

Abstract
The idiopathic inflammatory myopathies (IIM) are acquired muscle diseases characterized by muscle weakness and inflammation on muscle biopsy. Clinicoserologic classifications do not take muscle histology into account to distinguish the subsets of IIM. Our objective was to determine the pathologic features of each serologic subset of IIM and to correlate muscle biopsy results with the clinicoserologic classification defined by Troyanov et al, and with the final diagnoses. We retrospectively studied a cohort of 178 patients with clinicopathologic features suggestive of IIM with the exclusion of inclusion body myositis. At the end of follow-up, 156 of 178 cases were still categorized as IIM: pure dermatomyositis, n = 44; pure polymyositis, n = 14; overlap myositis, n = 68; necrotizing autoimmune myopathy, n = 8; cancer-associated myositis, n = 18; and unclassified IIM, n = 4. The diagnosis of IIM was ruled out in the 22 remaining cases. Pathologic dermatomyositis was the most frequent histologic picture in all serologic subsets of IIM, with the exception of patients with anti-Ku or anti-SRP autoantibodies, suggesting that it supports the histologic diagnosis of pure dermatomyositis, but also myositis of connective tissue diseases and cancer-associated myositis. Unspecified myositis was the second most frequent histologic pattern. It frequently correlated with overlap myositis, especially with anti-Ku or anti-PM-Scl autoantibodies. Pathologic polymyositis was rare and more frequently correlated with myositis mimickers than true polymyositis. The current study shows that clinicoserologic and pathologic data are complementary and must be taken into account when classifying patients with IIM patients. We propose guidelines for diagnosis according to both clinicoserologic and pathologic classifications, to be used in clinical practice.
INTRODUCTION
The idiopathic inflammatory myopathies (IIM) are a heterogeneous group of acquired muscle diseases characterized by muscle weakness and inflammatory infiltrates in skeletal muscle. Among them, polymyositis (PM) and dermatomyositis (DM) were defined in 1975 by Bohan and Peter2,3 with the following diagnostic criteria: 1) symmetric proximal muscle weakness, 2) elevation of serum skeletal muscle enzymes, 3) electromyographic changes, 4) muscle biopsy abnormalities (necrosis, regeneration, perifascicular atrophy, inflammatory exudates), 5) typical skin rash of dermatomyositis (heliotrope rash, Gottron sign, and Gottron papules). A diagnosis of definite/probable/possible PM requires respectively 4, 3, or 2 criteria among criteria 1 through 4. Definite/probable/possible DM is diagnosed if skin abnormalities (criteria 5) are present in addition to respectively 3, 2, or 1 of the other criteria. Additionally, Bohan and Peter2,3 pointed out the associated occurrence with connective tissue diseases (CTDs) and malignancies. This historical classification has become subject to increasing debate1,10,21,25,26 because DM is differentiated from PM by skin changes only, and overlap myositis (OM) is loosely defined, leading to misclassification of patients, overdiagnosis of PM, and nonrecognition of OM.26,27
An important advance in understanding the pathogenesis of IIM over the past 20 years has been the identification of myositis-associated autoantibodies (MAAs) and myositis-specific autoantibodies (MSAs) as markers of clinical subsets, disease prognosis, and treatment response.5,6,12,18,23,26 MAAs are not specific but may be found in patients with myositis in the context of overlap syndrome, especially those with features of systemic sclerosis (SS).16 They are directed to nuclear or nucleolar antigens, such as PM-Scl, Ku, U1-RNP, Ro60/SSA, and La/SSB.23 MSAs appear to be more clinically relevant and include antibodies directed against aminoacyl-tRNA synthetases (Jo-1, PL-7, PL-12, EJ, OJ, JS, and KS), signal recognition particle (SRP), nuclear helicase Mi-2, and p155.5,13,17,23
As a result, even if the Bohan and Peter classification is still useful as an approach for diagnosing myositis, it is obsolete as a basis for distinguishing the different subsets of diseases. Troyanov et al26 developed an interesting clinicoserologic classification where overlap clinical features as well as MAA and MSA were positioned at the core of the classification system. They found that myositis with overlap features (OM) was the most common IIM. Anti-Mi-2, which is not associated with overlap clinical features, was considered to be specific for pure DM. This new classification was proven to predict the response to prednisone and IIM course.26 However, we note that neither the Bohan and Peter classification nor the new clinicoserologic classification considers histopathologic findings to distinguish the subsets of IIM.
In contrast, numerous pathologists have a different approach, and consider that muscle biopsy is the most sensitive tool to diagnose IIM.9,10 Muscle histology allows 4 main subtypes of IIM to be distinguished on the basis on distinct immunopathologic features: DM, PM, sporadic inclusion body myositis (IBM), and necrotizing autoimmune myositis (NAM).9
DM is considered to be a complement-mediated microangiopathy leading to destruction of capillaries and hypoperfusion of the perifascicular regions. Muscle biopsy shows perivascular/perimysial inflammation often associated with perifascicular atrophy or microinfarcts. The major histocompatibility complex, class 1 (MHC-1) antigen is upregulated especially in perifascicular areas, and immune complex deposition in the vessels are common.8–10
PM and IBM are characterized by the presence of cytotoxic T-lymphocytic endomysial infiltrates that focally surround and invade nonnecrotic muscle fibers, with relative sparing of the vasculature.9,10 The MHC-1 antigen is ubiquitously upregulated on the surface of most fibers. In IBM, rimmed vacuoles plus or minus inclusions are present. Currently, the pathophysiologic bases of IBM are still under debate, and degenerative processes have been incriminated in addition to possible immunopathologic events. IBM was not included in the new clinicoserologic classification of IIM by Troyanov et al,26 or in the last European Neuromuscular Centre (ENMC) international workshop on adult IIM.15
NAM, also called immune-mediated necrotizing myopathy, is an increasingly recognized subacute myopathy characterized by very high creatine kinase (CK) levels, moderate to severe subacute muscle weakness, and numerous necrotic fibers invaded by macrophages on muscle biopsy, usually in the absence of both lymphocytic inflammation and diffuse MHC-1 overexpression.4,7,9,22 NAM should be triggered by statins, viral infections, cancer, or autoimmunity. It is noteworthy that antibodies against SRP22 have been identified in NAM, and, more recently, anti-HMG-CoA reductase has been identified in patients with statin-triggered autoimmune myopathy.20 In view of these histologic considerations, we note that although OM is probably the most frequent subtype of IIM, pathologists have difficulty identifying it and usually do not propose this diagnosis on the basis of muscle biopsy.
This lack of consensus among physicians, immunologists, and pathologists leads to confusion regarding the real incidence of the different subtypes of IIM and their association with autoantibodies. To our knowledge, no study has been reported that describes clinical and immunologic data in comparison with pathologic features in a large series of patients with IIM.
We conducted the current study to determine the pathologic features of each serologic subset of IIM and to correlate muscle biopsy results with the clinicoserologic classification defined by Troyanov et al26 and the final diagnoses.
PATIENTS AND METHODS
Patient Cohort, Clinical Data, and CK Level
We retrospectively reviewed a cohort of adult patients (aged 16 yr or older at diagnosis) referred to our institution between 2001 and 2011 with a diagnosis of definite/probable/possible myositis according to the Bohan and Peter classification2,3 and muscle biopsy abnormalities suggestive of myositis as defined by the last ENMC international workshop on IIM.15 Patients with muscle biopsy results suggestive of IBM or a diagnosis other than IIM were excluded. Finally, 178 patients were included in our cohort.
The following data were recorded: age, sex, typical skin rash of DM, clinical overlap features, association with cancer, and CK level before corticosteroid therapy. Clinical overlap features were defined as described by Troyanov et al26 (Table 1). Because no patient in that study’s cohort had primary Sjögren syndrome, it was not included as an associated CTD in their clinicoserologic classification. However, because 5 patients in our series had clinically symptomatic and biopsy-proven Sjögren syndrome before the occurrence of myositis, always with both anti-SSA and anti-SSB autoantibodies, we have chosen to consider the Sjögren syndrome as a supplementary overlap feature and have classified these cases as OM.
TABLE 1.
Clinicoserologic Classification of Troyanov et al26

Serologic Characterization
Antinuclear antibodies (ANA), anti-Sm, and anti-native DNA (commonly associated with systemic lupus erythematosus [SLE]) as well as the following MMAs and MSAs were screened in all patients’ serum: for MMAs, anti-PM-Scl and anti-Scl-70 (commonly associated with SS), anti-U1-RNP (commonly associated with SLE and Sharp syndrome), anti-SSA and anti-SSB (commonly associated with SLE and Sjögren), anti-Ku; and for MSAs, anti-synthetases (Jo-1, PL-7, PL-12), anti-Mi-2, and anti-SRP.
Total ANA were detected by indirect immunofluorescence on HEp-2 cells (Bio-Rad, Hercules, CA) with a screening dilution of 1:100. Extractable nuclear antigens (ENA) were detected by ELISA using ENA ELIA test (Phadia, Freiburg, Germany). MMA/MSA (anti-Mi-2, Ku, PM-Scl, SRP, Jo-1, PL-7, PL-12) were researched by using a dot blot (Orgentec, Mainz, Germany).
Troyanov et al26 grouped antibodies directed against PM-Scl, Scl-70, U1-RNP, Ku, synthetases, and SRP under the term “overlap antibodies” (associated with OM). To obtain more detailed findings on the pathologic features of each clinicoserologic subgroup, we classified serologic profiles as follows: 1) lack of specific autoantibodies, 2) autoantibodies associated with SLE, Sharp and Sjögren syndromes, called SLE/MCTD/GS antibodies (native DNA, Sm, RNP, SSA plus SSB), 3) anti-PM-Scl, 4) anti-Ku, 5) anti-synthetase (Jo-1, PL-7, PL-12), 6) anti-SRP, 7) anti-Mi-2. Patients with anti-SSA alone were considered to have a nonspecific profile (group 1). Patients with both MSA (anti-synthetase, SRP, or Mi-2) and 1 or several MAAs were attached to their MSA subgroup.
Muscle Biopsy
Muscle specimens were frozen in liquid nitrogen cooled by isopentane, and serial 5 μ-thick sections were prepared using conventional histologic stains and histochemical reactions. Automated immunohistochemical detection of MHC-1 antigen (HLA-ABC, clone B 9.12.1, dilution 1/200, Immunotech, Marseille, France) and membrane attack complex C5b-9 (clone aE11, dilution 1/100, DAKO France, Trappes, France), was performed on frozen sections on Ventana BenchMarks.
For each muscle biopsy, the following parameters were recorded: perifascicular atrophy, microinfarcts, necrotic fibers (more than 3 per 1000 muscle fibers), perivascular/perimysial mononuclear cell infiltrates, endomysial inflammation, and invasion of nonnecrotic muscle fibers (Figure 1A-F). MHC-1 expression was defined as follows: absent (no expression), focal (only sparse fibers or groups of fibers expressed MHC-1), diffuse (MHC-1 was upregulated on the surface of most/all fibers), perifascicular or diffuse with reinforcement in the perifascicular region (MHC-1 expression was stronger in perifascicular fibers), both referred to as “dermatomyositic pattern” (DM pattern) of MHC-1 expression (Figure 1G-I).15 C5b-9 deposits (microthrombi of C5b-9) were searched for in intramuscular capillaries (Figure 1J).
FIGURE 1.
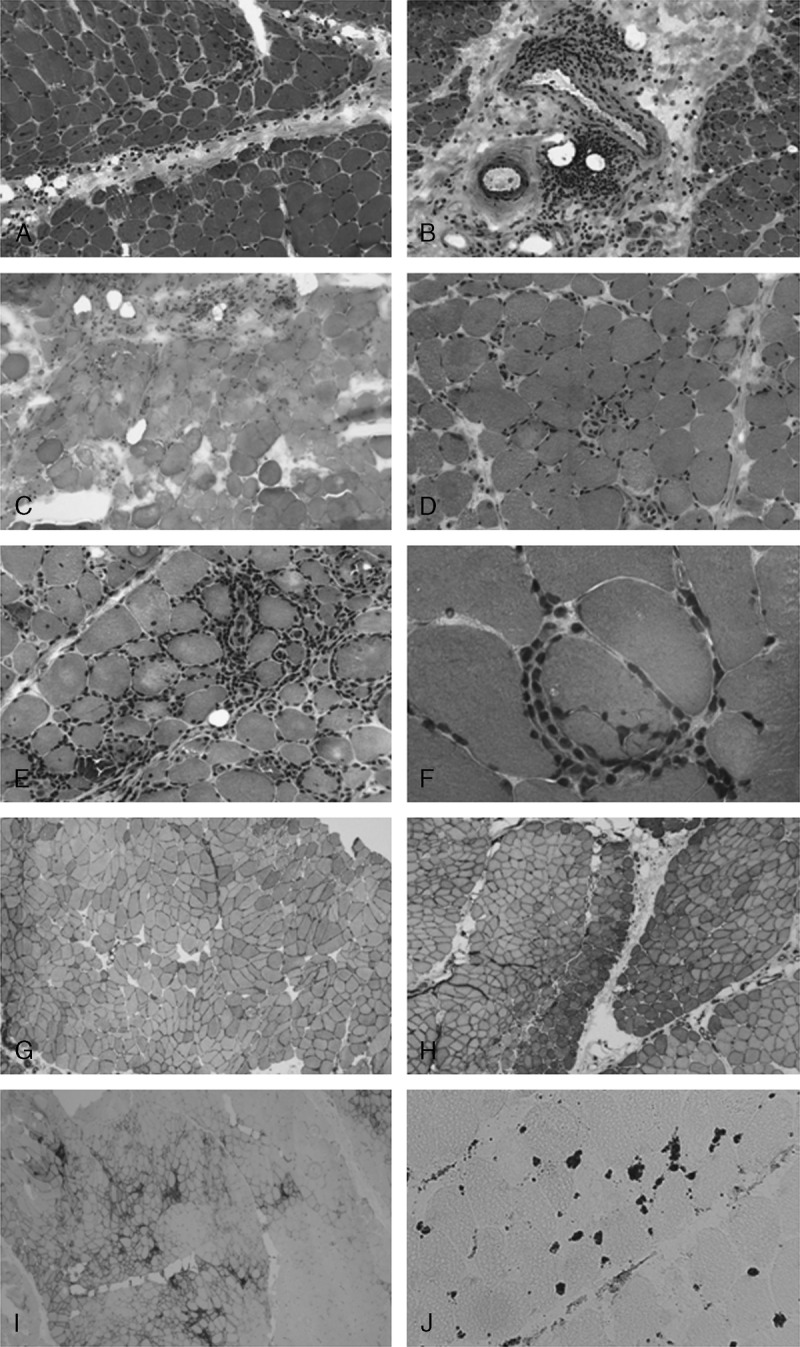
Pathologic features of idiopathic inflammatory myopathy. A–C. Pathologic dermatomyositis: perifascicular atrophy (A), perivascular lymphocytic infiltrates (B), and microinfarct (C). D. Necrotizing myopathy: numerous necrotic and regenerative fibers. E–F. Pathologic aspects suggestive of polymyositis: endomysial infiltrates (E) and invasion of a nonnecrotic muscle fiber by lymphocytes (F). G–I. Examples of MHC-1 overexpression: diffuse (G), diffuse with perifascicular reinforcement (H), focal (I). J. Microthrombi of C5b-9 in intramuscular capillaries. (A–F: Hematoxylin-eosin stain, G–J: immunohistochemistry. A–C original magnification × 100, D–E × 200, F × 630, G × 80, H–I × 40, J × 250.).
Definition of Clinicoserologic Subgroups, Pathologic Subgroups, and Final Diagnosis
To classify the patients on the basis of clinical and immunologic data, we used the clinicoserologic classification proposed by Troyanov et al26 (see Table 1). As suggested, cases where patients had both cancer and MMA or MSA were categorized as OM.
To classify the patients based on pathologic findings, we summarized consensual histologic features usually used for the diagnosis of myositis in a synthetic table inspired by the 119th ENMC international workshop on adult idiopathic inflammatory myopathies15 (Table 2). To distinguish DM/PM diagnosed pathologically based on muscle biopsy from DM/PM diagnosed based on clinicoserologic features, we named the categories “pathologic DM” (pDM) and “pathologic PM” (pPM) (by analogy with the TNM classification of malignant tumors system). The following pathologic categories were defined 1) pDM, definite, probable or possible, 2) pPM, 3) unspecified myositis, 4) necrotizing myopathy, 5) diffuse MHC-1 expression only (with normal muscle histology or minimal changes).
TABLE 2.
Pathologic Classification of IIM
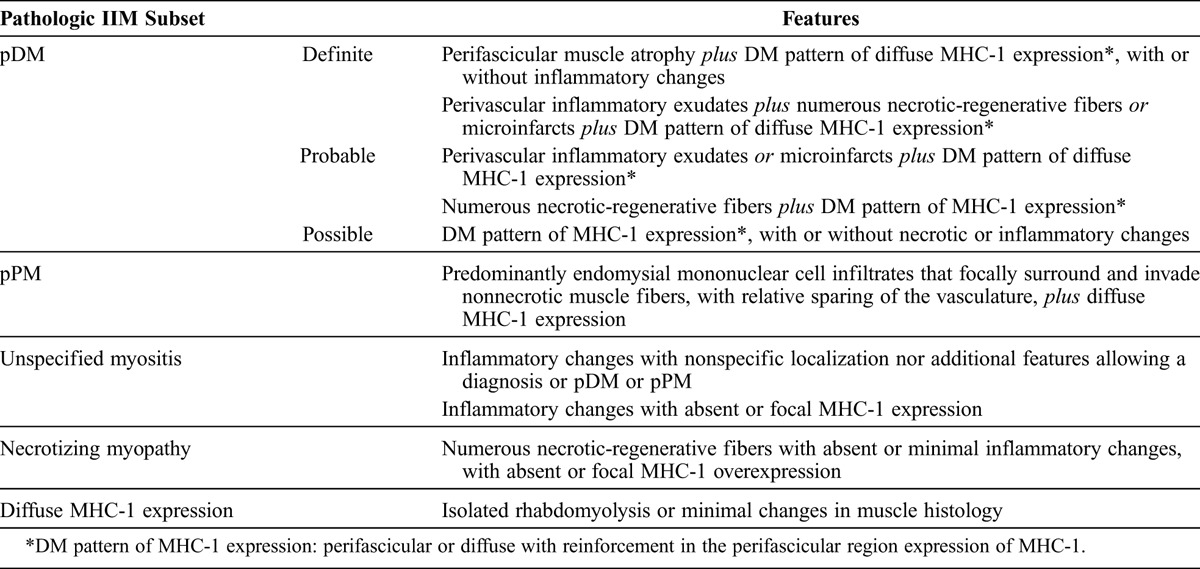
The final diagnosis was established at the end of follow-up on the basis of clinical, serologic, and pathologic features; additional investigations considered necessary by the clinicians (depending on clinical presentation); clinical course; and response to corticosteroid therapy.
RESULTS
Clinical Data, Clinicoserologic Classification, and Final Diagnoses
There were 117 females and 61 males (female to male ratio = 1.9) (Table 3). The mean age at diagnosis was 55.1 years (range, 17–85 yr). The overall frequency of DM rash and overlap features was respectively 51% and 34%. Mean CK level was 13.2 × normal (N) (N < 170 UI/L).
TABLE 3.
Overview of Clinical, Serologic and Pathologic Features of 178 Patients
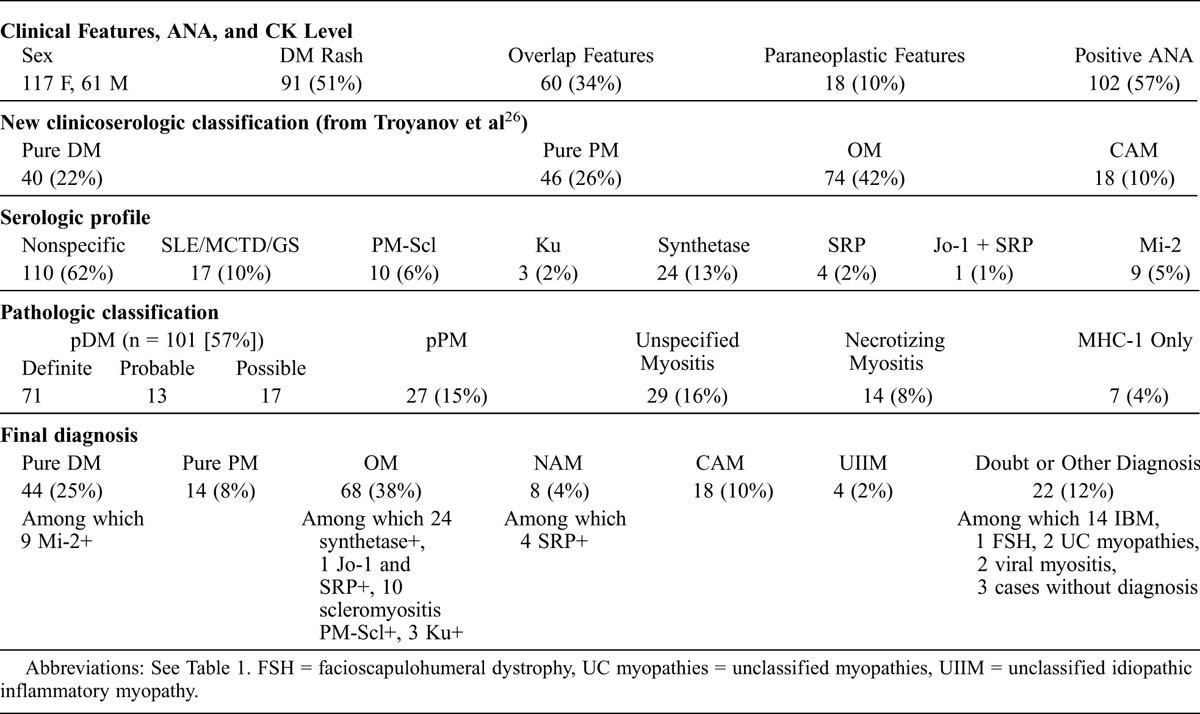
According to the clinicoserologic classification established by Troyanov et al,26 OM was the most frequent entity encountered at diagnosis (74 cases, 42%), followed by pure PM (46 cases, 26%) and pure DM (40 cases, 22%). Eighteen cases (10%) were classified as cancer-associated myositis (CAM), with the following cancers: breast, n = 4; ovary, lung, n = 2 each; and n = 1 each for upper aerodigestive tract, stomach, colon, endometrium, lymphoma, prostate, malignant germinal tumor, mesothelioma plus colonic carcinoma, thymic carcinoma, and metastatic carcinoma of unknown origin. Fifteen other patients had a history of malignancy, but their clinical course did not support the diagnosis of CAM.
At the end of follow-up (final diagnosis), 156 of 178 cases were still categorized as IIM: pure DM, n = 44; pure PM, n = 14; OM, n = 68; NAM, n = 8; CAM, n = 18; unclassified IIM, n = 4. We note that 2 cases were associated with myasthenia gravis. Based on additional investigations and/or follow-up, we excluded the diagnosis of IIM in the 22 remaining cases (12% of the initial cohort), called the “non-IIM subgroup.” For these patients, the following diagnoses were retained: IBM, n = 14; unclassified (noninflammatory) myopathy, n = 2; viral myositis (1 case of myositis associated with human immunodeficiency virus (HIV) and 1 case of myositis associated with primary cytomegalovirus infection), n = 2; facioscapulohumeral dystrophy (proven by genetics), n = 1. For 3 patients, no diagnosis was convincing at the end of follow-up. An overview of baseline patient characteristics and final diagnoses is given in Table 3.
Detailed Serologic Data
ANA were positive in 102 patients (57% of cases). One or several specific autoantibodies were identified in 73 patients (41% of cases). The most common autoantibodies were anti-SSA and anti-Jo-1 (n = 24 each), followed by anti-RNP (n = 14), anti-PM-Scl and anti-SSB (n = 10 each), anti-Mi-2 (n = 9), and anti-Sm (n = 7). Anti-native DNA (n = 5), anti-SRP (n = 5), anti-Ku (n = 3), and anti-PL-7 (n = 1) were rare. Anti-Scl-70 and anti-PL-12 were not recorded. Only 1 specific autoantibody was detected in the majority of sera (50 of 73 cases); 2, 3, or 4 specific autoantibodies were detected in respectively, 12, 6, and 5 sera. Anti-SSA was the autoantibody most frequently associated with 1 or several other ones, predominantly anti-SSB (10 cases) and anti-synthetases (7 cases). Overall, anti-native DNA, anti-Sm, and MAAs were commonly associated with each other, except for anti-PM-Scl and anti-Ku, which were always found to be isolated. MSAs were found to be exclusive, except for 1 serum containing both anti-Jo-1 and anti-SRP antibodies.
According to the serologic profiles defined above, the 178 patients were classified as follows: 1) lack of specific autoantibodies, n = 110 (62%); 2) SLE/mixed connective tissue disease/GS (SLE/MCTD/GS) autoantibodies, n = 17 (10%); 3) anti-PM-Scl, n = 10 (6%); 4) anti-Ku, n = 3 (2%); 5) anti-synthetase, n = 24 (13%); 6) anti-SRP, n = 4 (2%); association of anti-Jo-1 plus anti-SRP, n = 1; 7) anti-Mi-2, n = 9 (5%).
Clinical Features in Relation to Serologic Profile
The mean age at diagnosis was the lowest in the anti-SRP (mean age, 37 yr) and the SLE/MCTD/GS (mean age, 42.1 yr) subgroups. Mean age at diagnosis in the other categories was as follows: for anti-PM-Scl, 56 years; anti-Ku, 59.3 years; anti-Mi-2, 53.4 years; no specific autoantibody, 56.5 years. CAM and the non-IIM subgroup represented the oldest categories (mean age, 63.2 yr and 62.2 yr, respectively). The female to male sex ratio was especially high in patients with anti-Mi-2 (SR = 8), anti-SRP (SR = 4), and anti-synthetase (SR = 3). DM rash was common in the case of anti-Mi-2 autoantibodies, CAM, anti-synthetase, and in the absence of specific autoantibodies (respectively 100%, 83%, 62%, and 56% of cases). It was less frequent in the SLE/MCTD/GS, PM-Scl, and Ku subgroups (respectively 47%, 40%, and 33% of cases), and was never observed in patients with anti-SRP and in the non-IIM subgroup. Overlap signs were common in the SLE/MCTD/GS, anti-synthetase, and anti-PM-Scl subgroups (100%, 83%, and 80% of cases, respectively). They were rare in the case of anti-Ku or anti-Mi-2 autoantibodies, as well as in the absence of specific autoantibodies (respectively 33%, 11%, and 19% of cases). Overlap signs were never observed in association with anti-SRP or CAM. CK level was highest (>20N) in the anti-synthetase and anti-SRP subgroups, at an intermediate level (5–20N) in the remaining serologic subgroups of IIM, and lowest in the non-IIM subgroup (3.3N).
Histopathologic Features in Relation to Serologic Profile
According to the pathologic classification (see Table 2), patients were predominantly considered as having pDM (101 cases = 57%). The next most common pathologic aspects were unspecified myositis (29 cases = 16%) and pPM (27 cases = 15%). Necrotizing myopathy and isolated diffuse MHC-1 expression were rare (respectively 8% and 4% of cases) (see Table 3).
Muscle biopsy results were correlated with the serologic profile (Table 4). pDM was the most frequent pathologic picture in all serologic subsets of IIM, except for anti-Ku and anti-SRP; in contrast, it was not observed in the non-IIM subgroup. It was observed in 92%, 60%, and 70% of patients with anti-synthetase, anti-PM-Scl, and SLE/MCTD/GS serologic profiles, respectively. The most typical forms of pDM (definite pDM) were seen in association with anti-Mi-2, anti-synthetase, and cancer: in these cases, there was a high frequency of perifascicular atrophy, microinfarct, and DM pattern of MHC-1 expression.
TABLE 4.
Pathologic Features of IIM Related to Serologic Profiles
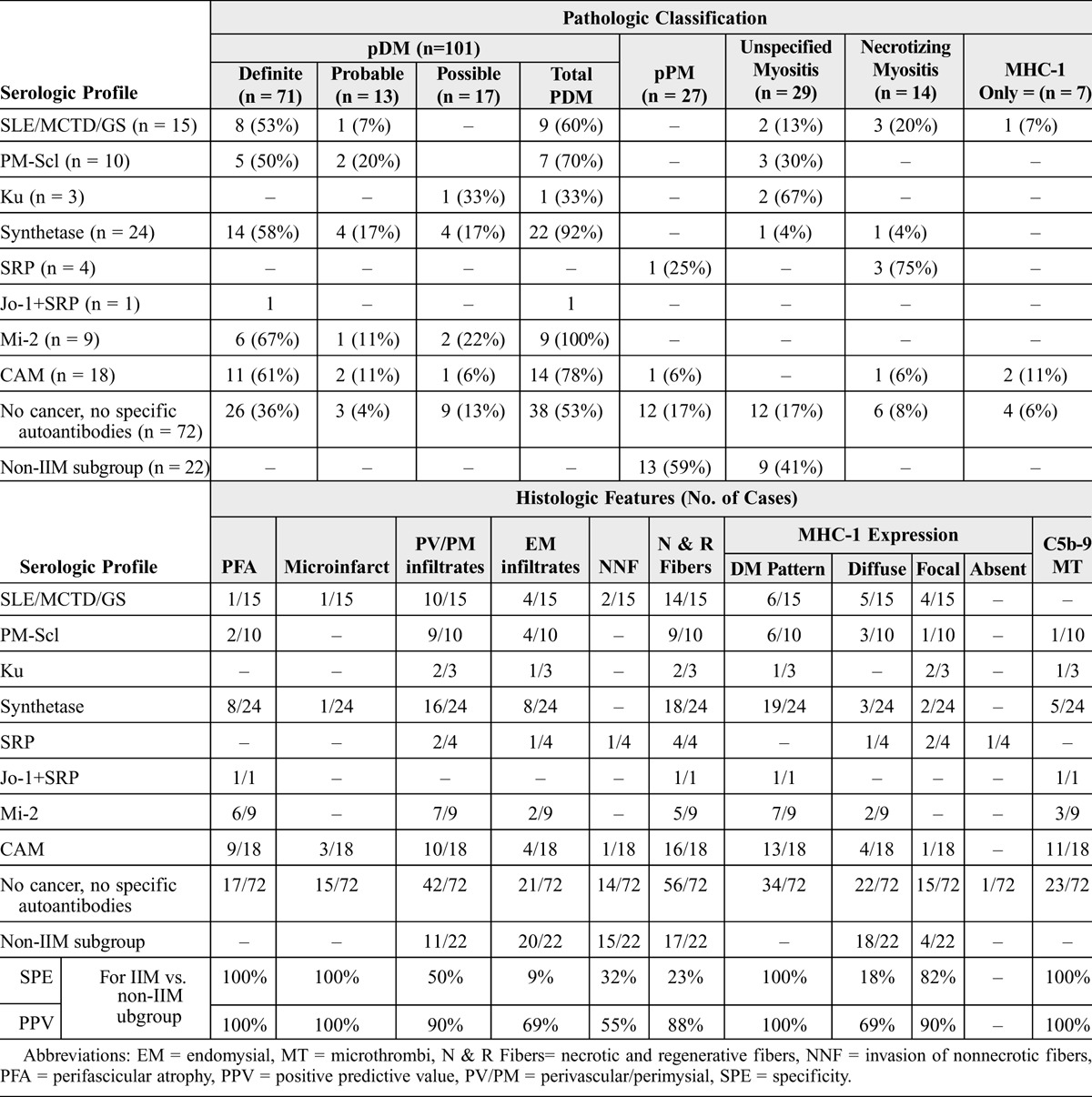
Almost all patients with pPM had a nonspecific serologic profile, as MAAs/MSAs were found in only 1 of 27 cases (1 patient with an anti-SRP). pPM corresponded to the most frequent pathologic pattern in the non-IIM subgroup (59% of cases).
Unspecified myositis was the most frequent pathologic expression of anti-Ku antibodies (2 of 3 cases) and was observed in 30% of patients with anti-PM-Scl autoantibodies. It represented the second most frequent pathologic pattern of the non-IIM subgroup (41% of cases).
Necrotizing myopathy was the pathologic hallmark of anti-SRP antibody (3 of 4 cases), but it also represented 20% of the SLE/MCTD/GS subgroup. Two patients belonging to the non-IIM subgroup showed a SLE/MCTD/GS serologic profile (1 anti-RNP and 1 SSA + SSB).
Perifascicular atrophy, microinfarcts, DM pattern of MHC-1 expression, and microthrombi of C5b-9 were the only pathologic features that had a specificity and a positive predictive value of 100% for IIM (and more specifically DM) versus the non-IIM subgroup (see Table 3). In contrast, perimysial/perivascular infiltrates, endomysial infiltrates, and necrosis were seen in all groups, whatever the serologic profile (except in the single case with anti-Jo-1 plus anti-SRP) or the final diagnosis, IIM or not. Specificity and positive predictive value of each pathologic feature for the diagnosis of IIM versus the non-IIM subgroup are given in Table 3.
Correlation Between Pathologic Features, Clinicoserologic Classification, and Final Diagnosis
When muscle biopsy showed features of pDM (101 cases), cases were classified, in accordance with the clinicoserologic classification system, as OM (49% of cases), pure DM (34% of cases), CAM (14% of cases), and, rarely, pure PM (4% of cases) (Table 5). These latter cases corresponded to 4 patients with IIM without DM rash, overlap features, or specific autoantibodies. Findings suggestive of DM on muscle biopsy enabled us to correct the diagnosis, and at the end of follow-up cases were classified as follows: OM (n = 49, 49% of cases, including 22 patients with anti-synthetase, 1 patient with anti-Jo-1 plus anti-SRP, 7 with scleromyositis with anti-PM-Scl, and 1 patient with anti-Ku); pure DM (n = 38, 38% of cases, including 9 patients with anti-Mi-2); and CAM (n = 14, 14% of cases).
TABLE 5.
Correlation Between Pathologic and Clinicoserologic Classifications and Final Diagnoses
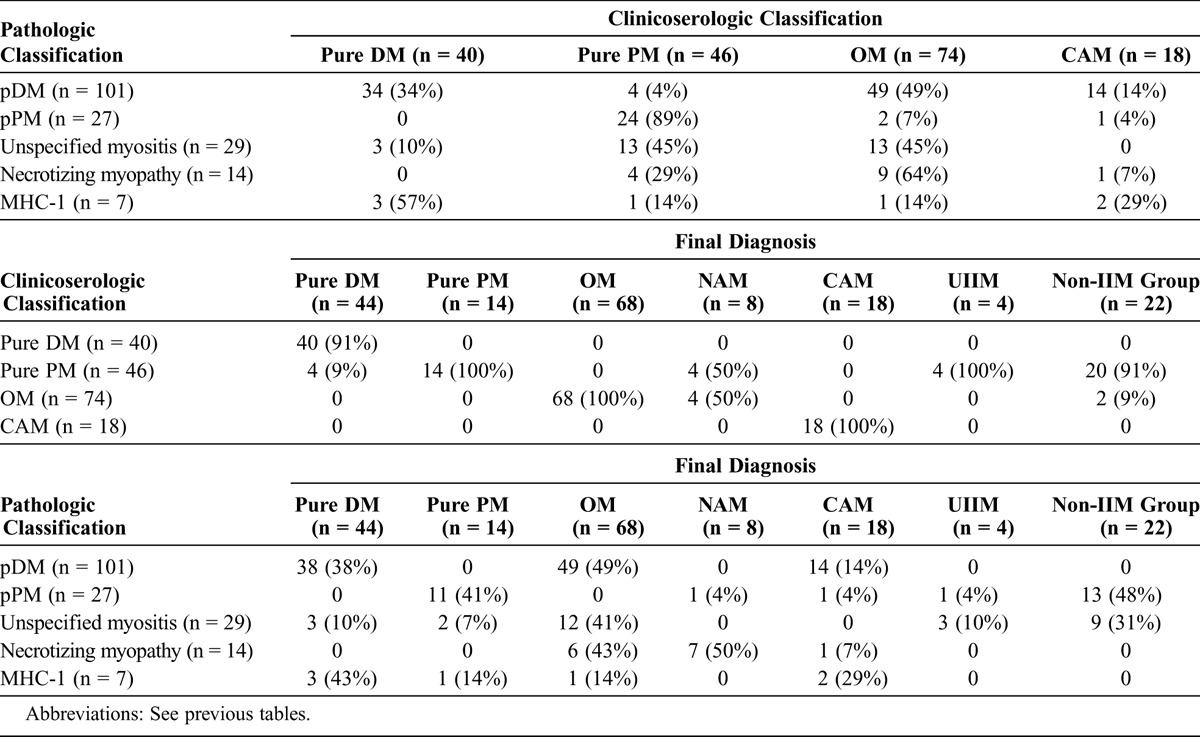
When muscle biopsy demonstrated pPM (27 cases), almost all cases were classified as pure PM using the clinicoserologic classification (89% of cases). But at the end of follow-up, only 52% (14 cases) were still considered as IIM; 11 of them were still considered PM (41% of cases), 1 patient with anti-SRP was considered as NAM, 1 patient with a history of cancer with paraneoplastic features was reclassified as CAM, and 1 patient was considered to have unclassified myositis. Among the 13 remaining patients for whom IIM was ruled out, IBM was the most frequent alternative diagnosis (10 cases). One patient was found to have viral myositis, and for the 2 remaining patients, no diagnosis was convincing at the end of follow-up.
When muscle biopsy demonstrated unspecified myositis (29 cases), cases were classified, in accordance with the clinicoserologic classification system, as pure PM (n = 13, 45% of cases), OM (n = 13, 45% of cases), and pure DM (n = 3, 10% of cases). At the end of follow-up, 41% of patients (n = 12) were considered to have OM (1 patient with overlap features was finally classified as IBM), among them 3 patients with anti-PM-Scl, 2 patients with anti-Ku, and 1 patient with anti-synthetase. Three patients (10%) were considered to have pure DM. The diagnosis of pure PM was maintained in 2 patients only (7% of cases); various diagnoses were made instead in the other patients (unclassified IIM, 3 cases; IBM, 3 cases; unclassified [noninflammatory] myopathy, 2 cases; viral myositis, 1 case; facioscapulohumeral dystrophy, 1 case; no diagnosis, 1 case).
Necrotizing myopathies on muscle biopsy were classified as NAM (50% of cases, n = 7, among them 3 patients with anti-SRP), OM (n = 6, among them 1 patient with anti-synthetase), and CAM (n = 1) at the end of follow-up. Isolated diffuse expression of MHC-1 (that is, with normal muscle biopsy, minimal changes, or mild rhabdomyolysis) was a rare occurrence; it supported the diagnosis of IIM but did not enable us to distinguish the different subtypes.
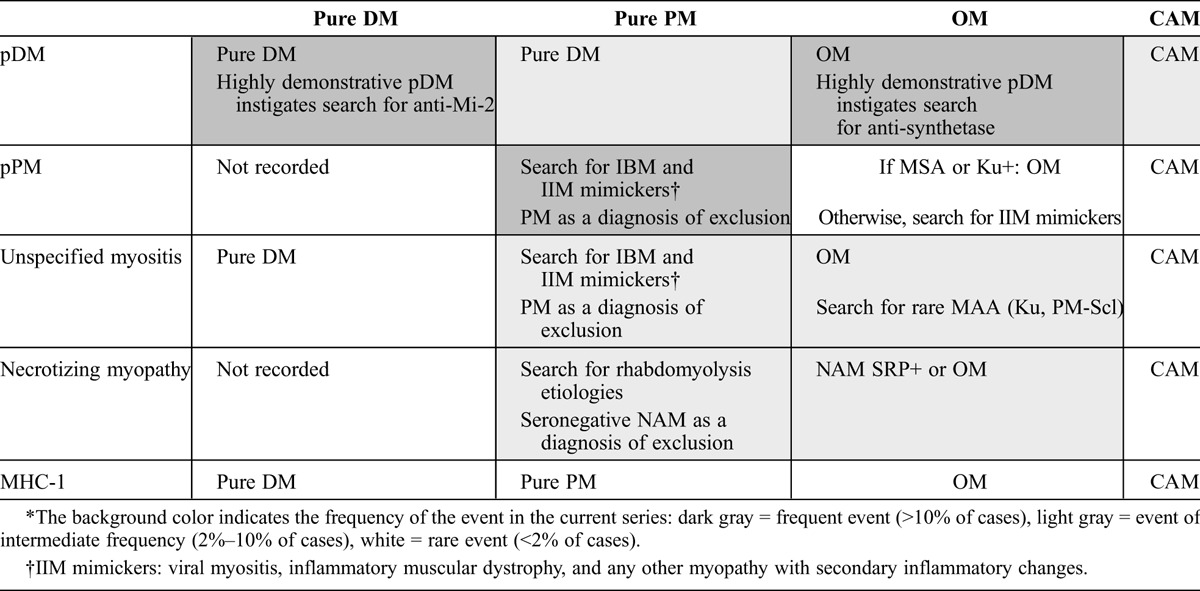
Based on the correlation between the clinicoserologic classification of Troyanov et al and the final diagnoses (see Table 5), we found that the classification system enabled us to correctly classify 79% of patients. This percentage reached 86% if the results of muscle biopsy were taken into account, as muscle histology facilitated the diagnosis of DM and NAM. The diagnosis was improved further if we considered that the finding of pPM and unspecified myositis at muscle biopsy instigated the search for rare MAA/MSA and IIM mimickers. All this shows that clinicoserologic and pathologic factors are complementary. Guidelines for diagnosis according to both classifications are given in Table 6.
TABLE 6.
Guidelines for Diagnosis as a Function of Clinicoserologic and Pathologic Classifications∗
DISCUSSION
To our knowledge we report here the largest recorded series of IIM (with the exception of IBM) that includes in the same study detailed clinical, immunologic, and histologic data. This work complements the findings of Troyanov et al26 and van der Meulen et al,27 studies that made significant headway in the field of IIM but did not account for, respectively, pathologic features and serologic profiles of patients.
Clinical and Immunologic Features of the Current Series Are in Accordance With Recent Literature
In 2005, Troyanov et al26 proposed a new clinicoserologic classification for IIM with overlap features and serologic profiles at the core of the system.2,3 Whereas PM was the most common IIM according to the classification of Bohan and Peter (45% of the cohort, versus 28% for DM and 24% for OM), using their new classification system the frequency of OM rose to 67%, the frequency of DM remained stable, and the frequency of PM fell to 10%.26 In the current series, the distribution pattern of IIM at the end of follow-up was similar (OM, 44%; pure DM, 28%; pure PM, 9%). The overall frequency of specific autoantibodies was 42% in our cohort of IIM and 78% in the subset of OM. Anti-Jo-1 were the most common overlap autoantibodies (24 cases). These results are similar to those of previous studies.5,12,23,26
pDm Is the Most Frequent Histologic Pattern, but Is Common to Pure DM, OM, and CAM
In the current series, pDM was the most frequent histologic pattern but was seen with various conditions: pure DM, OM, and CAM. So it is likely that pDM represents the histologic support for the diagnosis of pure DM, but also myositis of CTD, and almost all of CAM. Muscle biopsy by itself was not able to distinguish the different serologic subgroups reliably, even if typical pDM is suggestive of anti-Mi-2, anti-synthetase, or CAM.
In this context, the correlation between the pathologic classification and the clinicoserologic classification of Troyanov et al26 shows that the 2 classifications are complementary: on one hand, the muscle biopsy shows features of pDM, supporting the diagnosis of pure DM, OM, or CAM; on the other hand, the clinicoserologic data make it possible to distinguish these 3 clinicopathologic entities. Moreover, when a case is classified as pure PM using the clinicoserologic classification (no DM rash, no overlap features, no specific autoantibodies, no cancer), if muscle biopsy shows features of pDM, the case should be classified as pure DM.
pPM Is Rare, and Other Diagnoses Must Be Ruled out Before Making This Diagnosis
In the group of cases classified as PM with both the clinicoserologic classification and the pathologic classification (24 cases), less than half will be classified as PM at the end of follow-up. In this group, IBM was the most frequent diagnostic pitfall (14 cases). Our results are similar to those of van der Meulen et al,27 who concluded in a retrospective study of 165 Dutch myositis patients that pure PM was rare and overdiagnosed, accounting for only 2% of their patients. We conclude that because neither of the 2 classification systems is stringent enough for the diagnosis of PM, this diagnosis can be retained only after the exclusion of IIM mimickers. In the same way, if a MAA is identified in association with pPM on muscle biopsy, the diagnosis of OM must not be systematically made, because the prevalence of MAA in the general population is high, and their main pathologic expression is pDM, not pPM. In the current series, 1 patient with anti-RNP and 1 patient with anti-SSA plus anti-SSB and Sjögren syndrome, both with pPM at muscle biopsy, were finally considered as having IBM and unclassified myopathy.
In Case of Unspecified Myositis and Necrotizing Myopathy, Unusual Autoantibodies Must Be Sought
Unspecified myositis was the second most frequently observed histologic pattern, after pDM. If the case is classified as pure PM according to Troyanov et al (no DM rash, no overlap features, no specific autoantibodies, no cancer), the diagnosis must be questioned as it will be finally retained in only 15% of cases (2 of 13 cases in the current series). In other cases, the clinicoserologic classification provides a basis for classifying a subgroup of cases as pure DM and OM, when cutaneous signs suggestive of DM or a specific autoantibody are present. Interestingly, in the setting of OM, a histologic aspect of unspecified myositis was suggestive of an unusual autoantibody such as anti-Ku or anti-PM-Scl. For these peculiar autoantibodies, only small series of patients with muscle biopsy have been reported, and the pathologic phenotype remains to be refined. Anti-Ku might be associated with rimmed vacuole formation.28
The current study confirms that necrotizing myositis gives grounds for searching for an anti-SRP autoantibody.11,14,22,24 As NAM was not recorded as an entity in the clinicoserologic classification, most cases were classified as OM or pure PM with this classification. It is important to remember that the diagnosis of NAM can be retained only when exposure to myotoxic drugs or toxins, endocrinopathy, or family history of a neuromuscular disease have been ruled out.9
Classification Schemes Should Be Updated Periodically Because of the Identification of Novel Autoantibodies
During the last years, a number of groups have reported the identification of clinically significant novel MSAs, such as anti-CADM-140 (identified in Japanese patients with cutaneous features of DM and rapidly progressive interstitial pneumonia but no clinically significant muscle disease), anti-SAE (in adult patients who present with clinically amyopathic dermatomyositis first and then progress to develop myositis with a high frequency of systemic features), anti-p155/140 (present in 10%–20% of myositis patients, especially adult patients with severe cutaneous involvement and increased risk of malignancy, and juvenile dermatomyositis), anti-p140 (juvenile myositis with calcinosis), and anti-HMG-CoA reductase (patients with statin-associated necrotizing myopathy).13,19,20 Detection of these novel autoantibodies is not yet widely available, and consequently, the classification schemes will need to be regularly updated according to the scientific advances in the field of myopathology and immunology.
In conclusion, the current study shows that the clinicoserologic and pathologic classification systems are complementary, and both should be used to better classify patients suffering from IIM. Guidelines for diagnosis according to both clinicoserologic and pathologic data are given in Table 6; we recommend applying these guidelines in clinical practice.
Footnotes
Abbreviations: ANA = antinuclear antibodies, CAM = cancer-associated myositis, CK = creatine kinase, CTD = connective tissue disease, DM = dermatomyositis, IBM = inclusion body myositis, IIM = idiopathic inflammatory myopathies, MAA = myositis-associated autoantibody, MCTD = mixed connective tissue disease, MHC-1 = major histocompatibility complex, class 1, N = normal, NAM = necrotizing
The authors have no funding or conflicts of interest to disclose.
REFERENCES
- 1.Amato AA, Griggs RC. Unicorns, dragons, polymyositis, and other mythological beasts. Neurology. 2003; 61: 288–289. [DOI] [PubMed] [Google Scholar]
- 2.Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975; 292: 344–347. [DOI] [PubMed] [Google Scholar]
- 3.Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. 1975; 292: 403–407. [DOI] [PubMed] [Google Scholar]
- 4.Bronner IM, Hoogendijk JE, Wintzen AR, van der Meulen MF, Linssen WH, Wokke JH, de Visser M. Necrotising myopathy, an unusual presentation of a steroid-responsive myopathy. J Neurol. 2003; 250: 480–485. [DOI] [PubMed] [Google Scholar]
- 5.Brouwer R, Hengstman GJ, Vree Egberts W, Ehrfeld H, Bozic B, Ghirardello A, Grondal G, Hietarinta M, Isenberg D, Kalden JR, Lundberg I, Moutsopoulos H, Roux-Lombard P, Vencovsky J, Wikman A, Seelig HP, van Engelen BG, van Venrooij WJ. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis. 2001; 60: 116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chinoy H, Fertig N, Oddis CV, Ollier WE, Cooper RG. The diagnostic utility of myositis autoantibody testing for predicting the risk of cancer-associated myositis. Ann Rheum Dis. 2007; 66: 1345–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum. 2010; 62: 2757–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Civatte M, Schleinitz N, Krammer P, Fernandez C, Guis S, Veit V, Pouget J, Harle JR, Pellissier JF, Figarella-Branger D. Class I MHC detection as a diagnostic tool in noninformative muscle biopsies of patients suffering from dermatomyositis (DM). Neuropathol Appl Neurobiol. 2003; 29: 546–552. [DOI] [PubMed] [Google Scholar]
- 9.Dalakas MC. Review: an update on inflammatory and autoimmune myopathies. Neuropathol Appl Neurobiol. 2011; 37: 226–242. [DOI] [PubMed] [Google Scholar]
- 10.Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003; 362: 971–982. [DOI] [PubMed] [Google Scholar]
- 11.Dimitri D, Andre C, Roucoules J, Hosseini H, Humbel RL, Authier FJ. Myopathy associated with anti-signal recognition peptide antibodies: clinical heterogeneity contrasts with stereotyped histopathology. Muscle Nerve. 2007; 35: 389–395. [DOI] [PubMed] [Google Scholar]
- 12.Ghirardello A, Zampieri S, Tarricone E, Iaccarino L, Bendo R, Briani C, Rondinone R, Sarzi-Puttini P, Todesco S, Doria A. Clinical implications of autoantibody screening in patients with autoimmune myositis. Autoimmunity. 2006; 39: 217–221. [DOI] [PubMed] [Google Scholar]
- 13.Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology (Oxford). 2009; 48: 607–612. [DOI] [PubMed] [Google Scholar]
- 14.Hengstman GJ, ter Laak HJ, Vree Egberts WT, Lundberg IE, Moutsopoulos HM, Vencovsky J, Doria A, Mosca M, van Venrooij WJ, van Engelen BG. Anti-signal recognition particle autoantibodies: marker of a necrotising myopathy. Ann Rheum Dis. 2006; 65: 1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoogendijk JE, Amato AA, Lecky BR, Choy EH, Lundberg IE, Rose MR, Vencovsky J, de Visser M, Hughes RA. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10–12 October 2003, Naarden, The Netherlands. Neuromuscul Disord. 2004; 14: 337–345. [DOI] [PubMed] [Google Scholar]
- 16.Koenig M, Fritzler MJ, Targoff IN, Troyanov Y, Senecal JL. Heterogeneity of autoantibodies in 100 patients with autoimmune myositis: insights into clinical features and outcomes. Arthritis Res Ther. 2007; 9: R78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Limaye VS, Blumbergs P, Roberts-Thomson PJ. Idiopathic inflammatory myopathies. Intern Med J. 2009; 39: 179–190. [DOI] [PubMed] [Google Scholar]
- 18.Love LA, Leff RL, Fraser DD, Targoff IN, Dalakas M, Plotz PH, Miller FW. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore). 1991; 70: 360–374. [DOI] [PubMed] [Google Scholar]
- 19.Mammen AL. Autoimmune myopathies: autoantibodies, phenotypes and pathogenesis. Nat Rev Neurol. 2011; 7: 343–354. [DOI] [PubMed] [Google Scholar]
- 20.Mammen AL, Chung T, Christopher-Stine L, Rosen P, Rosen A, Casciola-Rosen LA. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme a reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum. 2011; 63: 713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller FW, Rider LG, Plotz PH, Isenberg DA, Oddis CV. Diagnostic criteria for polymyositis and dermatomyositis. Lancet. 2003; 362: 1762–1763. [DOI] [PubMed] [Google Scholar]
- 22.Miller T, Al-Lozi MT, Lopate G, Pestronk A. Myopathy with antibodies to the signal recognition particle: clinical and pathological features. J Neurol Neurosurg Psychiatry. 2002; 73: 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sordet C, Goetz J, Sibilia J. Contribution of autoantibodies to the diagnosis and nosology of inflammatory muscle disease. Joint Bone Spine. 2006; 73: 646–654. [DOI] [PubMed] [Google Scholar]
- 24.Takada T, Hirakata M, Suwa A, Kaneko Y, Kuwana M, Ishihara T, Ikeda Y. Clinical and histopathological features of myopathies in Japanese patients with anti-SRP autoantibodies. Mod Rheumatol. 2009; 19: 156–164. [DOI] [PubMed] [Google Scholar]
- 25.Targoff IN, Miller FW, Medsger TA, Jr, Oddis CV. Classification criteria for the idiopathic inflammatory myopathies. Curr Opin Rheumatol. 1997; 9: 527–535. [DOI] [PubMed] [Google Scholar]
- 26.Troyanov Y, Targoff IN, Tremblay JL, Goulet JR, Raymond Y, Senecal JL. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore). 2005; 84: 231–249. [DOI] [PubMed] [Google Scholar]
- 27.van der Meulen MF, Bronner IM, Hoogendijk JE, Burger H, van Venrooij WJ, Voskuyl AE, Dinant HJ, Linssen WH, Wokke JH, de Visser M. Polymyositis: an overdiagnosed entity. Neurology. 2003; 61: 316–321. [DOI] [PubMed] [Google Scholar]
- 28.Yamanishi Y, Maeda H, Katayama S, Ishioka S, Yamakido M. Scleroderma-polymyositis overlap syndrome associated with anti-Ku antibody and rimmed vacuole formation. J Rheumatol. 1996; 23: 1991–1994. [PubMed] [Google Scholar]