

Supplemental Digital Content is available in the text
Keywords: antiretroviral therapy, apoptosis, elite controllers, HCV/HIV, hepatic fibrosis, immune activation, TH-17
Abstract
HCV and HIV independently lead to immune dysregulation. The mechanisms leading to advanced liver disease progression in HCV/HIV coinfected subjects remain unclear.
In this cross-sectional study, we assessed the association of HCV viremia, liver fibrosis, and immune response patterns in well-characterized HIV phenotypes: Elite controllers (Elites), HIV controlled (ARTc), and HIV uncontrolled (ARTuc) matched by age and race. Groups were stratified by HCV RNA status. Regulatory T-cell frequencies, T-cell activation (HLADR+CD38+), apoptosis (Caspase-3+), and intracellular cytokines (interferon-γ, IL-2, IL-17) were assessed using multiparametric flow-cytometry. Liver fibrosis was scored by AST to platelet ratio index (APRI).
We found liver fibrosis (APRI) was 50% lower in Elites and ARTc compared to ARTuc. Higher liver fibrosis was associated with significantly low CD4+ T cell counts (P < 0.001, coefficient r = −0.463). Immune activation varied by HIV phenotype but was not modified by HCV viremia. HCV viremia was associated with elevated CD8 T-cell Caspase-3 in Elites, ARTuc, and HIV− except ARTc. CD8 T-cell Caspase-3 levels were significantly higher in HCV RNA+ Elites (P = 0.04) and ARTuc (P = 0.001) and HIV− groups (P = 0.02) than ARTc. Importantly, ARTuc HCV RNA+ had significantly higher CD4 T-cell interleukin-17 levels than ARTuc HCV RNA− (P = 0.005).
HIV control was associated with lower liver fibrosis in HCV/HIV co-infected women. HCV viremia is associated with an inflammatory CD4 TH-17 phenotype in absence of HIV control and higher frequency of pro-apoptosis CD8 T-cells critical to avert progression of HIV and HCV disease that is attenuated in ART controllers. Elite controllers with HCV viremia are more prone to CD8 T-cell apoptosis than ART controllers, which could have negative consequences over time, highlighting the importance of ART control in HCV/HIV coinfected individuals.
1. Introduction
Globally, 185 million individuals are estimated to be infected with Hepatitis C virus (HCV)[1] and 3.4 to 4.4 million people in the United States.[2] An estimated 25% of individuals infected with human immunodeficiency virus (HIV) are co-infected with HCV.[3] Life-threatening infections owing to AIDS have declined with the advent of highly active antiretroviral therapy (HAART), yet the incidence of morbidity and mortality from liver disease has increased, and end-stage liver disease has become the leading cause of non-AIDS-related death in HIV patients.[4] HCV/HIV co-infected patients not receiving ART have more advanced liver fibrosis.[5] Advanced fibrosis leads to poorer response to HCV therapy[6] and increased rates of cirrhosis, liver failure, and hepatocellular carcinoma.[7] The mechanisms leading to advanced liver disease progression in HCV/HIV co-infected subjects remain unclear.
Chronic immune activation is a hallmark of HIV disease and is associated with CD4 decline and HIV disease progression.[8,9] Studies examining the effects of HCV co-infection on immune activation are contradictory with some reporting an association with CD8 T cell activation not confirmed in others.[10,11] HCV progression in HIV infection has been reported to be owing to HIV-mediated loss of CD4 helper T cells.[12] Liver damage in chronic infection is predominantly from non-HCV-specific inflammatory T cells infiltrating the liver.[13,14] To better understand the immune mechanisms in HCV/HIV co-infected, we examined the association of HCV viremia, hepatic fibrosis, and immune response patterns namely CD4 numbers, T regulatory cell frequency, immune activation, apoptosis, and T cell function in well characterized HIV phenotypes: Elite controllers, ART-controlled, ART-uncontrolled, and HIV negatives. Liver disease progression was assessed by the AST to platelet ratio index (APRI). We hypothesized that HCV viremia affects both hepatic fibrosis and T cell phenotype and function in HIV and HCV co-infected subjects.
2. Materials and methods
2.1. Patient population
The cross-sectional study included HCV seropositive women enrolled in the Women's Interagency HIV Study (WIHS), an ongoing NIH multisite prospective study that includes HIV-infected and HIV-uninfected women followed longitudinally semiannually with clinical, laboratory assessment and collection of biological specimens.[15] Overall, 32% of HIV-seropositive women were co-infected with HCV, acquired before enrolment in WIHS. Women who received HCV therapy were excluded. All women gave written, informed consent for participation in WIHS. The WIHS study was approved by all Institutional Review Boards of participating institutions. HCV seropositivity was documented by antibody to HCV by second-generation or third-generation EIA (Ortho-Diagnostic Systems) and viremia by RT-PCR (COBAS Amplicor HCV Detection Kit, Roche Diagnostic Systems). HIV-RNA was measured using the isothermal nucleic acid sequence-based amplification (NASBA/Nuclisens) method (bioMerieaux) with limit of detection of <50 HIV RNA copies/mL.
The study population was comprised of homogenous, well-characterized HIV phenotypes stratified by serum HCV RNA-positive (HCV RNA+) or -negative (HCV RNA− <50 IU/mL). The HCV RNA-negative women did not have chronic HCV as they had spontaneously cleared HCV infection and were compared with those who had chronic HCV infection (detectable HCV RNA in serum). Women were matched by age, race/ethnicity, HCV RNA status within the following groups: Elite controllers (Elites, n = 19), defined as ART-naïve, CD4 >500 cells/mm3, VL <50 HIV RNA copies/mL, for at least 18 months; HIV-controlled on antiretroviral therapy (ART) ([ARTc, n = 20], individuals who are currently on ART CD4 >350 cells/mm3, VL <50 HIV RNA copies/mL, for at least 18 months); HIV-uncontrolled on ART (ARTuc, n = 21, individuals who are currently on ART but with detectable HIV RNA for at least 18 months); and HIV-negative (HIV−, n = 18) women.
2.2. Assessment of liver fibrosis
Liver fibrosis was assessed by the AST to platelet ratio index (APRI), a noninvasive serum marker that has been shown to reflect hepatic fibrosis accurately among chronic hepatitis C patients with HIV.[16,17] APRI can be easily obtained in the clinical setting, as it is calculated using serum aspartate aminotransferase (upper limit of normal used 40 IU/L) and platelet count (×109 cells/L) and was used for this analysis as described elsewhere.[16]
2.3. Assessment of Immune Markers
Immune activation and pro-apoptosis marker Caspase-3 in CD4 & CD8 T cells was measured on single cell level using multiparametric flow cytometry on cryopreserved peripheral blood mononuclear cells (PBMCs). PBMCs were washed and stained with LIVE/DEAD Fixable Aqua Dead Cell Stain to differentiate viable cells and stained with fluorochrome conjugated antibodies against the following cell surface markers CD3, CD4, CD8, CD38, and HLA-DR. Following permeabilization, cells were stained intracellularly with fluorochrome conjugated anti-Caspase-3. PBMCs were fixed and acquired on a LSR-II flow cytometer (BD Biosciences), and analyzed using FlowJo (Tree Star Inc, Ashland, OR). Immune activation was defined as co-expression of HLADR+CD38+ and intracellular Caspase-3 expression was assessed as a measure of cells prone to apoptosis in live CD3+CD4+ and CD3+CD8+ T cells. Regulatory T cells (T-regs) were enumerated in thawed PBMCs by measuring transcription factor Forkhead box Protein 3 (FoxP3) expression intracellularly on CD25+CD4+CD3+T cells, stained using antibodies as described above. Regulatory T cells were defined as CD4+CD25+FoxP3+ T cells. For assessing T cell functionality, thawed PBMCs were stimulated with PMA (100 ng/mL) and Ionomycin (1 μg/mL) for 6 hours; Brefeldin A was used to block cytokine egress. Stimulated PBMCs were stained for T cell subtyping using fluorochrome conjugated antibodies against CD3, CD4, and CD8. Cells were permeabilized and stained intracellularly with fluorochrome conjugated anti interferon [IFN]-γ, interleukin [IL]-2, and IL-17. PBMCs were fixed, acquired as above.
2.4. Statistical analyses
Demographic and clinical characteristics at the time of the patient visit were evaluated with proportions for categorical variables and medians (IQRs) for continuous variables within these HIV phenotypic and HCV viremia subgroups. For easy of interpretability, we report the median (IQR), although variables lacking a normal distribution were transformed to achieve normality (log10 or square root) for use in more powerful parametric tests. Measures of fibrosis and immune markers were compared across HCV viremia subgroups using the t test and compared across HIV phenotypic subgroups using one-way analysis of variance with post-hoc tests by the Tukey–Kramer method. Associations were assessed with Spearman correlation coefficients. P values <0.05 were considered statistically significant. All analyses were completed with SAS v9.3 (SAS Institute, Cary, NC).
3. Results
3.1. Patients demographics
A total of 78 HCV antibody-positive women were included in this study of whom 41 had chronic HCV (HCV RNA+) and 37 had spontaneously cleared HCV (HCV RNA−). Study participants’ characteristics are described in Table 1: 19 Elite Controllers, 20 ART-controlled, 21 ART-uncontrolled, and 18 HIV-negatives; and by the presence of HCV viremia. HCV viremia across the RNA+ groups was comparable (median Log10 6.1 [5.6–6.4 IQR]). The median age was 49 years and 56% were African American.
Table 1.
Selected Patient Characteristics and Demographics.
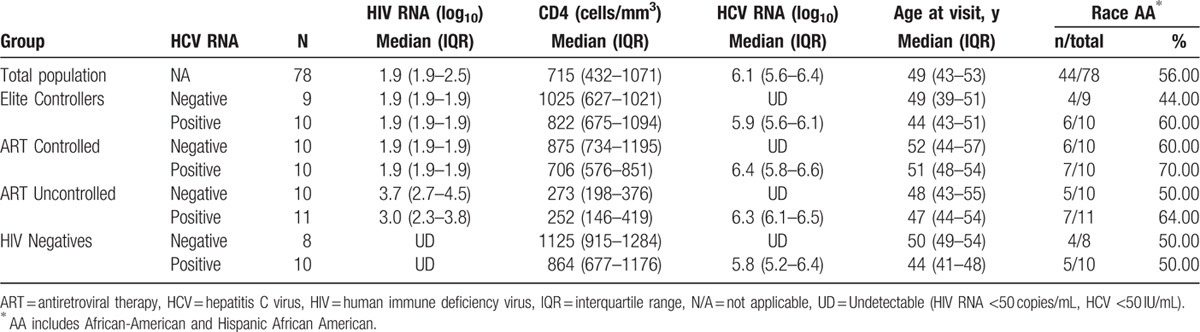
3.2. Hepatic fibrosis varies by HIV phenotype
The presence of HCV viremia (chronic HCV) was significantly associated with an increase in liver fibrosis marker as expected (APRI median HCV RNA+ 0.40 vs. HCV RNA− 0.21, P < 0.001) (Fig. 1A). We examined whether liver fibrosis measured by APRI differed in various HIV phenotypes. As shown in Fig. 1B, liver fibrosis varied significantly by HCV viremia in HIV phenotypes. HCV viremia was significantly associated with higher Log10 APRI compared to HCV RNA− in each of the HIV phenotype groups: Elites (P = 0.004), ARTc (P = 0.02), and ARTuc (P = 0.02). Importantly, the median APRI scores in Elites (0.31) and ARTc (0.37) with HCV RNA+ were 50% lower compared to ARTuc HCV RNA+ (0.74). This clinically lower fibrosis score was observed with HIV viral control, either in natural Elite control or via ART.
Figure 1.
Correlates of Liver fibrosis. (A) Liver fibrosis was associated with HCV viremia; fibrosis serum marker APRI was used to assess liver fibrosis in study participants. Significantly higher fibrosis was observed in women with HCV viremia. Significance between groups as depicted using t test. (B) Hepatic fibrosis varies by HIV Phenotype: APRI Scores in Elites, ARTc, and ARTuc in HCV RNA+ and HCV RNA− groups. Boxes represent 25th, 75th percentiles of the distribution (interquartile range [IQR]) and whiskers represent minimum and maximum values. Median value is shown as the solid line. Significance as depicted (C) Liver fibrosis was associated with low CD4 numbers: Higher liver fibrosis (Log10 APRI) correlated significantly with lower CD4 numbers. Spearman correlation coefficient between CD4 numbers and Log10 APRI as depicted.
We further investigated the immune correlates of liver fibrosis in HCV/HIV coinfected subjects. In this study, we found low CD4 numbers to be significantly associated with liver fibrosis (Log10 APRI) (P < 0.001, coefficient r = −0.463) (Fig. 1C). We sought to examine whether liver fibrosis was associated with alterations in systemic levels of CD4 T regulatory cells. In this study, although not statistically significant, we found a positive correlation between liver fibrosis scores Log10 APRI and T-reg frequencies (P = 0.06, r = 0.237).
3.3. Higher immune activation and Caspase-3 levels are associated with lower CD4 numbers
We investigated whether association of liver fibrosis with low CD4 T cell count found in this study was attributable to chronic immune activation (via activation induced cell death) or to apoptotic loss of T cells. Overall, in all subjects, we found the latter: lower CD4 numbers were associated with higher CD4 and CD8 T cell activation (P < 0.001 and P < 0.001, respectively, Supplemental Fig. 1A and B) and CD4 and CD8 T cell Caspase-3 levels (P = 0.06 and p = 0.005, respectively, Supplemental Fig. 1C and D).
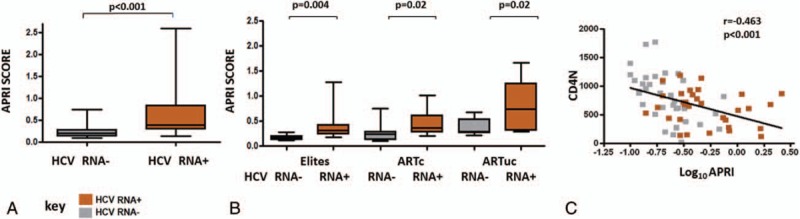
3.4. Immune Activation varies with HIV phenotypes and is not modified by HCV viremia
We examined whether immune activation levels varied with the presence of HCV viremia in the different HIV phenotypes. We found, CD4 T cell activation varied by HIV phenotype, but did not differ significantly by HCV viremia. Significant differences in CD4 T cell activation were observed in HIV phenotypes. Differences in CD4 T cell activation levels are shown in Figure 2A. ARTuc subjects had significantly higher CD4 T cell activation levels compared to HIV− in HCV RNA+ (P = 0.02). Furthermore, ARTuc subjects had significantly higher CD4 T cell activation levels compared to all other HIV phenotypic groups within HCV RNA− subjects (ARTc, P = 0.02; Elite, P = 0.001; and HIV−, P = 0.01). Similarly, CD8 T cell activation varied by HIV phenotype and HCV viremia did not modify CD8 T cell activation within any HIV group. Significant differences in CD8 T cell activation were observed in HIV phenotypes. Differences in CD8 T cell activation levels are shown in Figure 2B. ARTuc subjects had significantly higher % CD8 CD38+HLADR+ compared to ARTc, and HIV−, in both HCV RNA+ (P = 0.008 and P = 0.003, respectively) and HCV RNA− groups (P = 0.046 and P = 0.01, respectively). The lack of a detectable association between HCV viremia and the activation of CD4 and CD8 T cells potentially suggests HCV pathogenesis may involve pathways distinct from immune activation.
Figure 2.
Immune activation varies with HIV phenotypes and is not modified with HCV viremia, T-cell activation levels in HIV phenotypes by HCV RNA status. (A) CD4 T-cell activation (B) CD8 T-cell activation in Elites, ARTc, ARTuc, and HIV-negatives in HCV RNA+ and HCV RNA− groups. Boxes represent 25th, 75th percentiles of the distribution (interquartile range IQR]) and whiskers represent minimum and maximum values. Median value is shown as the solid line. Significance as depicted, t test was used across HCV viremia subgroups and 1-way analysis of variance across HIV phenotypes.
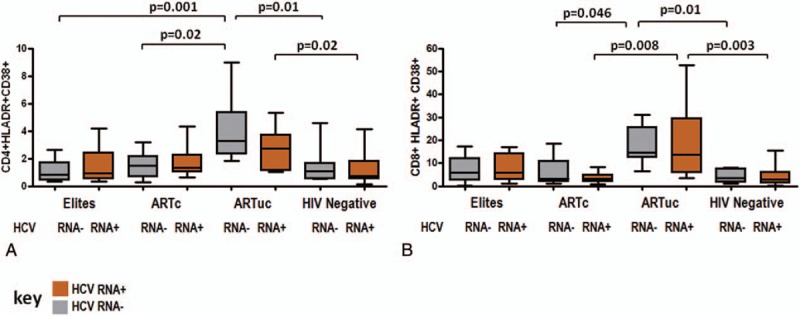
3.5. HCV viremia is associated with elevated levels of CD8 T cell Caspase-3 which is significantly lower in ARTc; Elites with HCV viremia are more prone to CD8 T cell apoptosis than ARTc
To evaluate potential mechanisms of the interaction between HCV and HIV disease progression, we investigated constitutive intracellular Caspase-3, a pro-apoptotic marker in CD4 and CD8 T cells. In this study, we found Caspase-3 expression varied by HIV phenotypes and by HCV viremia. Differences were observed across HIV phenotypes with lowest CD4 T cell Caspase-3 in ARTc HCV RNA+ group. Differences in CD4 T cell Caspase-3 are shown in Figure 3A. ARTc HCV RNA+ group had significantly lower CD4 Caspase-3 compared to ARTuc HCV RNA+ (P = 0.02). Similarly, differences were observed across HIV phenotypes with lowest CD8 T cell Caspase-3 in ARTc HCV RNA+ compared to all groups. Differences in CD8 T-cell Caspase-3 are shown in Figure 3B. CD8 T-cell Caspase-3 was significantly higher in HCV RNA+ Elites (P = 0.04), ARTuc (P = 0.001), and HIV− (P = 0.02) compared to ARTc HCV RNA+. Importantly, Elites HCV RNA+ subjects had significantly higher CD8 T cell Caspase-3 compared to ARTc HCV RNA+.
Figure 3.
CD8 T-cell Caspase-3 level varies with HCV viremia and is ameliorated in ARTc, Caspase-3 levels in HIV phenotypes by HCV RNA status. (A) CD4 T-cell Caspase-3 levels and (B) CD8 T-cell Caspase-3 levels in Elites, ARTc, ARTuc, and HIV negatives in HCV RNA+ and HCV RNA− groups. Boxes represent 25th, 75th percentiles of the distribution (interquartile range [IQR]) and whiskers represent minimum and maximum values. Median value is shown as the solid line. Significance as depicted, t test was used across HCV viremia subgroups and 1-way analysis of variance across HIV phenotypes.
HCV viremia was associated with elevated CD8 T cell Caspase-3 in Elites, ARTuc, and HIV− (P = 0.049) as compared to HCV RNA− in each group except ARTc. In ARTc, however, HCV viremia was associated with significantly lower Caspase-3 expression (P = 0.03) compared to HCVRNA−. Thus, HCV viremia could result in CD8 T cell loss via apoptosis; this is lower in HCV RNA+ ARTc. The more pronounced decrease in this group could be attributed to better CD8 T-cell survival owing to high IL-2 production.
3.6. ART augments CD8 IL-2 production in HCV viremic subjects
We assessed intracellular production of IL-2 and IFN-γ in CD8 T cells. The highest production of CD8 IL-2 was observed in ARTc RNA+ compared to ARTc HCV RNA− group (P = 0.06), a group with the lowest CD8 T-cell caspase-3 levels. Elites, ARTuc, and HIV− did not differ significantly by HCV RNA status in CD8 IL-2+, IFN-γ+, and IFN-γ+IL-2+-producing T cells.
The mechanism by which ART impacts cell survival is likely by augmenting IL-2 production. We found an inverse correlation of CD4 and CD8 T -ell caspase-3 with IL-2 production (P = 0.05, r = −0.677) and (P = 0.050, r = −0.683), respectively (data not shown), in the ARTc RNA+ group, suggesting that ART indirectly attenuates effects of HCV viremia by improving immune dysregulation in HCV/HIV co-infected women.
3.7. HCV viremia in absence of HIV control is associated with a proinflammatory CD4 Th-17 phenotype
Patterns of cytokines in CD4 T cells in HIV phenotypes by HCV RNA status are shown in Figure 4. Elites and ARTc did not differ in their frequency of CD4 IFN-γ+, IL-2+, IFN-γ+IL-2+, and IL-17+-producing cells by HCV RNA status. CD4 IFN-γ+ (TH-1) cells were significantly elevated in HIV-negative HCV viremia compared to HIV-negative HCV RNA− subjects (P = 0.02). HCV viremia was associated with significantly higher IL-17 in ARTuc subjects compared to ARTuc HCV RNA− (P = 0.005). HCV viremia is associated with an increase in proinflammatory IL17 CD4 (TH-17) phenotype in absence of HIV control. Thus, replication of both HIV and HCV viruses appears to skew to a proinflammatory TH-17 phenotype.
Figure 4.
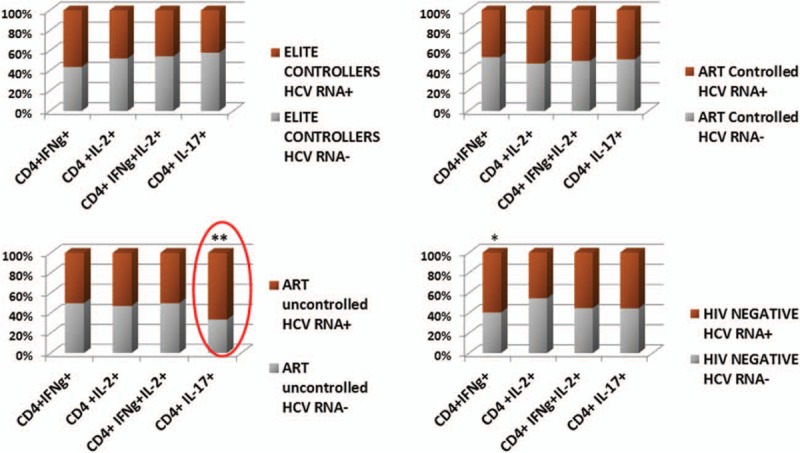
HCV viremia is associated with a proinflammatory CD4 TH-17 phenotype in absence of HIV control, CD4 T-cell intracellular cytokine production in HIV phenotypes by HCV RNA status: IL-17, IFN-γ, IL-2, and IFN-γ and IL-2 production in (A) Elites (upper left), (B) ARTc (upper right), (C) ARTuc (lower left), and (d) HIV-negative (lower right). The Bar graphs compare percentage of each value of HCV RNA+ and RNA− to the total and are displayed as 100% stacked columns.
4. Discussion
In this study, we addressed the hypothesis that coinfection with HCV and HIV affects both hepatic fibrosis and T cell phenotype and function. We report, in well characterized HIV phenotypes, the effects of chronic HCV (presence of HCV viremia) on liver fibrosis, CD4 T cell numbers, T regulatory cell frequency, immune activation, apoptosis, and T cell cytokine production. APRI was used for assessment of liver fibrosis. The prognostic utility of APRI has been assessed in this cohort earlier and found to be independently associated with all-cause and liver mortality in HCV/HIV-co-infected women.[18] We found HCV viremia to be significantly associated with worse liver fibrosis as expected. Contrary to earlier reports,[19] recent reports suggest that rates of advanced fibrosis and liver disease progression in persons with controlled HIV are similar to the rates of those with HCV alone.[20,21] The difference in liver fibrosis with HIV control was compelling: 50% lower with HIV control by ART or in Elites as compared to ARTuc.
Chronic HCV likely alters T-cell frequencies as well as phenotype and function. HCV/HIV coinfection is known to be associated with lower CD4 counts and a smaller CD4-cell recovery[22,23] attributed to homeostatic alterations in CD4 subsets or thymic defects.[24–26] Our data show a significant association of lower CD4 numbers with worse fibrosis suggesting that immune failure is a strong correlate of liver fibrosis in HCV/HIV coinfected patients.
HIV infection is associated with chronic immune activation[27] and activation-induced cell death is one of the proposed mechanisms of immune decline.[28] HIV control usually requires long-term ART, but it is also seen in Elite controllers, who do not require ART. However, some individuals do not achieve HIV control despite ART (ARTuc). Our study is the first to study immune function in women with chronic HCV and well characterized different HIV status and control. Our data show that immune activation in HCV/HIV infection varies by HIV phenotype but does not differ significantly by HCV viremia.
Previous studies have shown CD8 T-cell activation to be elevated in HCV/HIV coinfected subjects and persisted despite antiretroviral control of HIV.[10,29] More recent studies have reported CD8 T-cell activation is not enhanced in HCV/HIV coinfected[11,24,30] and HCV/HIV coinfected subjects have activated CD4 T cells.[11,24] An earlier study in the WIHS cohort by Kovacs et al[29] reported elevated CD8 T-cell activation in HCV/HIV coinfected subjects, wherein HIV-1 disease in most women was at an early stage: almost 50% were untreated and only 7.7% were receiving HAART. In contrast, a more recent study in WIHS cohort by Kuniholm et al[24] assessing T-cell phenotype in HIV-infected women stratified by HCV RNA did not find CD8 T-cell activation in HCV/HIV coinfected women. A Scandinavian study by Gonzalez et al[10] showed immune activation as determined by CD38 expression in T cells to be more pronounced despite successful suppression of HIV in HCV/HIV coinfected subjects than in the appropriate monoinfected controls. Participants in the Scandinavian study were on combination of pegylated IFN-α (IFNα) and ribavirin, with 64% of patients reaching a sustained virological response to treatment.[10] The differences in our results from the Scandinavian study could be because of differences in definition of immune activation (CD38+HLADR+ vs. CD38+) and that our participants were not on IFN-based HCV therapy. IFN-α is known to contribute to CD8 T cell activation as was shown in a study in untreated HIV-infected subjects receiving IFN therapy.[31] None of these previous reports studied carefully selected different HIV phenotypes but rather, chose unselected HIV-positive subjects. Although there are fewer subjects in each group, HIV Elites are uncommon and our groups were carefully chosen for HIV status with matched HCV viremia within groups.
Apoptosis or programmed cell death can be induced by a variety of stimuli including HIV and HCV. A study by Nunez et al showed using Annexin V labeling that T-cell apoptosis was significantly higher in HCV/HIV coinfected than in monoinfected patients. Plasma HCV RNA levels were not associated with the levels of apoptosis in CD4+ or in CD8+ T cells.[32] Another study by Korner et al using the TUNEL assay to measure apoptosis reported that HCV co-infection disproportionately increased rates of apoptosis in CD4+ T-cells from untreated HIV-positive patients that was correlated with HIV, but not HCV, viral loads.[33] In our study using constitutive intracellular Caspase-3 as a measure of cells prone to apoptosis, we show CD8 T cell Caspase-3 is elevated in all groups: Elites, ARTuc, and HIV− except ARTc. Elites are known to have higher CD8 T-cell Caspase-3 compared to HIV−.[34] In this study, importantly we show that presence of HCV viremia can result in higher CD8 T cell apoptosis in Elites than Elites HCV RNA− and significantly higher compared to ARTc with HCV viremia. Thus, Elites with HCV viremia could have negative consequences over time. CD8 T-Cell Caspase-3 was lowest in ART controllers. The role of CD8 T cells in viral clearance is known and recent reports have shown that clearance of chronic HCV infection can occur after exposing HIV/HCV coinfected patients to HAART.[35–37] In our subjects, ART appears to attenuate HCV-mediated immune dysregulation likely by improving T-cell survival.
HIV infection is associated with an imbalance in T-helper cell subsets.[38] The imbalance of T-helper lymphocytes is also known to play an important role in immune pathogenesis of chronic hepatitis C virus (HCV) infection.[39] Our data suggest presence of HCV viremia alone is associated with frequency of TH-1 (IFN-γ) cells, as observed in HIV-negative subjects with HCV viremia. Importantly, we report significantly higher frequency of IL-17+ cells in subjects with HIV and HCV replication, representing an inflammatory CD4 TH-17 cell phenotype. Hepatitis C virus promotes T-helper (TH) 17 responses through thymic stromal lymphopoietin production by infected hepatocytes.[40] IL-17-producing CD4+ TH-17 cells have been reported to trigger tissue inflammation and are important contributors to hepatic inflammation and liver cirrhosis[41] and may be important contributors to hepatic inflammation and liver fibrosis and cirrhosis in HCV/HIV co-infected subjects. Our data show in absence of HIV control that HCV viremia is associated with CD4 TH-17 phenotype, highlighting the importance of HIV control in HCV/HIV coinfected individuals.
The data presented in this study should be interpreted in light of limitations of the study. HCV and HIV pathogenesis studies are predominantly conducted in men and this study was conducted in women only. Although this may seem a limitation, the WIHS cohort, of HIV- and HCV- infected and uninfected women, allowed us to study effects of HCV viremia in unique well characterized HIV phenotypes including the least prevalent (<1%) elite controllers that were matched for age and race. We used noninvasive markers of fibrosis APRI, which can be utilized retrospectively. As this was a retrospective analysis, liver biopsy and Fibroscan, while more sensitive for fibrosis, were not available in these subjects. Age has been used as a proxy for length of infection[42], which may affect the results, but our HIV groups were matched by age. The subjects may migrate from one HIV phenotype to another over time, but these women were carefully elected in this cross-sectional study to be sustained in the HIV phenotype for a period of at least 18 months. Although our sample size was small, this study is unique in the careful study design of extremely well characterized HIV and HCV phenotypes. Importantly, the study participants were comprised of homogenous, well characterized HIV phenotypes stratified by HCV RNA-negative and –positive groups, not on HCV therapy. Prospective investigations are needed to establish the contribution of these findings including the role of IL-17 in liver disease progression.
5. Conclusions
Hepatic fibrosis varied by HCV viremia and HIV phenotype. HIV control Elite or via ART was associated with lower liver fibrosis in HCV/HIV coinfected women. Chronic HCV in HIV coinfected individuals affects T-cell phenotype and function. Immune activation varied within distinct HIV phenotypes, but did not differ significantly with HCV viremia. HCV viremia was associated with elevated levels of CD8 T-cell Caspase-3 in Elites, ART–uncontrolled, and HIV-negative except in ART-controlled women. Importantly, we report that HCV viremia is associated with an inflammatory CD4 TH-17 phenotype in absence of HIV control, which has implications to hepatic injury and higher frequency of pro-apoptotic CD8 T cells critical to avert progression of HIV and HCV diseases. Elite controllers with HCV viremia are more prone to CD8 T-cell apoptosis than ART controllers, which could have negative consequences over time.
Supplementary Material
Acknowledgements
The authors thank the participants of this study.
Footnotes
Abbreviations: APRI = AST to platelet ratio index, ART = antiretroviral therapy, Elites = Elite controllers, HCV = Hepatitis C Virus, HIV = human immunodeficiency virus, IFN-γ = interferon-gamma, IL = interleukin, IQR = interquartile range, PMA = phorbol myristate acetate, TH-17 = T helper 17 cell, T-regs = regulatory T cells, WIHS = Women's Interagency HIV Study.
Data in this manuscript were collected by the Women's Interagency HIV Study (WIHS). The contents of this publication are solely the responsibility of the authors and do not represent the official views of the National Institutes of Health (NIH). WIHS (Principal Investigators): UAB-MS WIHS (Michael Saag, Mirjam-Colette Kempf, and Deborah Konkle-Parker), U01-AI-103401; Atlanta WIHS (Ighovwerha Ofotokun and Gina Wingood), U01-AI-103408; Bronx WIHS (Kathryn Anastos), U01-AI-035004; Brooklyn WIHS (Howard Minkoff and Deborah Gustafson), U01-AI-031834; Chicago WIHS (Mardge Cohen), U01-AI-034993; Metropolitan Washington WIHS (Mary Young), U01-AI-034994; Miami WIHS (Margaret Fischl and Lisa Metsch), U01-AI-103397; UNC WIHS (Adaora Adimora), U01-AI-103390; Connie Wofsy Women's HIV Study, Northern California (Ruth Greenblatt, Bradley Aouizerat, and Phyllis Tien), U01-AI-034989; WIHS Data Management and Analysis Center (Stephen Gange and Elizabeth Golub), U01-AI-042590; Southern California WIHS (Alexandra Levine and Marek Nowicki), U01-HD-032632 (WIHS I–WIHS IV). The WIHS is funded primarily by the National Institute of Allergy and Infectious Diseases (NIAID), with additional co-funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), the National Cancer Institute (NCI), the National Institute on Drug Abuse (NIDA), and the National Institute on Mental Health (NIMH). Targeted supplemental funding for specific projects is also provided by the National Institute of Dental and Craniofacial Research (NIDCR), the National Institute on Alcohol Abuse and Alcoholism (NIAAA), the National Institute on Deafness and other Communication Disorders (NIDCD), and the NIH Office of Research on Women's Health. WIHS data collection is also supported by UL1-TR000004 (UCSF CTSA) and UL1-TR000454 (Atlanta CTSA).
Author contributions: SD wrote the manuscript, conducted literature search, data analysis, data interpretation, and computed the figures. MP was involved in all aspects of the study: planning, study design, selection of subjects and worked with SD on data analysis, interpretation and writing. JD provided statistical expertise to the study, conducted data analysis and data interpretation. AL provided scientific input, contributed to data interpretation and editing. MG, PL, MV, AF, SG, RG critically reviewed the manuscript and provided scientific input.
The authors report no conflicts of interest.
Supplemental Digital Content is available for this article.
References
- 1.Mohd Hanafiah K, Groeger J, Flaxman AD, et al. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology 2013; 57:1333–1342. [DOI] [PubMed] [Google Scholar]
- 2.Chak E, Talal AH, Sherman KE, et al. Hepatitis C virus infection in USA: an estimate of true prevalence. Liver Int 2011; 31:1090–1101. [DOI] [PubMed] [Google Scholar]
- 3.Maier I, Wu GY. Hepatitis C and HIV co-infection: a review. World J Gastroenterol 2002; 8:577–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lewden C, Salmon D, Morlat P, et al. Causes of death among human immunodeficiency virus (HIV)-infected adults in the era of potent antiretroviral therapy: emerging role of hepatitis and cancers, persistent role of AIDS. Int J Epidemiol 2005; 34:121–130. [DOI] [PubMed] [Google Scholar]
- 5.Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis 2005; 5:558–567. [DOI] [PubMed] [Google Scholar]
- 6.Ghany MG, Strader DB, Thomas DL, et al. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology 2009; 49:1335–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strader DB. Coinfection with HIV and hepatitis C virus in injection drug users and minority populations. Clin Infect Dis 2005; 41 Suppl 1:S7–S13. [DOI] [PubMed] [Google Scholar]
- 8.Deeks SG, Kitchen CM, Liu L, et al. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood 2004; 104:942–947. [DOI] [PubMed] [Google Scholar]
- 9.Hazenberg MD, Otto SA, van Benthem BH, et al. Persistent immune activation in HIV-1 infection is associated with progression to AIDS. Aids 2003; 17:1881–1888. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez VD, Falconer K, Blom KG, et al. High levels of chronic immune activation in the T-cell compartments of patients coinfected with hepatitis C virus and human immunodeficiency virus type 1 and on highly active antiretroviral therapy are reverted by alpha interferon and ribavirin treatment. J Virol 2009; 83:11407–11411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hodowanec AC, Brady KE, Gao W, et al. Characterization of CD4(+) T-cell immune activation and interleukin 10 levels among HIV, hepatitis C virus, and HIV/HCV-coinfected patients. J Acquir Immune Defic Syndr 2013; 64:232–240. [DOI] [PubMed] [Google Scholar]
- 12.Kim AY, Lauer GM, Ouchi K, et al. The magnitude and breadth of hepatitis C virus-specific CD8+ T cells depend on absolute CD4+ T-cell count in individuals coinfected with HIV-1. Blood 2005; 105:1170–1178. [DOI] [PubMed] [Google Scholar]
- 13.Klenerman P, Thimme R. T cell responses in hepatitis C: the good, the bad and the unconventional. Gut 2012; 61:1226–1234. [DOI] [PubMed] [Google Scholar]
- 14.Dustin LB, Cashman SB, Laidlaw SM. Immune control and failure in HCV infection–tipping the balance. J Leukoc Biol 2014; 96:535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barkan SE, Melnick SL, Preston-Martin S, et al. The Women's Interagency HIV Study. WIHS Collaborative Study Group. Epidemiology 1998; 9:117–125. [PubMed] [Google Scholar]
- 16.Wai CT, Greenson JK, Fontana RJ, et al. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology 2003; 38:518–526. [DOI] [PubMed] [Google Scholar]
- 17.Sterling RK, Lissen E, Clumeck N, et al. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology 2006; 43:1317–1325. [DOI] [PubMed] [Google Scholar]
- 18.Bambha K, Pierce C, Cox C, et al. Assessing mortality in women with hepatitis C virus and HIV using indirect markers of fibrosis. AIDS 2012; 26:599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benhamou Y, Bochet M, Di Martino V, et al. Liver fibrosis progression in human immunodeficiency virus and hepatitis C virus coinfected patients. The Multivirc Group. Hepatology 1999; 30:1054–1058. [DOI] [PubMed] [Google Scholar]
- 20.Brau N, Salvatore M, Rios-Bedoya CF, et al. Slower fibrosis progression in HIV/HCV-coinfected patients with successful HIV suppression using antiretroviral therapy. J Hepatol 2006; 44:47–55. [DOI] [PubMed] [Google Scholar]
- 21.Sterling RK, Wegelin JA, Smith PG, et al. Similar progression of fibrosis between HIV/HCV-infected and HCV-infected patients: Analysis of paired liver biopsy samples. Clin Gastroenterol Hepatol 2010; 8:1070–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greub G, Ledergerber B, Battegay M, et al. Clinical progression, survival, and immune recovery during antiretroviral therapy in patients with HIV-1 and hepatitis C virus coinfection: the Swiss HIV Cohort Study. Lancet 2000; 356:1800–1805. [DOI] [PubMed] [Google Scholar]
- 23.Sajadi MM, Shakeri N, Talwani R, et al. Hepatitis C infection in HIV-1 natural viral suppressors. AIDS 2010; 24:1689–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuniholm MH, Xie X, Anastos K, et al. Association of chronic hepatitis C infection with T-cell phenotypes in HIV-negative and HIV-positive women. J Acquir Immune Defic Syndr 2014; 67:295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shmagel KV, Saidakova EV, Korolevskaya LB, et al. Influence of hepatitis C virus coinfection on CD4(+) T cells of HIV-infected patients receiving HAART. AIDS 2014; 28:2381–2388. [DOI] [PubMed] [Google Scholar]
- 26.Yonkers NL, Sieg S, Rodriguez B, et al. Reduced naive CD4 T cell numbers and impaired induction of CD27 in response to T cell receptor stimulation reflect a state of immune activation in chronic hepatitis C virus infection. J Infect Dis 2011; 203:635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giorgi JV, Hultin LE, McKeating JA, et al. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J Infect Dis 1999; 179:859–870. [DOI] [PubMed] [Google Scholar]
- 28.Badley AD, Pilon AA, Landay A, et al. Mechanisms of HIV-associated lymphocyte apoptosis. Blood 2000; 96:2951–2964. [PubMed] [Google Scholar]
- 29.Kovacs A, Al-Harthi L, Christensen S, et al. CD8(+) T cell activation in women coinfected with human immunodeficiency virus type 1 and hepatitis C virus. J Infect Dis 2008; 197:1402–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhuang Y, Wei X, Li Y, et al. HCV coinfection does not alter the frequency of regulatory T cells or CD8+ T cell immune activation in chronically infected HIV+ Chinese subjects. AIDS Res Hum Retroviruses 2012; 28:1044–1051. [DOI] [PubMed] [Google Scholar]
- 31.Manion M, Rodriguez B, Medvik K, et al. Interferon-alpha administration enhances CD8+ T cell activation in HIV infection. PLoS One 2012; 7:e30306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nunez M, Soriano V, Lopez M, et al. Coinfection with hepatitis C virus increases lymphocyte apoptosis in HIV-infected patients. Clin Infect Dis 2006; 43:1209–1212. [DOI] [PubMed] [Google Scholar]
- 33.Korner C, Kramer B, Schulte D, et al. Effects of HCV co-infection on apoptosis of CD4+ T-cells in HIV-positive patients. Clin Sci (Lond) 2009; 116:861–870. [DOI] [PubMed] [Google Scholar]
- 34.Ronquillo RE, Desai SN, Norris PJ, et al. Elevated caspase-3 expression and T-cell activation in elite suppressors. J Acquir Immune Defic Syndr 2010; 54:110–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Torti C, Barnes E, Quiros-Roldan E, et al. Suppression of hepatitis C virus replication is maintained long term following HAART therapy, in an individual with HCV/HIV co-infection. Antivir Ther 2004; 9:139–142. [PubMed] [Google Scholar]
- 36.Zeitoun JD, Mallet V, Chaix ML, et al. Stable recovery from HCV in HIV-HCV co-infection under antiretroviral therapy. J Clin Virol 2007; 40:71–73. [DOI] [PubMed] [Google Scholar]
- 37.Endo T, Fujimoto K, Nishio M, et al. Case report: clearance of hepatitis C virus after changing the HAART regimen in a patient infected with hepatitis C virus and the human immunodeficiency virus. J Med Virol 2009; 81:979–982. [DOI] [PubMed] [Google Scholar]
- 38.Clerici M, Shearer GM. The Th1-Th2 hypothesis of HIV infection: new insights. Immunol Today 1994; 15:575–581. [DOI] [PubMed] [Google Scholar]
- 39.Takaki A, Wiese M, Maertens G, et al. Cellular immune responses persist and humoral responses decrease two decades after recovery from a single-source outbreak of hepatitis C. Nat Med 2000; 6:578–582. [DOI] [PubMed] [Google Scholar]
- 40.Lee HC, Sung SS, Krueger PD, et al. Hepatitis C virus promotes T-helper (Th)17 responses through thymic stromal lymphopoietin production by infected hepatocytes. Hepatology 2013; 57:1314–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang JY, Zhang Z, Lin F, et al. Interleukin-17-producing CD4(+) T cells increase with severity of liver damage in patients with chronic hepatitis B. Hepatology 2010; 51:81–91. [DOI] [PubMed] [Google Scholar]
- 42.Bacchetti P, Boylan R, Astemborski J, et al. Progression of biopsy-measured liver fibrosis in untreated patients with hepatitis C infection: non-Markov multistate model analysis. PLoS One 2011; 6:e20104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.