

Abstract
Cancer-associated fibroblasts (CAFs) are major components of the surrounding stroma of carcinomas that emerge in the tumor microenvironment as a result of signals derived from the cancer cells. Biochemical cross-talk between cancer cells and CAFs as well as mechanical remodeling of the stromal extracellular matrix (ECM) by CAFs are important contributors to tumor cell migration and invasion, which are critical for cancer progression from a primary tumor to metastatic disease. In this review, we discuss key paracrine signaling pathways between CAFs and cancer cells that promote cancer cell migration and invasion. In addition, we discuss physical changes that CAFs exert on the stromal ECM to facilitate migration and invasion of cancer cells.
Introduction
Carcinomas are heterogeneous tissues composed of cancer cells and stromal cells, such as fibroblasts, immune cells, proinflammatory cells, pericytes, and endothelial cells, as well as noncellular constituents including the extracellular matrix (ECM) and soluble growth factors, which are collectively referred to as the tumor microenvironment [1]. One of the most prominent cell types in the tumor stroma is fibroblasts, which are spindle-shaped cells of connective tissue that deposit ECM, regulate inflammation, and mediate wound healing [2]. Fibroblasts that are found in the carcinoma micro-environment can display an altered phenotype that is characterized by increased expression of certain markers, such as α-smooth muscle actin (α-SMA) and fibroblast activation protein (FAP), and increased secretion of ECM proteins, including fibronectin and type I collagen [3]. These fibroblasts that become ‘activated’ in tumors, through interactions with cancer cells, are referred to as cancer-associated fibroblasts (CAFs) (Figure 1). CAFs can promote tumor progression to malignancy and metastatic spread by facilitating underlying processes such as cancer cell migration and invasion (Figure 1). In this mini-review, we discuss the central role that CAFs play in mediating cancer cell migration and invasion through secretion of soluble factors and modification of the stromal ECM composition and the architecture (Figure 2).
Figure 1. Cross-talk between cancer cells and CAFs in the tumor microenvironment.
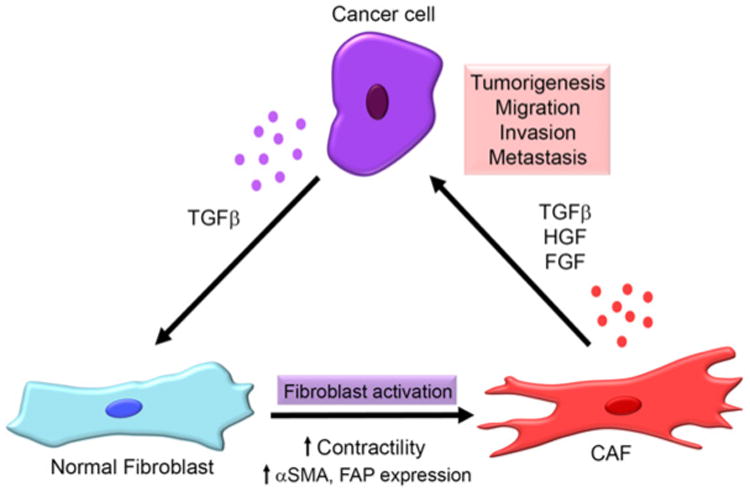
Secretion of growth factors such as TGF-β from cancer cells can activate stromal fibroblasts into CAFs, increasing their contractility and expression of CAF markers, including α-SMA and FAP. CAFs, in turn, secrete higher amounts of growth factors such as TGF-β, HGF and FGF, which further stimulate tumorigenesis, migration, invasion and metastasis of cancer cells.
Figure 2. Schematic of CAF-mediated cancer cell migration and invasion.
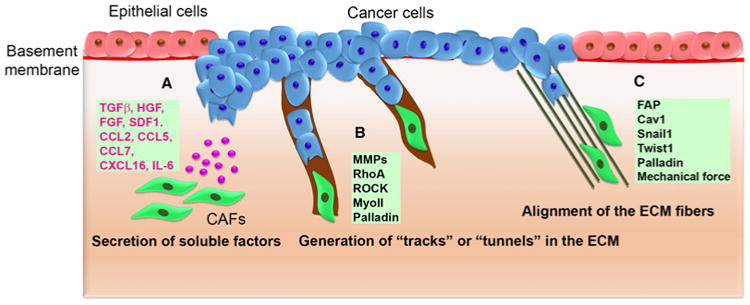
(A) CAFs modulate cancer cell migration and invasion by altering the tumor microenvironment via secretion of growth factors and cytokines such as TGF-β, HGF, FGF, SDF1, CCL2, CCL5, CCL7, CXCL16, and IL-6. (B) CAFs modify the stromal ECM through MMP, RhoA, ROCK, MyoII, and palladin, generating ‘tracks’ or ‘tunnels’ in the matrix for cancer cells to follow. (C) CAFs alter the matrix architecture in the tumor stroma, organizing the ECM into parallel fibers that facilitate cancer cell migration and invasion. FAP, Cav1, Snail1, Twist1, palladin, and mechanical force play a role in ECM alignment.
The role of CAF-secreted factors in cancer cell migration and invasion
Migration and invasion of cancer cells into the adjacent tissue is an important initial step in metastasis, which can result in the formation of tumors at secondary sites [4]. As cancer progresses, tumor cells obtain the ability to break down the underlying basement membrane and invade into the surrounding tissue. Some cancer cells attain this invasive behavior through acquisition of a mesenchymal phenotype, known as the epithelial-to-mesenchymal transition (EMT) [5]. CAFs may induce the EMT of cancer cells through paracrine signaling with growth factors and pro-migratory cytokines (Figure 2A). One of the major pro-metastatic factors derived from CAFs is the cytokine transforming growth factor-β (TGF-β) [6]. CAFs have been shown to secrete increased levels of TGF-β [6,7]. In addition, TGF-β present in CAF-conditioned media stimulates EMT in different stage breast cancer cell lines, which showed increased expression of mesenchymal markers, including vimentin, fibronectin, and matrix metalloproteinases 2 and 9 (MMP2 and MMP9), enhanced migratory behavior, and increased invasion into Matrigel [6]. Inhibition of the TGF-β pathway in cancer cells negated the effects of CAF-conditioned media on cancer cells [6]. Similar pro-migratory effects of TGF-β, secreted from CAFs, were also observed in other cancer types such as gastric, colorectal, and bladder cancer [8–11]. These data suggest that targeting TGF-β in stromal fibroblasts impedes the invasive behavior of cancer cells. Intriguingly, other studies reported that blocking TGF-β signaling in CAFs enhances invasion of carcinoma cells by increasing the expression of hepatocyte growth factor (HGF) by CAFs [12,13]; TGF-β have been shown to negatively regulate HGF expression [14,15]. Deletion of TGF-β type II receptor in mouse fibroblasts [12] or treatment of CAFs with the TGF-β type I receptor inhibitor [13] increased HGF expression, thus increasing cancer cell invasion. However, how TGF-β–HGF cross-talk is regulated in different cancers and CAFs are currently not well understood.
HGF, also called scatter factor (SF/HGF), is primarily secreted by stromal cells and regulates various cell functions, such as differentiation, proliferation, and migration in epithelial cells, which express its cell-surface receptor c-Met [16]. CAFs secrete high levels of HGF and the ECM glycoprotein tenascin-C, which were necessary for invasion of colon cancer cells into Matrigel and type I collagen gels [17]. Interestingly, these two factors individually were not sufficient to promote the invasion of cancer cells, underscoring the interdependent roles of the ECM and soluble factors in modulating cancer cell behavior [17]. HGF secreted by hepatocellular carcinoma (HCC) CAFs was shown to enrich the tumor-initiating cell population in HCC cells and increase their migration and invasion abilities [18]. In organotypic assays, HGF released by CAFs promoted invasion of esophageal squamous cell carcinoma cells, which could be inhibited by siRNAs targeting HGF in fibroblasts or by altering c-Met expression or function in carcinoma cells [19]. Similarly, impairment of the HGF–cMet interaction with the monoclonal antibody ficlatuzumab inhibited head and neck squamous cell carcinoma (HNSCC) cell migration and invasion in response to treatment with CAF-conditioned media [20]. Intriguingly, when HNSCC cells were injected with CAFs into the floor of the mouth of athymic mice, much larger tumors were formed, compared with cancer cells alone, and increased metastasis to lungs and regional lymph nodes was observed. The ability of CAFs to promote HNSCC cell migration and invasion was attributed to increased secretion of HGF [21], suggesting that targeting HGF signaling in cancers is beneficial in inhibiting tumor progression. Phase I clinical trials are currently underway using the anti-HGF antibody ficlatuzumab for the treatment of HNSCC and non-small-cell lung cancers [16].
TGF-β and HGF signaling have been shown to play a critical role in the CAF-mediated invasion of cancer cells. However, soluble factors, such as fibroblast growth factor (FGF), are also important in the CAF-mediated migration and invasion of cancer cells. Henriksson et al. showed that treating fibroblasts with conditioned media from colon cancer cells transformed the fibroblasts into CAFs [22]. Conditioned media from these CAFs, which produced increased levels of FGF1, enhanced the migration and invasion of colon cancer cells in Boyden chambers and in three-dimensional organotypic invasion assays. Furthermore, blocking the interaction of FGF1 with its receptor FGF receptor-3 (FGFR3) abolished the pro-invasive effects of the CAF-conditioned media on colon cancer cells [22]. CAFs also express other FGF isoforms such as the surface-associated growth factor FGF2, which is typically not secreted into the extracellular space. When CAFs expressing FGF2 came into direct contact with colorectal cancer cells, the cancer cells became elongated, migrated longer distances, and invaded into the fibroblast-supplemented Matrigel [23]. These effects were attributed to the activation of FGFR in cancer cells that bound to FGF2 on CAFs, which could be counteracted by treatment with FGFR inhibitors [23]. This study also showed that the nonreceptor tyrosine kinase, Src, is activated downstream of FGFR activation in cancer cells and that cancer cells utilize αvβ5 integrin to adhere to fibroblasts.
Another group of soluble molecules that are important in the regulation of tumor progression and cancer cell–stroma interactions is in the cytokine/chemokine family (Table 1 and Figure 2A) [24]. One of these chemokines is the stromal cell-derived factor-1 (SDF1), also known as C–X–C motif chemokine 12 (CXCL12), which interacts with the chemokine receptor-4 (CXCR4). Orimo et al. [25] demonstrated that breast carcinoma CAFs express high levels of SDF1 that recruit CXCR4-expressing endothelial precursor cells to tumors, resulting in augmented angiogenesis and tumor growth. Increased SDF1α expression was also observed in endometrial cancer (EC)-derived CAFs, which increased cancer cell migration and invasion [26]. The treatment of EC cells with AMD3100 (a CXCR4 antagonist), in the presence of CAF-conditioned media, blocked CAF-induced EC cell migration and invasion [26]. In this study, SDF1 was shown to increase the expression of MMP2 and MMP9 in CAFs through an autocrine mechanism, which enhanced EC cell invasion in CAF-modified Matrigel. SDF1, secreted by CAFs, also acts in a paracrine manner on EC cells inducing their proliferation by activating PI3K/Akt and MAP kinase signaling pathways [26].
Table 1. Soluble factors released by CAFs and their receptors in cancer cells.
Two independent studies of oral squamous cell carcinomas showed that CAFs secrete increased levels of the chemokine (C–C motif) ligand 2 (CCL2), also known as monocyte chemoattractant protein 1 (MCP1). CCL2 promoted cancer cell migration and invasion through activation of focal adhesion kinase and galectin-1 [27,28]. CAFs also secrete higher levels of the chemokines CCL2, CCL5, CCL7 and CXCL16 compared with peritumoral fibroblasts [29]. All of these chemokines increased the migration of HCC cells; however, only CCL7 and CXCL16 also promoted HCC invasion. CAF-derived CCL2 and CCL5 activate the Hedgehog (Hh) pathway, whereas CCL7 and CXCL16 activate TGF-β signaling in HCC cells; activation of these pathways can induce EMT in cancer cells. Indeed, CAFs no longer stimulated the expression of EMT markers, when these four chemokines were inhibited from interacting with their receptors in HCC cells [29].
In addition to chemokines, CAFs also secrete cytokines from the interleukin family. Interleukin-6 (IL-6) in CAF-conditioned media promoted the invasion of melanoma cells from tumor spheroids into the surrounding type I collagen matrix [30]. Co-culturing CAFs with melanoma cells induced IL-8 secretion by CAFs, and inhibition of IL-6 and IL-8 abrogated the invasion of melanoma spheroids [30]. Retinoic acid, a lipophilic molecule derived from vitamin A, is thought to exert therapeutic effects by decreasing the secretion of IL-6 by CAFs, thereby limiting pancreatic cancer cell migration and induction of EMT [31,32].
Paracrine communication between CAFs and tumor cells plays a significant role in mediating cancer cell migration, invasion, and metastasis. Some growth factors and cytokines that were shown to induce cancer cell migration and invasion are listed in Table 1. Targeting these growth factor/cytokine signaling pathways in CAFs could have therapeutic benefits for cancer patients by impairing the tumor-supportive roles of CAFs. However, additional studies are needed to better discern the role of paracrine communication between CAFs and cancer cells in vivo.
CAFs promote cancer cell invasion by modifying the stromal ECM
Fibroblasts are indispensable components of connective tissue, and their major function is deposition and assembly of the ECM. They secrete multiple components of the stromal ECM, including fibronectin as well as types I, III, and V collagens [33]. Fibroblasts also maintain ECM homeostasis by facilitating its turnover and degradation through secretion of enzymes such as MMPs [34]. CAFs exhibit abnormal activity in terms of ECM regulation; they secrete increased levels of ECM proteins, such as fibronectin and type I collagen, and express oncofetal isoforms of fibronectin. In addition, CAFs modify the stromal ECM by enhanced expression and activation of MMPs [33].
Changes in the matrix environment, implemented by CAFs, can facilitate cancer cell migration and invasion. Gaggioli et al. [35] observed that CAFs deformed type I collagen matrices, generating ‘tracks’, thereby enabling SCC cells to invade as a collective chain, following the lead of the CAFs (Figure 2B). They further showed that the leading CAFs utilized MMPs and Rho-ROCK-mediated contractile force to create the tracks in the ECM. This study provided a new model of CAF-mediated cancer cell invasion, in which CAFs physically remodel the matrix, allowing cancer cells to invade while still maintaining their epithelial properties [35]. A recent study made similar observations using salivary gland adenoid cystic carcinoma (ACC) cells [36]. Utilizing a microfluidic device, they co-cultured ACC cells and CAFs in a chamber that was separated from the chemoattractant (20% FBS) by Matrigel. CAFs invaded the Matrigel using MMP activity and ACC cells followed behind them (Figure 2B). However, in this study, blocking the CXCL12/CXCR4 pathway inhibited cancer cell invasion into the tracks generated by CAFs, suggesting that signaling between CAFs and cancer cells drives ACC cell invasion [36].
Another study examined the role of mechanical regulation of the ECM by CAFs focusing on caveolin-1 (Cav1). Cav1 is a major component of the caveolar membrane, which also activates the small GTPase Rho by regulating its inhibitor p190RhoGAP (p190) [37]. Cav1 is enriched in the stroma of breast and renal carcinomas, and it is overexpressed by breast cancer CAFs. Interestingly, Cav1-expressing mouse embryonic fibroblasts (MEFs) organized fibronectin into parallel fibers that supported cancer cell migration with increased velocity and directionality (Figure 2C) [37]. The remodeling of the fibronectin matrix by Cav1-expressing mouse fibroblasts was dependent on cellular contractility mediated by Rho. Furthermore, orthotopic injection of cancer cells into the mammary glands of wild-type (WT) or Cav1 knockout mice resulted in differences in the tumor stromal ECM. The loss of Cav1 slightly decreased tumor growth and metastasis, and more importantly, these tumors were minimally invasive with fewer collagen fibers. In contrast, tumors in WT mice consisted of radially aligned collagen fibers with interacting tumor cells along them, suggesting a more invasive tumor microenvironment [37]. Mechanical remodeling of the ECM through increased contractility of CAFs was also observed in scirrhous gastric carcinoma (SGC) [38]. Co-culture of SGC cells and CAFs generated ‘invasive foci’ in Matrigel. These invasive foci were comprised of CAFs, which reorganized the matrix through actin–myosin contractility, and cancer cells that surrounded the CAFs invaded into Matrigel. Interestingly, treatment with the myosin-II (MyoII) inhibitor blebbistatin significantly reduced the invasion of these foci, whereas treatment with the broad MMP inhibitor GM6001 did not perturb the matrix remodeling and invasion phenotype [38].
FAP, which is a serine protease that is overexpressed in CAFs, also contributes to matrix organization [39]. FAP-expressing mouse 3T3 fibroblasts organize the ECM into an aligned orientation, which promotes the directional migration and invasion of pancreatic cancer cells (Figure 2C) [39]. Another protein that is involved in CAF-medicated matrix organization is Snail1 [40]. Snail1 is a transcriptional regulator that is responsive to TGF-β signaling and regulates EMT in cancer cells [41]. It is expressed by a subset of CAFs and mediates anisotropic fiber organization and increased matrix stiffness by up-regulating α-SMA expression and RhoA activation in response to TGF-β treatment, suggesting that changes in cellular contractility through Snail1 support CAF-mediated changes in the ECM architecture [40]. Snail1 knockout MEFs do not generate a matrix with anisotropic fiber orientation and, thus, do not support directional cell migration and invasion [40]. Twist1, another transcriptional regulator of the EMT, is also expressed by CAFs [42]. Expression of Twist1 in immortalized human fibroblasts induced CAF-like properties, including increased collagen contraction and alignment, stiffening of matrix fibers, and stimulation of cancer cell invasion. These effects of Twist1 resulted from up-regulation of expression of type VI collagen α1 and palladin [42]. Brentnall et al. [43] demonstrated that increased palladin expression in CAFs correlates with increased invadopodia formation, RhoA activation, matrix degradation, and cell invasion into Matrigel. Pancreatic cancer cells that are co-cultured with palladin-expressing fibroblasts invade through Matrigel by following ‘tunnels’ created by the fibroblasts. Increased palladin expression by pancreatic CAFs is associated with Cdc42 GTPase activation and invadopodia formation, which facilitated CAF-led cancer cell invasion into Matrigel [44].
Aside from the biochemical changes governed by CAFs, mechanical tension has also been shown to induce CAF-like properties in normal fibroblasts. Ao et al. [45] showed that following mechanical stretching, normal prostate fibroblasts align fibronectin, similar to prostate CAFs. Furthermore, mechanical stretching augmented the expression of platelet-derived growth factor receptor α (PDGFRα); mechanical tension can activate signaling pathways in cells, such as PDGFRα signaling, which may have a role in matrix organization. Our study also showed that the aligned fibronectin produced by stretched fibroblasts promoted the directionally persistent migration of cancer cells, suggesting that changes in mechanical force can induce biogenesis of CAFs (Figure 2C) [45].
Conclusions
The supporting role of the tumor microenvironment is emerging as a critical contributor to cancer progression. Activated fibroblasts, CAFs, have been observed in many types of carcinomas as one of the most prominent components of the tumor stroma. As discussed in this review, many studies indicate that CAF-secreted soluble factors sustain the invasive properties of cancer cells. In addition, CAFs modify the surrounding ECM generating a niche that supports cancer cell invasion. Some of the mechanisms discussed in this review converge on TGF-β pathways in CAFs (Figures 1 and 3). Activation of TGF-β in CAFs results in increased actomyosin contractility and concomitant changes in the ECM organization by CAFs that induce migration and invasion of cancer cells (Figure 3). Therefore, targeting TGF-β signaling in the tumor microenvironment could prove to be beneficial in alleviating cancer cell–stroma interaction and CAF-mediated changes in the ECM.
Figure 3. TGF-β secreted by cancer cells activate the noncanonical pathway in CAFs leading to RhoA activation and ROCK-mediated MyoII contractility.
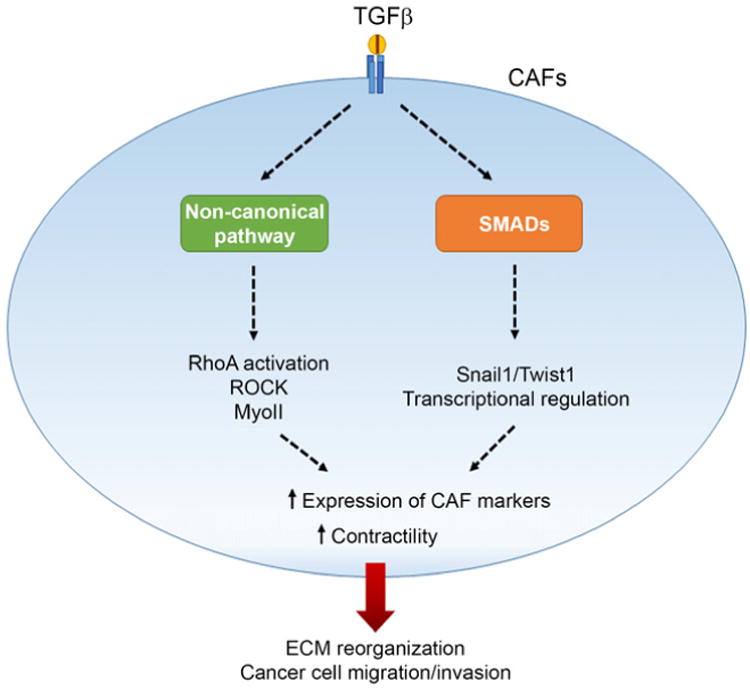
In addition, activation of the TGF-β canonical pathway can induce transcriptional regulation of Snail1 and Twist1 target genes, resulting in increased contractility of CAFs. Activation of both pathways leads to an altered ECM environment that enhances cell migration and invasion of cancer cells.
CAFs can promote an initial step of metastasis, which is the local migration and invasion of cancer cells. Moreover, recent reports indicate that CAFs are present at secondary tumor sites [46,47]. However, more studies are needed to better understand the roles of CAFs in metastatic tumors. The factors involved in CAF– cancer cell interaction are promising potential targets for cancer therapies. One should also keep in mind that CAFs are not a homogeneous population of cells, and different subtypes of CAFs can differentially regulate cancer cell response to treatment [48]. Nevertheless, further studies are needed to decipher the complex molecular interactions between cancer cells and CAFs, to pave the way for developing effective therapeutic strategies against cancer.
Acknowledgments
Funding: This work was supported by the National Institutes of Health Grant [GM117916] to D.J.W. and by Vanderbilt University.
Abbreviations
- ACC
adenoid cystic carcinoma
- CAF
cancer-associated fibroblasts
- Cav1
caveolin-1
- CCL
C–C motif chemokine
- CXCL
C–X–C motif chemokine
- CXCR4
chemokine receptor-4
- EC
endometrial cancer
- ECM
extracellular matrix
- EMT
epithelial-to-mesenchymal transition
- FAP
fibroblast activation protein
- FGF
fibroblast growth factor
- FGFR
fibroblast growth factor receptor
- HCC
hepatocellular carcinoma
- HGF
hepatocyte growth factor
- Hh
Hedgehog
- HNSCC
head and neck squamous cell carcinoma
- IL
interleukin
- MCP1
monocyte chemoattractant protein 1
- MEF
mouse embryonic fibroblast
- MMP
matrix metalloproteinase
- MyoII
non-muscle myosin-II
- PDGFRα
platelet-derived growth factor receptor-α
- SCC
squamous cell carcinoma
- SDF1
stromal cell-derived factor-1
- SGC
scirrhous gastric carcinoma
- TGF-β
transforming growth factor β
- WT
wild type
- α-SMA
α-smooth muscle actin
Footnotes
Competing Interests: The Authors declare that there are no competing interests associated with the manuscript.
References
- 1.Lorusso G, Rüegg C. The tumor microenvironment and its contribution to tumor evolution toward metastasis. Histochem Cell Biol. 2008;130:1091–1103. doi: 10.1007/s00418-008-0530-8. [DOI] [PubMed] [Google Scholar]
- 2.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 3.Xing F, Saidou J, Watabe K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front Biosci. 2010;15:166–179. doi: 10.2741/3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–292. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalluri R, Weinberg RA. The basics of epithelial–mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu Y, Xiao CH, Tan LD, Wang QS, Li XQ, Feng YM. Cancer-associated fibroblasts induce epithelial–mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br J Cancer. 2014;110:724–732. doi: 10.1038/bjc.2013.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ao M, Franco OE, Park D, Raman D, Williams K, Hayward SW. Cross-talk between paracrine-acting cytokine and chemokine pathways promotes malignancy in benign human prostatic epithelium. Cancer Res. 2007;67:4244–4253. doi: 10.1158/0008-5472.CAN-06-3946. [DOI] [PubMed] [Google Scholar]
- 8.Fuyuhiro Y, Yashiro M, Noda S, Matsuoka J, Hasegawa T, Kato Y, et al. Cancer-associated orthotopic myofibroblasts stimulates the motility of gastric carcinoma cells. Cancer Sci. 2012;103:797–805. doi: 10.1111/j.1349-7006.2012.02209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimao Y, Nabeshima K, Noue T, Oono M. Role of fibroblasts in HGF/SF-induced cohort migration of human colorectal carcinoma cells: fibroblasts stimulate migration associated with increased fibronectin production via upregulated TGF-beta1. Int J Cancer. 1999;82:449–458. doi: 10.1002/(sici)1097-0215(19990730)82:3<449::aid-ijc20>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 10.Kim SA, Lee EK, Kuh HJ. Co-culture of 3D tumor spheroids with fibroblasts as a model for epithelial–mesenchymal transition in vitro. Exp Cell Res. 2015;335:187–196. doi: 10.1016/j.yexcr.2015.05.016. [DOI] [PubMed] [Google Scholar]
- 11.Zhuang J, Lu Q, Shen B, Huang X, Shen L, Zheng X, et al. TGFβ1 secreted by cancer-associated fibroblasts induces epithelial–mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci Rep. 2015;5:11924. doi: 10.1038/srep11924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng N, Chytil A, Shyr Y, Joly A, Moses HL. Transforming growth factor-β signaling-deficient fibroblasts enhance hepatocyte growth factor signaling in mammary carcinoma cells to promote scattering and invasion. Mol Cancer Res. 2008;6:1521–1533. doi: 10.1158/1541-7786.MCR-07-2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oyanagi J, Kojima N, Sato H, Higashi S, Kikuchi K, Sakai K, et al. Inhibition of transforming growth factor-β signaling potentiates tumor cell invasion into collagen matrix induced by fibroblast-derived hepatocyte growth factor. Exp Cell Res. 2014;326:267–279. doi: 10.1016/j.yexcr.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 14.Matsumoto K, Tajima H, Okazaki H, Nakamura T. Negative regulation of hepatocyte growth factor gene expression in human lung fibroblasts and leukemic cells by transforming growth factor-beta 1 and glucocorticoids. J Biol Chem. 1992;267:24917–24920. [PubMed] [Google Scholar]
- 15.Mungunsukh O, Day RM. Transforming growth factor-β1 selectively inhibits hepatocyte growth factor expression via a micro-RNA-199-dependent posttranscriptional mechanism. Mol Biol Cell. 2013;24:2088–2097. doi: 10.1091/mbc.E13-01-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rizwani W, Allen A, Trevino J. Hepatocyte growth factor from a clinical perspective: a pancreatic cancer challenge. Cancers (Basel) 2015;7:1785–1805. doi: 10.3390/cancers7030861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Wever O, Nguyen QD, Van Hoorde L, Bracke M, Bruyneel E, Gespach C, et al. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004;18:1016–1018. doi: 10.1096/fj.03-1110fje. [DOI] [PubMed] [Google Scholar]
- 18.Lau EYT, Lo J, Cheng BYL, Ma MKF, Lee JMF, Ng JKY, et al. Cancer-associated fibroblasts regulate tumor-initiating cell plasticity in hepatocellular carcinoma through c-Met/FRA1/HEY1 signaling. Cell Rep. 2016;15:1175–1189. doi: 10.1016/j.celrep.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 19.Grugan KD, Miller CG, Yao Y, Michaylira CZ, Ohashi S, Klein-Szanto AJ, et al. Fibroblast-secreted hepatocyte growth factor plays a functional role in esophageal squamous cell carcinoma invasion. Proc Natl Acad Sci USA. 2010;107:11026–11031. doi: 10.1073/pnas.0914295107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar D, Kandl C, Hamilton CD, Shnayder Y, Tsue TT, Kakarala K, et al. Mitigation of tumor-associated fibroblast-facilitated head and neck cancer progression with anti-hepatocyte growth factor antibody ficlatuzumab. JAMA Otolaryngol Neck Surg. 2015;141:1133. doi: 10.1001/jamaoto.2015.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wheeler SE, Shi H, Lin F, Dasari S, Bednash J, Thorne S, et al. Enhancement of head and neck squamous cell carcinoma proliferation, invasion, and metastasis by tumor-associated fibroblasts in preclinical models. Head Neck. 2014;36:385–392. doi: 10.1002/hed.23312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henriksson ML, Edin S, Dahlin AM, Oldenborg PA, Öberg Å, Van Guelpen B, et al. Colorectal cancer cells activate adjacent fibroblasts resulting in FGF1/FGFR3 signaling and increased invasion. Am J Pathol. 2011;178:1387–1394. doi: 10.1016/j.ajpath.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knuchel S, Anderle P, Werfelli P, Diamantis E, Rüegg C. Fibroblast surface-associated FGF-2 promotes contact-dependent colorectal cancer cell migration and invasion through FGFR-SRC signaling and integrin αvβ5-mediated adhesion. Oncotarget. 2015;6:14300–14317. doi: 10.18632/oncotarget.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mezawa Y, Orimo A. The roles of tumor- and metastasis-promoting carcinoma-associated fibroblasts in human carcinomas. Cell Tissue Res. 2016;365:675–689. doi: 10.1007/s00441-016-2471-1. [DOI] [PubMed] [Google Scholar]
- 25.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 26.Teng F, Tian WY, Wang YM, Zhang YF, Guo F, Zhao J, et al. Cancer-associated fibroblasts promote the progression of endometrial cancer via the SDF-1/CXCR4 axis. J Hematol Oncol. 2016;9:8. doi: 10.1186/s13045-015-0231-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Min A, Zhu C, Wang J, Peng S, Shuai C, Gao S, et al. Focal adhesion kinase knockdown in carcinoma-associated fibroblasts inhibits oral squamous cell carcinoma metastasis via downregulating MCP-1/CCL2 expression. J Biochem Mol Toxicol. 2015;29:70–76. doi: 10.1002/jbt.21669. [DOI] [PubMed] [Google Scholar]
- 28.Wu MH, Hong HC, Hong TM, Chiang WF, Jin YT, Chen YL. Targeting galectin-1 in carcinoma-associated fibroblasts inhibits oral squamous cell carcinoma metastasis by downregulating MCP-1/CCL2 expression. Clin Cancer Res. 2011;17:1306–1316. doi: 10.1158/1078-0432.CCR-10-1824. [DOI] [PubMed] [Google Scholar]
- 29.Liu J, Chen S, Wang W, Ning BF, Chen F, Shen W, et al. Cancer-associated fibroblasts promote hepatocellular carcinoma metastasis through chemokine-activated hedgehog and TGF-β pathways. Cancer Lett. 2016;379:49–59. doi: 10.1016/j.canlet.2016.05.022. [DOI] [PubMed] [Google Scholar]
- 30.Jobe NP, Rösel D, Dvořánková B, Kodet O, Lacina L, Mateu R, et al. Simultaneous blocking of IL-6 and IL-8 is sufficient to fully inhibit CAF-induced human melanoma cell invasiveness. Histochem Cell Biol. 2016;146:205–217. doi: 10.1007/s00418-016-1433-8. [DOI] [PubMed] [Google Scholar]
- 31.Guan J, Zhang H, Wen Z, Gu Y, Cheng Y, Sun Y, et al. Retinoic acid inhibits pancreatic cancer cell migration and EMT through the downregulation of IL-6 in cancer associated fibroblast cells. Cancer Lett. 2014;345:132–139. doi: 10.1016/j.canlet.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 32.Carapuça EF, Gemenetzidis E, Feig C, Bapiro TE, Williams MD, Wilson AS, et al. Anti-stromal treatment together with chemotherapy targets multiple signalling pathways in pancreatic adenocarcinoma. J Pathol. 2016;239:286–296. doi: 10.1002/path.4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miles FL, Sikes RA. Insidious changes in stromal matrix fuel cancer progression. Mol Cancer Res. 2014;12:297–312. doi: 10.1158/1541-7786.MCR-13-0535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cunha GR, Hayward SW, Wang YZ, Ricke WA. Role of the stromal microenvironment in carcinogenesis of the prostate. Int J Cancer. 2003;107:1–10. doi: 10.1002/ijc.11335. [DOI] [PubMed] [Google Scholar]
- 35.Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 36.Li J, Jia Z, Kong J, Zhang F, Fang S, Li X, et al. Carcinoma-associated fibroblasts lead the invasion of salivary gland adenoid cystic carcinoma cells by creating an invasive track. PLoS ONE. 2016;11:e0150247. doi: 10.1371/journal.pone.0150247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goetz JG, Minguet S, Navarro-Lérida I, Lazcano JJ, Samaniego R, Calvo E, et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. 2011;146:148–163. doi: 10.1016/j.cell.2011.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamaguchi H, Yoshida N, Takanashi M, Ito Y, Fukami K, Yanagihara K, et al. Stromal fibroblasts mediate extracellular matrix remodeling and invasion of scirrhous gastric carcinoma cells. PLoS ONE. 2014;9:1–12. doi: 10.1371/journal.pone.0085485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee HO, Mullins SR, Franco-Barraza J, Valianou M, Cukierman E, Cheng JD. FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC Cancer. 2011;11:245. doi: 10.1186/1471-2407-11-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stanisavljevic J, Loubat-Casanovas J, Herrera M, Luque T, Pena R, Lluch A, et al. Snail1-expressing fibroblasts in the tumor microenvironment display mechanical properties that support metastasis. Cancer Res. 2015;75:284–295. doi: 10.1158/0008-5472.CAN-14-1903. [DOI] [PubMed] [Google Scholar]
- 41.Zhang J, Tian XJ, Xing J. Signal transduction pathways of EMT induced by TGF-β, SHH, and WNT and their crosstalks. J Clin Med. 2016;5:41. doi: 10.3390/jcm5040041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.García-Palmero I, Torres S, Bartolomé RA, Peláez-García A, Larriba MJ, Lopez-Lucendo M, et al. Twist1-induced activation of human fibroblasts promotes matrix stiffness by upregulating palladin and collagen α1(VI) Oncogene. 2016;35:5224–5236. doi: 10.1038/onc.2016.57. [DOI] [PubMed] [Google Scholar]
- 43.Brentnall TA, Lai LA, Coleman J, Bronner MP, Pan S, Chen R. Arousal of cancer-associated stroma: overexpression of palladin activates fibroblasts to promote tumor invasion. PLoS ONE. 2012;7:e30219. doi: 10.1371/journal.pone.0030219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goicoechea SM, García-Mata R, Staub J, Valdivia A, Sharek L, McCulloch CG, et al. Palladin promotes invasion of pancreatic cancer cells by enhancing invadopodia formation in cancer-associated fibroblasts. Oncogene. 2014;33:1265–1273. doi: 10.1038/onc.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ao M, Brewer BM, Yang L, Franco Coronel OE, Hayward SW, Webb DJ, et al. Stretching fibroblasts remodels fibronectin and alters cancer cell migration. Sci Rep. 2015;5:8334. doi: 10.1038/srep08334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duda DG, Duyverman AMMJ, Kohno M, Snuderl M, Steller EJA, Fukumura D, et al. Malignant cells facilitate lung metastasis by bringing their own soil. Proc Natl Acad Sci USA. 2010;107:21677–21682. doi: 10.1073/pnas.1016234107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Del Valle PR, Milani C, Brentani MM, Katayama MLH, de Lyra EC, Carraro DM, et al. Transcriptional profile of fibroblasts obtained from the primary site, lymph node and bone marrow of breast cancer patients. Genet Mol Biol. 2014;37:480–489. doi: 10.1590/S1415-47572014000400002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brechbuhl HM, Finlay-Schultz J, Yamamoto T, Gillen A, Cittelly DM, Tan AC, et al. Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-15-2851. [DOI] [PMC free article] [PubMed] [Google Scholar]