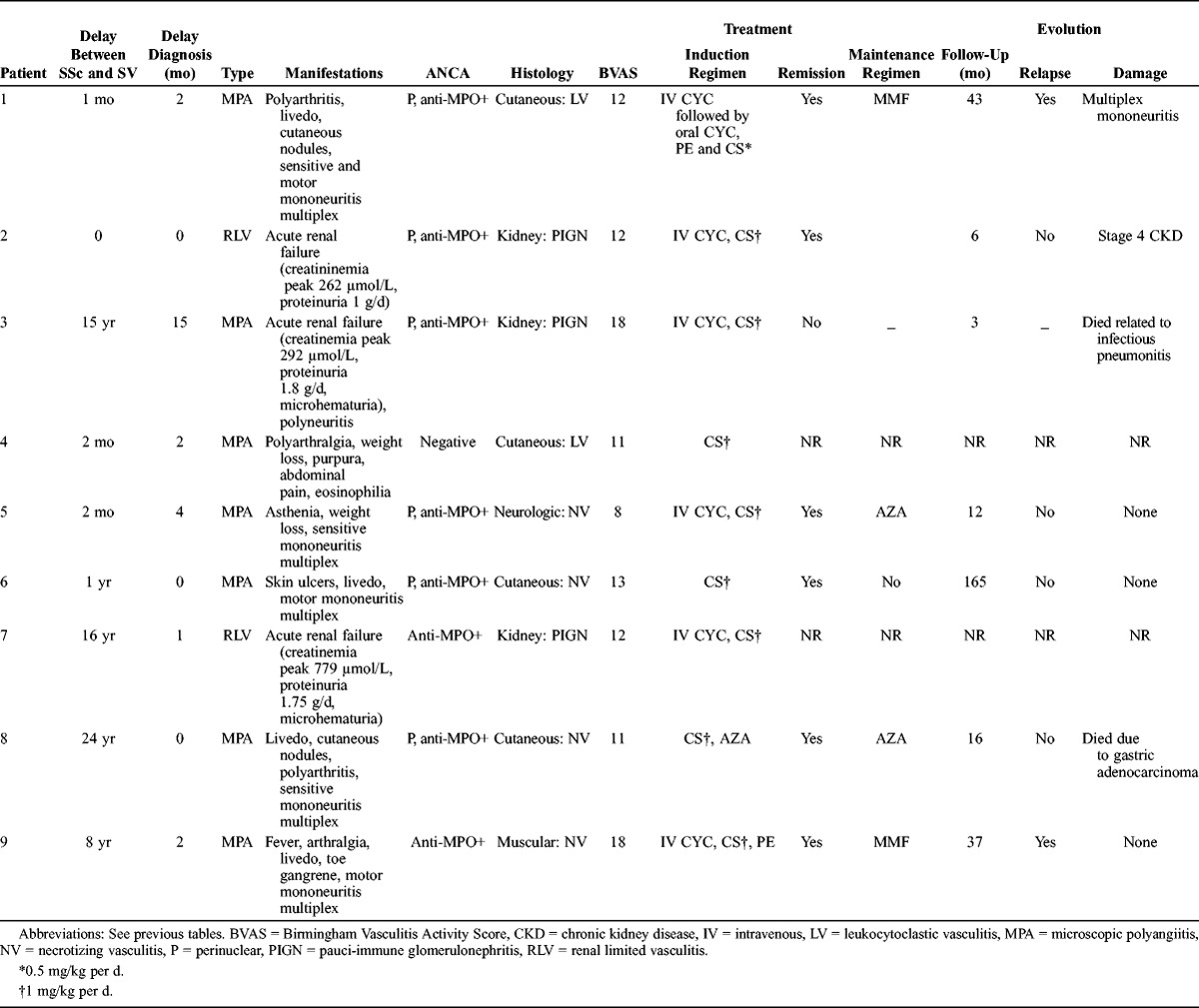
At the time of AASV diagnosis, multiple mononeuropathy was confirmed in 5 patients. Three patients presented with acute renal failure (creatininemia 266, 292, 779 μmol/L, respectively) related to pauci-immune glomerulonephritis. Kidney biopsy did not reveal the features of microvascular involvement usually observed in SSc (mucoid intimal thickening with concentrically arranged myointimal cellular proliferation described as the “onion-skin” lesion). Other manifestations were cutaneous in 5 patients and articular in 3 patients. The Five Factor Score was above 1 in 3 of 9 patients.
p-ANCA were present in 6 of 7 patients, and anti-MPO antibodies in 8 of 9 patients. No c-ANCA or anti-PR3 antibodies were found in our patients.
Data on treatment for systemic vasculitis were available in 9 of 9 patients. The induction regimen for AASV was based on corticosteroids in 9 of 9 patients, cyclophosphamide in 6 of 9 patients, azathioprine in 1 of 8, and plasma exchanges in 2 of 8 patients. Follow-up data were available for 7 of 9 patients, with a mean follow-up of 40.3 months. One patient died due to bacterial pneumonitis while receiving oral corticosteroids during the first month of treatment. Remission was obtained in 6 of 7 patients. Two patients had a minor relapse during remission maintenance regimen with mycophenolate mofetil. No patient had end-stage renal failure. None of the patients developed scleroderma renal crisis after receiving high doses of corticosteroids. One patient died due to gastric adenocarcinoma 18 months after the onset of systemic vasculitis.
Data on the 42 cases from the literature review are described in Table 5, along with characteristics of the 9 patients from the current study. The delay between diagnosis of SSc and AASV, the type of AASV, and the frequency of MPO-ANCA are quite similar to what we found in the current study. To our knowledge, no anti-PR3 antibody-associated vasculitis in patients with SSc has been reported. In the literature, kidney involvement was more prevalent and neuropathy less frequent than in the patients of the current study.
TABLE 5.
AAVS Features, Present and Previous Reports
DISCUSSION
In the current study, the prevalence of vasculitis in SSc patients referred to tertiary centers was less than 1%. Previous prevalence studies of ANCA and therefore of AASV have provided conflicting results. Three English, Italian, and Japanese studies1,11,31 provided a prevalence of AASV close to our estimation of 0 to 1.3% in a cohort of 77-115 SSc patients, whereas Endo and collaborators14 found a high prevalence of 6% in a cohort of 100 Japanese patients.
The association of systemic vasculitis and SSc, although very unusual, is probably not fortuitous. In reviewing our cases and the literature cases with an association of systemic vasculitis and SSc, we differentiated between 2 types of systemic vasculitis: MCV and AASV.
Mixed cryoglobulinemia was first reported by Husson et al22 to be present in up to 50% of SSc patients (10/20 patients). However, none of these 10 patients presented with symptoms of systemic vasculitis.22 Then, in 1990, Buskila et al8 reported only a single case of mixed cryoglobulinemia in a cohort of 108 SSc patients tested for the presence of cryoglobulin. This patient had symptoms of cutaneous vasculitis. To our knowledge, until now, only 9 patients with SSc and MCV have been described.8,23,37 We can speculate that in these SSc patients, cryoglobulinemia is a manifestation of Sjögren syndrome linked to SSc.6 In 1987, Oddis et al37 had already shown that patients with SSc are at an increased risk of developing vasculitis when they show features of Sjögren syndrome. In our study, 2 of 3 patients with cryoglobulinemic vasculitis had Sjögren syndrome according to the American-European Consensus Group criteria. The clinical presentation and prognosis of MCV associated with SSc does not seem to differ from that of other types of MCV. Salliot et al42 have already reported that Sjögren syndrome associated with SSc has the same features as primary Sjögren syndrome. In the current study, the outcome of MCV was favorable under a corticosteroid and immunosuppressive regimen.
Antibodies against neutrophil cytoplasmic antigens are usually found in granulomatosis with polyangiitis (Wegener granulomatosis), microscopic polyangiitis, Churg-Strauss syndrome, and renal limited vasculitis. Few studies have investigated the presence and the role of ANCA in SSc: ANCA prevalence ranged from 0 to 11.7% in SSc (Table 6).1,11,14,15,26,31,34,36,41 In these prevalence studies, vasculitis was found in 11%-100% of SSc patients bearing p-ANCA/anti-MPO antibodies. However, these studies mainly used immunofluorescence examination for ANCA detection. ELISA tests to detect anti-MPO antibodies were performed only subsequently. This focus on anti-MPO antibodies was probably related to the constant perinuclear pattern described on immunofluorescence examination in SSc patients and because of the previously reported cases of anti-MPO-associated vasculitis in SSc patients. Ruffatti et al41 performed a prevalence study using both immunofluorescence and ELISA (both anti-PR3 and anti-MPO) screening tests. They demonstrated the presence of p-ANCA in only 5 of 115 SSc patients, with anti-PR3 antibodies in 2 patients, anti-MPO antibodies in 1 patient, and both anti-PR3 and anti-MPO antibodies in 2 patients. Negative results of a PR3 capture ELISA led the authors to suppose that these autoantibodies recognize different epitopes from those of sera of AASV patients. If we consider AASV and SSc, anti-MPO were the unique autoantibodies, found in 44 of 46 cases with no anti-PR3 antibodies.
TABLE 6.
Prevalence of ANCA, Anti-MPO Antibodies, and AASV in SSc Patients, Previous Reports
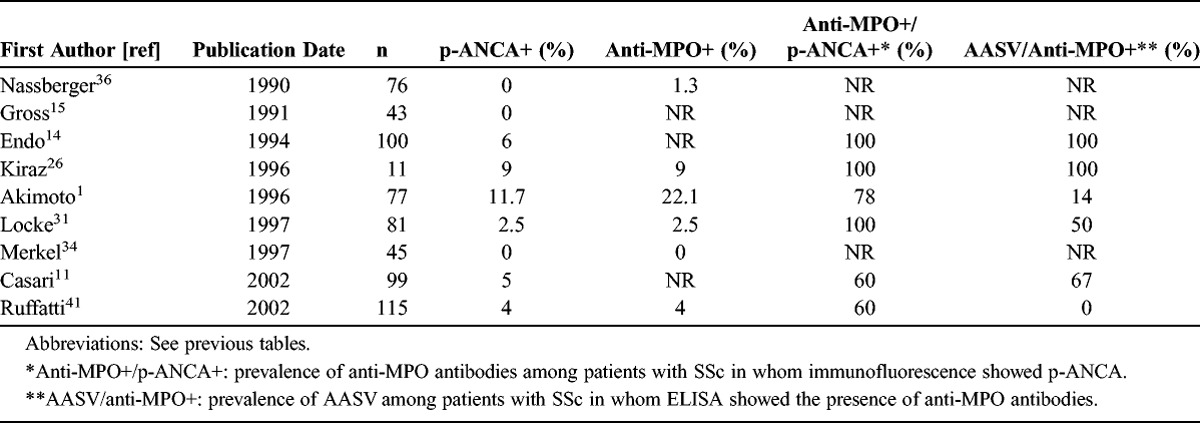
We reviewed 9 personal and 42 literature cases of SSc and AASV. SSc was mainly of the limited cutaneous type despite a high prevalence of anti-Scl-70 antibodies. In their review of literature cases, Rho et al40 suggested that anti-Scl-70 antibodies could play a role in the development of AASV in SSc patients and could be a significant predictor of the development of AASV in SSc patients. Interstitial lung disease was the main complication of SSc. It was the only feature that required an immunosuppressive regimen in 3 of 51 patients. Interestingly, pulmonary fibrosis has been described as a manifestation of microscopic polyangiitis with MPO-ANCA, sometimes occurring several years before the microscopic polyangiitis diagnosis.47 Such a manifestation has been attributed to recurrent alveolar hemorrhage, but also to an accelerated fibrotic process related to increased oxidative stress mediated by anti-MPO antibodies.17 Therefore, interstitial lung disease can also be a manifestation of anti-MPO-associated vasculitis rather than a component of SSc. AASV was diagnosed a mean 8 years after the diagnosis of SSc. Twenty-nine percent of patients (15/51) received a D-penicillamine regimen. Eight patients were receiving D-penicillamine at the time of AASV diagnosis, for a median duration of 5 years (range, 0–24 yr). D-penicillamine-induced AASV has already been reported in other diseases.7 So, in these 8 SSc patients, we cannot exclude the imputability of this therapy in the occurrence of AASV. On the other hand, 43 of 51 SSc patients developed AASV without a potentially inducing drug, suggesting other mechanisms for the development of systemic vasculitis in SSc.
The final diagnoses of AASV were microscopic polyangiitis in 31 of 51 patients, renal limited vasculitis in 19 of 51 patients, and other vasculitis in 1 of 51 patients. Immunofluorescence revealed p-ANCA in 41 of 42 cases, and ELISA revealed anti-MPO in 46 of 48 tested patients. Most of the previously reported cases of AASV associated with SSc were described as normotensive renal failure. Kidney involvement is reported to be found in up to 60% of patients with SSc.9 Scleroderma renal crisis occurs in nearly 10% of patients and encompasses progressive renal failure, thrombocytopenia, non-autoimmune hemolytic anemia, and high blood pressure.45 Such manifestations occur in the first years of SSc and are associated with diffuse SSc, arthritis, anti-Scl-70 antibodies, and use of corticosteroids. In 1989, Helfrich et al18 described 15 SSc patients with normotensive renal failure; these patients frequently showed signs of hemolytic anemia and thrombocytopenia. Six of the 15 patients, however, presented with alveolar hemorrhage. Thrombocytopenia was present in all 6 patients. The authors did not indicate whether a search for ANCA was performed. They described no unifying pulmonary pathologic feature in 4 of these 6 patients at autopsy. Thereafter, 42 patients with AASV and SSc were reported in the literature. Whereas 36 of 42 reported patients presented with rapidly pauci-immune glomerulonephritis (31/36 biopsy proven), only 3 of our 9 patients did. This might account for the poorer prognosis observed in the previously reported cases (13/38 died after a mean follow-up of 12.8 mo) than in our cases (2/7 died after a mean follow-up of 40.3 mo).
The current study does not allow us to draw conclusions about the treatment of AASV in SSc patients. We can only underline that no scleroderma renal crisis occurred in our patients or in the previously reported cases despite the use of high doses of corticosteroids. This could be related to the high prevalence of the limited cutaneous type of SSc in patients with AASV and SSc. Finally, the current study demonstrates that diffuse organ involvement (skin, nerves, kidney, lung) related to AASV can be observed in SSc patients.
Systemic vasculitis is a rare complication of SSc that can lead to misdiagnosis, especially in the face of confounding conditions, such as digital ischemia and SSc renal crisis. In the case of SSc renal crisis, normal blood pressure and the absence of microangiopathic hemolytic anemia should lead to ANCA testing and a kidney biopsy. In the event of vasculitic syndromes in SSc patients, 2 diagnoses should be considered: MCV potentially associated with Sjögren syndrome, but especially ANCA-associated vasculitis. This latter category can be responsible of severe major organ involvement such as that found in microscopic polyangiitis. Prompt recognition of vasculitis could lead to the early introduction of an appropriate immunosuppressive regimen and better renal and global survival.
ACKNOWLEDGMENTS
The authors thank Pr. J. Cabane and the “Groupe Français de Recherche sur la Sclérodermie” and the “Groupe français d’étude des Vascularites” for their help in this study.
Footnotes
Abbreviations: AASV = ANCA-associated systemic vasculitis, ANA = antinuclear antibodies, ANCA = antineutrophil cytoplasmic antibodies, anti-MPO = anti-myeloperoxidase, anti-PR3 = anti-proteinase 3, c-ANCA = cytoplasmic ANCA, dcSSc = diffuse cutaneous SSc, lcSSc = limited cutaneous SSc, MCV = mixed cryoglobulinemia vasculitis, p-ANCA = perinuclear ANCA, SSc = systemic sclerosis
The authors have no funding or conflicts of interest to disclose.
REFERENCES
- 1.Akimoto S, Ishikawa O, Tamura T, Miyachi Y. Antineutrophil cytoplasmic autoantibodies in patients with systemic sclerosis. Br J Dermatol. 1996; 134: 407–410. [PubMed] [Google Scholar]
- 2.Akkara Veetil BM, Schimmer BM. A case of limited systemic sclerosis with p-ANCA, complicated by multiple cerebral hemorrhages. Rheumatol Int. 2009; 29: 325–329. [DOI] [PubMed] [Google Scholar]
- 3.Anders HJ, Wiebecke B, Haedecke C, Sanden S, Combe C, Schlondorff D. MPO-ANCA-positive crescentic glomerulonephritis: a distinct entity of scleroderma renal disease? Am J Kidney Dis. 1999; 33: e3. [DOI] [PubMed] [Google Scholar]
- 4.Arad U, Balbir-Gurman A, Doenyas-Barak K, Amit-Vazina M, Caspi D, Elkayam O. Anti-neutrophil antibody associated vasculitis in systemic sclerosis. Semin Arthritis Rheum. 2011; 41: 223–229. [DOI] [PubMed] [Google Scholar]
- 5.Arnaud L, Huart A, Plaisier E, Francois H, Mougenot B, Tiev K, Kettaneh A, Ronco P, Rougier JP. ANCA-related crescentic glomerulonephritis in systemic sclerosis: revisiting the “normotensive scleroderma renal crisis.” Clin Nephrol. 2007; 68: 165–170. [DOI] [PubMed] [Google Scholar]
- 6.Avouac J, Airo P, Dieude P, Caramaschi P, Tiev K, Diot E, Sibilia J, Cappelli S, Granel B, Vacca A, Wipff J, Meyer O, Kahan A, Matucci-Cerinic M, Allanore Y. Associated autoimmune diseases in systemic sclerosis define a subset of patients with milder disease: results from 2 large cohorts of European Caucasian patients. J Rheumatol. 2010; 37: 608–614. [DOI] [PubMed] [Google Scholar]
- 7.Bienaimé F, Clerbaux G, Plaisier E, Mougenot B, Ronco P, Rougier JP. D-Penicillamine-induced ANCA-associated crescentic glomerulonephritis in Wilson disease. Am J Kidney Dis. 2007; 50: 821–825. [DOI] [PubMed] [Google Scholar]
- 8.Buskila D, Langevitz P, Lee P. The frequency of cryoglobulinaemia in systemic sclerosis (scleroderma). Br J Rheumatol. 1990; 29: 234. [DOI] [PubMed] [Google Scholar]
- 9.Cannon PJ, Hassar M, Case DB, Casarella WJ, Sommers SC, LeRoy EC. The relationship of hypertension and renal failure in scleroderma (progressive systemic sclerosis) to structural and functional abnormalities of the renal cortical circulation. Medicine (Baltimore). 1974; 53: 1–46. [DOI] [PubMed] [Google Scholar]
- 10.Carvajal I, Bernis C, Sanz P, Garcia A, Garcia-Vadillo A, Traver JA. Antineutrophil cytoplasmic autoantibodies (ANCA) and systemic sclerosis. Nephrol Dial Transplant. 1997; 12: 576–577. [DOI] [PubMed] [Google Scholar]
- 11.Casari S, Haeney M, Farrand S, Herrick A. Antineutrophil cytoplasmic antibodies a “red flag” in patients with systemic sclerosis. J Rheumatol. 2002; 29: 2666–2667. [PubMed] [Google Scholar]
- 12.Cheung G, Chew G, Wyndham R, Peters M, Riminton S. Myeloperoxidase-antineutrophil cytoplasmic antibody seroconversion and fulminant vasculitis in Scl-70-positive scleroderma. Intern Med J. 2007; 37: 205–207. [DOI] [PubMed] [Google Scholar]
- 13.Chifflot H, Fautrel B, Sordet C, Chatelus E, Sibilia J. Incidence and prevalence of systemic sclerosis: a systematic literature review. Semin Arthritis Rheum. 2008; 37: 223–235. [DOI] [PubMed] [Google Scholar]
- 14.Endo H, Hosono T, Kondo H. Antineutrophil cytoplasmic autoantibodies in 6 patients with renal failure and systemic sclerosis. J Rheumatol. 1994; 21: 864–870. [PubMed] [Google Scholar]
- 15.Gross WL, Schmitt WH, Csernok E. Antineutrophil cytoplasmic autoantibody-associated diseases: a rheumatologist’s perspective. Am J Kidney Dis. 1991; 18: 175–179. [DOI] [PubMed] [Google Scholar]
- 16.Guillevin L, Lhote F, Gayraud M, Cohen P, Jarrousse B, Lortholary O, Thibult N, Casassus P. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore). 1996; 75: 17–28. [DOI] [PubMed] [Google Scholar]
- 17.Guilpain P, Chereau C, Goulvestre C, Servettaz A, Montani D, Tamas N, Pagnoux C, Hachulla E, Weill B, Guillevin L, Mouthon L, Batteux F. The oxidation induced by antimyeloperoxidase antibodies triggers fibrosis in microscopic polyangiitis. Eur Respir J. 2011; 37: 1503–1513. [DOI] [PubMed] [Google Scholar]
- 18.Helfrich DJ, Banner B, Steen VD, Medsger TA., Jr Normotensive renal failure in systemic sclerosis. Arthritis Rheum. 1989; 32: 1128–1134. [DOI] [PubMed] [Google Scholar]
- 19.Herrera-Esparza R, Aguilar JL, Saucedo A, Gonzalez I, Lopez-Robles E, Avalos-Diaz E. Scleroderma with type III glomerulonephritis and MPO-ANCA antibodies in the serum. J Eur Acad Dermatol Venereol. 2005; 19: 617–620. [DOI] [PubMed] [Google Scholar]
- 20.Hidalgo-Tenorio C, Càliz R, Gallego M, Jaimez L, Jimenez-Alonzo J. Systemic sclerosis, pulmonary fibrosis and anti-MPO antibodies. Clinical Rheumatology. 2005; 24: 658–660. [DOI] [PubMed] [Google Scholar]
- 21.Hillis GS, Khan IH, Simpson JG, Rees AJ. Scleroderma, D-penicillamine treatment, and progressive renal failure associated with positive antimyeloperoxidase antineutrophil cytoplasmic antibodies. Am J Kidney Dis. 1997; 30: 279–281. [DOI] [PubMed] [Google Scholar]
- 22.Husson JM, Druet P, Contet A, Fiessinger JN, Camilleri JP. Systemic sclerosis and cryoglobulinemia. Clin Immunol Immunopathol. 1976; 6: 77–82. [DOI] [PubMed] [Google Scholar]
- 23.Invernizzi F, Galli M, Serino G, Monti G, Meroni PL, Granatieri C, Zanussi C. Secondary and essential cryoglobulinemias. Frequency, nosological classification, and long-term follow-up. Acta Haematol. 1983; 70: 73–82. [DOI] [PubMed] [Google Scholar]
- 24.Kamen DL, Wigley FM, Brown AN. Antineutrophil cytoplasmic antibody-positive crescentic glomerulonephritis in scleroderma—a different kind of renal crisis. J Rheumatol. 2006; 33: 1886–1888. [PubMed] [Google Scholar]
- 25.Katrib A, Sturgess A, Bertouch JV. Systemic sclerosis and antineutrophil cytoplasmic autoantibody-associated renal failure. Rheumatol Int. 1999; 19: 61–63. [DOI] [PubMed] [Google Scholar]
- 26.Kiraz S, Simsek H, Ertenli I, Benekli M, Kadayifci A. Antineutrophil cytoplasmic antibodies in systemic sclerosis. Clin Rheumatol. 1996; 15: 519–520. [DOI] [PubMed] [Google Scholar]
- 27.Kumakura S, Ishikura H, Kondo M, Murakawa Y, Kobayashi S. Hemophagocytosis associated with MPO-ANCA positive vasculitis in systemic sclerosis. Clin Exp Rheumatol. 2002; 20: 411–414. [PubMed] [Google Scholar]
- 28.Kyndt X, Ducq P, Bridoux F, Reumaux D, Makdassi R, Gheerbrant JD, Vanhille P. Extracapillary glomerulonephritis with anti-myeloperoxidase antibodies in 2 patients with systemic scleroderma treated with penicillamine D. Presse Med. 1999; 28: 67–70. [PubMed] [Google Scholar]
- 29.LeRoy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA, Jr, Rowell N, Wollheim F. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988; 15: 202–205. [PubMed] [Google Scholar]
- 30.Le Thi Huong D, Gatfosse M, Papo T, Beaufils H, Faucher C, Frances C, Godeau P. Wegener’s granulomatosis and CREST syndrome. Br J Rheumatol. 1994; 33: 1087–1088. [DOI] [PubMed] [Google Scholar]
- 31.Locke IC, Worrall JG, Leaker B, Black CM, Cambridge G. Autoantibodies to myeloperoxidase in systemic sclerosis. J Rheumatol. 1997; 24: 86–89. [PubMed] [Google Scholar]
- 32.Maes B, Van Mieghem A, Messiaen T, Kuypers D, Van Damme B, Vanrenterghem Y. Limited cutaneous systemic sclerosis associated with MPO-ANCA positive renal small vessel vasculitis of the microscopic polyangiitis type. Am J Kidney Dis. 2000; 36: E16. [DOI] [PubMed] [Google Scholar]
- 33.Marlier S, Gisserot O, Yao N, Hecht M, Paris JF, Carli P, Chagnon A. Extra-capillary glomerulonephritis induced by D-penicillamine therapy. Presse Med. 1999; 28: 689–690. [PubMed] [Google Scholar]
- 34.Merkel PA, Polisson RP, Chang Y, Skates SJ, Niles JL. Prevalence of antineutrophil cytoplasmic antibodies in a large inception cohort of patients with connective tissue disease. Ann Intern Med. 1997; 126: 866–873. [DOI] [PubMed] [Google Scholar]
- 35.Mimura I, Hori Y, Matsukawa T, Uozaki H, Tojo A, Fujita T. Noncrescentic ANCA-associated renal crisis in systemic sclerosis. Clin Nephrol. 2008; 70: 183–185. [DOI] [PubMed] [Google Scholar]
- 36.Nassberger L, Sjoholm AG, Jonsson H, Sturfelt G, Akesson A. Autoantibodies against neutrophil cytoplasm components in systemic lupus erythematosus and in hydralazine-induced lupus. Clin Exp Immunol. 1990; 81: 380–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oddis CV, Eisenbeis CH, Jr, Reidbord HE, Steen VD, Medsger TA., Jr Vasculitis in systemic sclerosis: association with Sjogren’s syndrome and the CREST syndrome variant. J Rheumatol. 1987; 14: 942–948. [PubMed] [Google Scholar]
- 38.Omote A, Muramatsu M, Sugimoto Y, Hosono S, Murakami R, Tanaka H, Watanabe Y, Sano H, Kato K. Myeloperoxidase-specific anti-neutrophil cytoplasmic autoantibodies—related scleroderma renal crisis treated with double-filtration plasmapheresis. Intern Med. 1997; 36: 508–513. [DOI] [PubMed] [Google Scholar]
- 39.Reveille JD, Solomon DH. American College of Rheumatology Ad Hoc Committee of Immunologic Testing Guidelines. Evidence-based guidelines for the use of immunologic tests: anticentromere, Scl-70, and nucleolar antibodies. Arthritis Rheum. 2003; 49: 399–412. [DOI] [PubMed] [Google Scholar]
- 40.Rho YH, Choi SJ, Lee YH, Ji JD, Song GG. Scleroderma associated with ANCA-associated vasculitis. Rheumatol Int. 2006; 26: 465–468. [DOI] [PubMed] [Google Scholar]
- 41.Ruffatti A, Sinico RA, Radice A, Ossi E, Cozzi F, Tonello M, Grypiotis P, Todesco S. Autoantibodies to proteinase 3 and myeloperoxidase in systemic sclerosis. J Rheumatol. 2002; 29: 918–923. [PubMed] [Google Scholar]
- 42.Salliot C, Mouthon L, Ardizzone M, Sibilia J, Guillevin L, Gottenberg JE, Mariette X. Sjogren’s syndrome is associated with and not secondary to systemic sclerosis. Rheumatology. 2007; 46: 321–326. [DOI] [PubMed] [Google Scholar]
- 43.Sol EB, Bonkain F, Vande Houtte K, Dratwa M. Systemic scleroderma associated to a glomerulonephritis with anti-MPO antibodies: a case report and literature review. Rev Med Brux. 2008; 29: 559–567. [PubMed] [Google Scholar]
- 44.Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972-2002. Ann Rheum Dis. 2007; 66: 940–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Teixeira L, Mouthon L, Mahr A, Berezne A, Agard C, Mehrenberger M, Noel LH, Trolliet P, Frances C, Cabane J, Guillevin L. Mortality and risk factors of scleroderma renal crisis: a French retrospective study of 50 patients. Ann Rheum Dis. 2008; 67: 110–116. [DOI] [PubMed] [Google Scholar]
- 46.Tomioka M, Hinoshita F, Miyauchi N, Akiyama Y, Saima S, Hiroe M. ANCA-related crescentic glomerulonephritis in a patient with scleroderma without marked dermatological change and malignant hypertension. Intern Med. 2004; 43: 496–502. [DOI] [PubMed] [Google Scholar]
- 47.Tzelepis GE, Kokosi M, Tzioufas A, Toya SP, Boki KA, Zormpala A, Moutsopoulos HM. Prevalence and outcome of pulmonary fibrosis in microscopic polyangiitis. Eur Respir J. 2010; 36: 116–121. [DOI] [PubMed] [Google Scholar]
- 48.Vazquez-Del Mercado M, Mendoza-Topete A, Best-Aguilera CR, Garcia-De La Torre I. Diffuse alveolar hemorrhage in limited cutaneous systemic sclerosis with positive perinuclear antineutrophil cytoplasmic antibodies. J Rheumatol. 1996; 23: 1821–1823. [PubMed] [Google Scholar]
- 49.Villaverde V, Balsa A, Cabezas JA, Fernandez-Prada M, Torre A, Mola EM. Normotensive renal failure in a patient with systemic sclerosis and p-antineutrophil cytoplasmic autoantibodies which developed into Paget’s disease of bone after immunosuppressive therapy. Rheumatology. 1999; 38: 190–191. [DOI] [PubMed] [Google Scholar]
- 50.Wong M, Ranganath VK, Clements PJ. Antineutrophil cytoplasmic antibody-positive digital necrosis in a patient with limited systemic sclerosis. J Rheumatol. 2010; 37: 214–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wutzl AL, Foley RN, O’Driscoll BR, Reeve RS, Chisholm R, Herrick AL. Microscopic polyangiitis presenting as pulmonary-renal syndrome in a patient with long-standing diffuse cutaneous systemic sclerosis and antibodies to myeloperoxidase. Arthritis Rheum. 2001; 45: 533–536. [DOI] [PubMed] [Google Scholar]
- 52.Yamashita K, Yorioka N, Kyuden Y, Naito T, Tanji C, Ueda C, Usui K, Shigemoto K, Harada S, Yamakido M. A case of CREST syndrome and myeloperoxidase-specific anti-neutrophil cytoplasmic autoantibody-associated glomerulonephritis. Clin Nephrol. 2000; 5: 296–300. [PubMed] [Google Scholar]