

Abstract
Autologous vein grafts are commonly used for coronary and peripheral artery bypass but have a high incidence of intimal hyperplasia (IH) and failure. We present a nanopolyplex (NP) approach that efficiently delivers a mitogen-activated protein kinase (MAPK)–activated protein (MAPKAP) kinase 2 inhibitory peptide (MK2i) to graft tissue to improve long-term patency by inhibiting pathways that initiate IH. In vitro testing in human vascular smooth muscle cells revealed that formulation into MK2i-NPs increased cell internalization, endosomal escape, and intracellular half-life of MK2i. This efficient delivery mechanism enabled MK2i-NPs to sustain potent inhibition of inflammatory cytokine production and migration in vascular cells. In intact human saphenous vein, MK2i-NPs blocked inflammatory and migratory signaling, as confirmed by reduced phosphorylation of the posttranscriptional gene regulator heterogeneous nuclear ribonucleoprotein A0, the transcription factor cAMP (adenosine 3′,5′-monophosphate) element–binding protein, and the chaperone heat shock protein 27. The molecular effects of MK2i-NPs caused functional inhibition of IH in human saphenous vein cultured ex vivo. In a rabbit vein transplant model, a 30-min intraoperative graft treatment with MK2i-NPs significantly reduced in vivo IH 28 days posttransplant compared with untreated or free MK2i–treated grafts. The decrease in IH in MK2i-NP–treated grafts in the rabbit model also corresponded with decreased cellular proliferation and maintenance of the vascular wall smooth muscle cells in a more contractile phenotype. These data indicate that nanoformulated MK2 inhibitors are a promising strategy for preventing graft failure.
INTRODUCTION
Coronary artery bypass with the patient’s own saphenous vein is the standard treatment for multivessel coronary heart disease. However, almost half of saphenous vein grafts fail within 18 months owing to intimal hyperplasia (IH) (1), and no current therapeutic approaches inhibit IH and improve graft patency in humans. Antithrombotic and antiplatelet agents, such as warfarin, clopidogrel, and aspirin, have little or no effect on IH (2). Two large clinical trials tested topical, ex vivo delivery of coronary and peripheral vascular vein grafts with an E2F transcription factor decoy designed to prevent smooth muscle proliferation, but these trials were unsuccessful at preventing graft failure (1, 3). However, the E2F decoy trials did establish the clinical feasibility of using a 30-min intraoperative window to treat the graft tissue ex vivo and support further development of prophylactic therapies, such as the one described here, that can be applied with precise dosing and negligible systemic drug exposure. Here, we sought to develop a therapeutic delivery system that achieves maximum potency and duration of action after the brief treatment time available ex vivo. Furthermore, the failure of E2F motivated our exploration of therapeutic targets that more broadly affect the processes that collectively cause IH, rather than focusing solely on cell proliferation.
The mechanical and biochemical stresses on the graft during harvest as well as posttransplant adaptation to arterial pressure activate the p38 mitogen-activated protein kinase (p38 MAPK) signaling pathway in vascular smooth muscle cells (VSMCs) (4), which causes downstream activation of multiple proinflammatory and profibrotic effectors implicated in IH (5–7). Unfortunately, inhibitors of p38 MAPK have failed clinical trials because of the adverse effects associated with blocking this pleiotropic, upstream mediator (8). p38 phosphorylation of MK2 triggers its translocation from the nucleus to the cytosol (9). Activated MK2 signals through downstream targets such as heat shock protein 27 (HSP27), heterogeneous nuclear ribonucleoprotein A0 (hnRNP A0), and cAMP (adenosine 3′,5′-monophosphate) response element–binding protein (CREB) to promote VSMC migration (10), proliferation (11), and inflammatory cytokine production (7), which combined lead to graft IH and failure. However, small-molecule inhibitors of MK2 have also failed to gain U.S. Food and Drug Administration approval primarily due to lack of specificity and solubility (12). A highly specific, cell-penetrating peptide (CPP)–based MK2 inhibitor (MK2i) has been developed (13). This MK2i peptide is currently in phase 1 clinical trials for treatment of idiopathic pulmonary fibrosis in Europe (initiated by Moerae Matrix Inc.) and shows potential to reduce IH in vein transplants (14). However, like many intracellular-acting biologics, MK2i bioavailability within the cytoplasm (where activated MK2 is localized) is limited by sequestration/degradation within late endosomes and early lysosomes (15).
Herein, we demonstrate a method for formulating endosomolytic, electrostatically complexed nanoparticles (nanopolyplexes or NPs) that efficiently deliver MK2i into vascular cells and tissues, enhancing peptide bioactivity by about an order of magnitude in vitro, ex vivo, and in vivo compared to the free peptide. MK2i-NPs represent a pharmaceutical approach that could enable clinical translation of intracellularly acting peptides, specifically to improve performance of vascular grafts after surgery by preventing IH and prolonging graft patency.
RESULTS
Synthesis and physicochemical characterization of MK2i-NPs
NPs were formed by mixing of the poly(acrylic acid) (PAA; fig. S1, B and D) or poly(propylacrylic acid) (PPAA; fig. S1, A and C) homopolymers with the MK2i peptide at pH 8.0, which is between the pKa (where Ka is the acid dissociation constant) of the primary amines on the MK2i peptide and the carboxylic acid moieties on both polymers, ensuring electrostatic complexation (Fig. 1A). PPAA was used in the lead MK2i-NP formulation because of its well-defined pH-dependent membrane-disruptive activity (Fig. 1B) (16) and safety in animals (17). PAA was used as a vector control because it is an anionic polymer with structural similarity to PPAA, but lacks pH responsiveness in a physiologically relevant range (pKa ~4.3) (Fig. 1C).
Fig. 1. MK2i-NP synthesis and characterization.
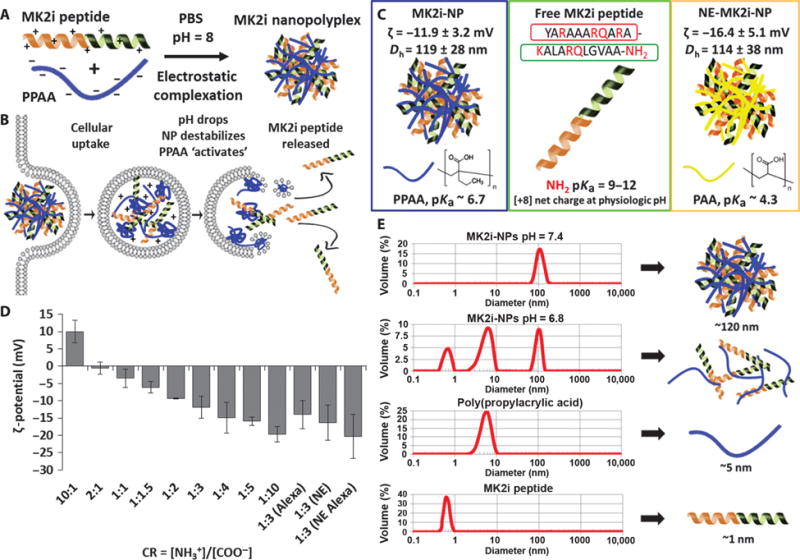
(A) MK2i-NP synthesis was achieved through electrostatic complexation of the MK2i peptide and PPAA. (B) Hypothesized MK2i-NP mechanism of endosomal escape. (C) Comparison of different MK2i formulations: endosomolytic MK2i-NPs formulated with PPAA, and NE-MK2i-NPs formulated with PAA. MK2i-NPs and NE-MK2i-NPs were formulated with the shown MK2i peptide sequence (red, modified TAT mimetic CPP sequence; green, MK2 inhibitory sequence). (D) ζ-potential of polyplexes prepared at different CRs ([NH3+]/[COO−]). “Alexa” denotes NPs formulated with an Alexa Fluor 488–conjugated MK2i peptide. NE denotes nonendosomolytic MK2i-NPs. Data are means ± SEM (n = 3 independent measurements). (E) Dynamic light scattering (DLS) analysis of MK2i-NP disassembly at pH values low enough to protonate the PPAA polymer.
To determine optimal NP formulation conditions, we prepared a library of MK2i-NPs at a range of charge ratios [CR = ([NH3+]MK2i: [COO−]PPAA)], and the size distribution (table S1) and ζ-potential were characterized. MK2i-NP ζ-potential was directly proportional to the CR, with an apparent isoelectric point at CR ~2:1 (Fig. 1D). A CR of 1:3 was chosen as the optimal formulation because this ratio yielded a unimodal size distribution with minimal particle size and polydispersity (fig. S2). Nonendosomolytic MK2i-NPs (NE-MK2i-NPs) were formulated with PAA as a vehicle control for biological studies. NE-MK2i-NPs prepared at CR 1:3 were equivalent in size and ζ-potential to the endosomolytic MK2i-NPs. MK2i-NPs dissociated as the pH was lowered from extracellular pH toward the pKa of the carboxylic acids (pH ~6.7) on PPAA, which corresponds to early endosomal conditions (Fig. 1E) and suggests that peptide cargo will unpackage after cellular uptake into acidifying endosomal pathways.
MK2i-NP cell internalization, endosome escape, intracellular retention, and tissue uptake
MK2i-NPs significantly increased peptide internalization and intracellular retention in human VSMCs compared with controls (Fig. 2A and fig. S3). NE-MK2i-NP uptake was equivalent to the free peptide, indicating that differences in cell internalization are due to NP composition and are independent of particle morphology and charge. Enhanced peptide delivery with the MK2i-NP formulation was also detected in endothelial cells (ECs), suggesting that the high level of uptake of this formulation is not cell type–specific (fig. S4, A and B).
Fig. 2. MK2i-NP formulation increases cellular uptake, extends intracellular retention, and reduces endolysosomal colocalization of MK2i.
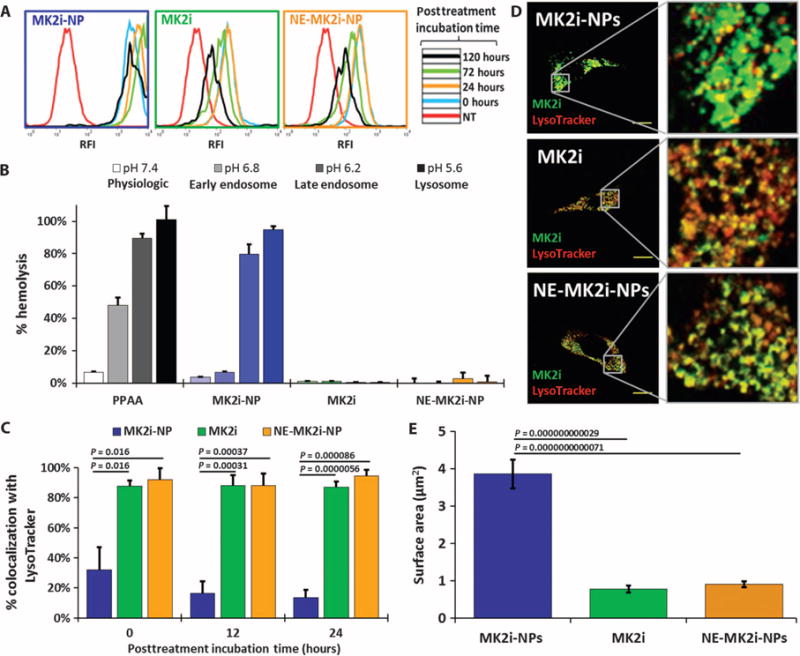
(A) Representative flow cytometry histograms of MK2i internalization when delivered as a free peptide or via NPs. (B) Red blood cell hemolysis assay for pH-dependent membrane-disruptive activity. Data are means ± SEM (n = 4 technical replicates). (C) MK2i Alexa Fluor 488–labeled MK2i colocalization with LysoTracker red in VSMCs over time after 2 hours of treatment determined by calculating Mander’s coefficients. Data are means ± SEM (n ≥ 3 separate images). P values determined by one-way analysis of variance (ANOVA). (D) Representative confocal microscopy images of Alexa Fluor 488–labeled MK2i colocalization with LysoTracker in VSMCs. Scale bars, 20 μm. (E) Quantification of intracellular compartment size of VSMCs treated with each MK2i formulation. Data are means ± SEM (n ≥ 50 separate intracellular compartments per treatment group). P values determined via single-factor ANOVA.
NPs increased the intracellular half-life of the MK2i peptide by more than an order of magnitude from 4 to 58 days (fig. S3B), likely owing to decreased rate of peptide degradation in the endolysosomal pathway and/or the rate of exocytotic recycling of the cell (18). A red blood cell hemolysis assay that measures pH-dependent membrane-disruptive activity as an indicator for endosomal escape function revealed that the PPAA polymer and MK2i-NPs, but not MK2i or NE-MK2i-NPs, disrupt erythrocyte membranes at pHs encountered in the endolysosomal pathway at or below the pKa of PPAA (~6.7) (Fig. 2B). Imaging and quantification of MK2i-NP endosomal escape in human VSMCs demonstrated that the MK2i-NP formulation significantly reduced MK2i endolysosomal colocalization (Fig. 2, C and D). MK2i delivered via MK2i-NPs was found within larger intracellular regions, potentially representative of the cytosol or disrupted endosomal vesicles (Fig. 2, D and E).
MK2i-NP uptake and bioactivity in human saphenous vein ex vivo
Assessment of MK2i delivery into intact human saphenous vein (HSV) showed that uptake for all formulations occurred in both smooth muscle (Fig. 3A) and endothelial (Fig. 3B) cells, with more concentrated delivery at the luminal and adventitial surfaces (red MK2i staining in Fig. 3). The MK2i-NPs had greater penetration and uptake throughout the cross section of the vessel compared to the free MK2i peptide or control NE-MK2i-NPs. The increased delivery into the vessel wall by MK2i-NPs relative to MK2i and NE-MK2i-NPs was visualized on the basis of colocalization with the smooth muscle marker α-smooth muscle actin (α-SMA) (Fig. 3A) and quantified through the pixel intensity distributions of MK2i-positive red fluorescence in each of the treated vessel cross sections (Fig. 3C).
Fig. 3. MK2i-NP formulation increases peptide delivery to human vein.
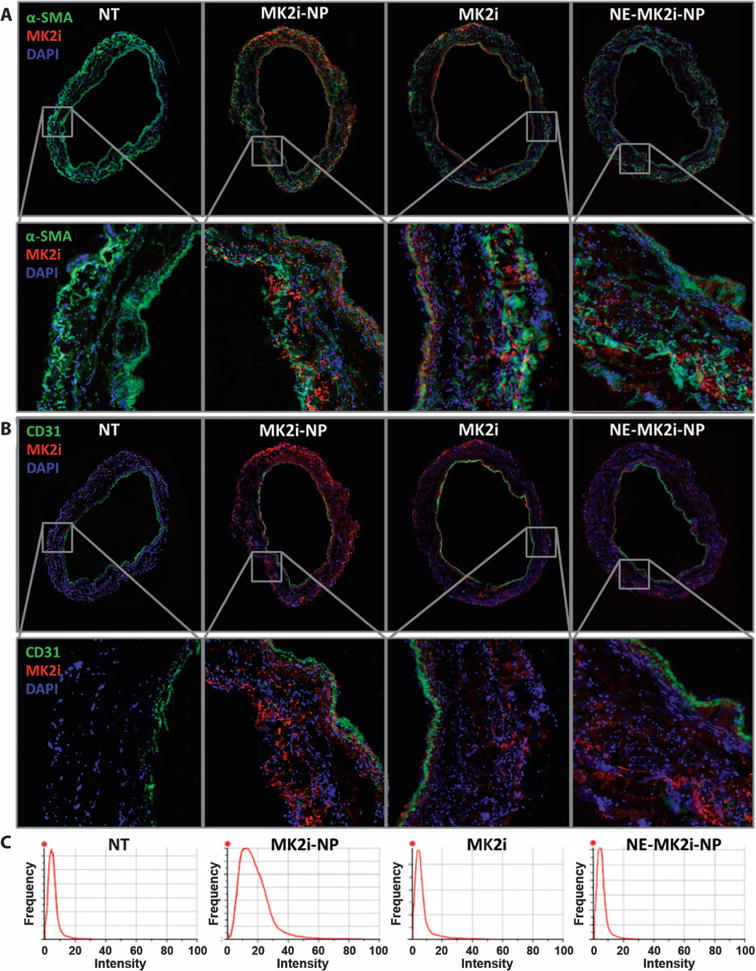
(A) Immunofluorescence microscopy images and zoomed insets of HSV cross sections treated with Alexa Fluor 568–labeled MK2i, MK2i-NPs, or NE-MK2i-NPs (all red) and stained for the VSMC marker α-SMA and the nuclear marker 4′,6-diamidino-2-phenylindole (DAPI). (B) Immunofluorescence microscopy images and zoomed insets of HSV cross sections treated with Alexa Fluor 568–labeled MK2i, MK2i-NPs, or NE-MK2i-NPs (all red) and stained for the endothelial marker CD31 and DAPI. (C) Pixel intensity distribution derived from the red fluorescent channel of entire (unzoomed) cross-sectional images in (A) and (B). Plots are representative of n = 3 HSV cross sections from a single donor.
Delivery of MK2i-NPs to intact HSV ex vivo modulated phosphorylation of several MK2 substrates relevant to graft IH, including hnRNP A0, CREB, and HSP27 (Fig. 4, A to E). hnRNP A0 stabilizes the mRNA of inflammatory cytokines, increasing their translation, and CREB binds to cAMP-responsive elements to promote expression of genes that induce smooth muscle cell migration, proliferation, and production of the inflammatory cytokines. Phosphorylation of HSP27 is also believed to contribute to the pathological VSMC migration characteristic of IH through control of actin dynamics. MK2i-NP treatment significantly inhibited neointima formation without affecting tissue viability (fig. S5) in HSV in a dose-dependent fashion, and at an order of magnitude less peptide than free MK2i (Fig. 4, F and G). Furthermore, MK2i-NP at 100 μM MK2i was the only treatment that fully abrogated IH, resulting in an intimal thickness after 2 weeks of culture that was statistically equivalent to control tissues (P = 0.49, single-factor ANOVA) (Fig. 4G).
Fig. 4. Treatment with MK2i-NP reduces neointima formation and alters phosphorylation of molecules downstream of MK2 in HSV ex vivo.
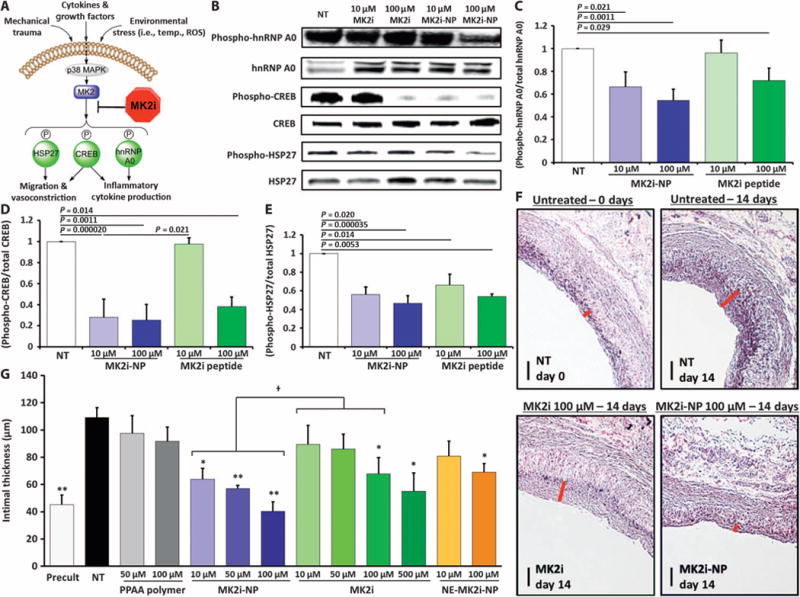
(A) p38 MAPK–MK2 signaling pathway in smooth muscle. (B to E) Representative Western blots showing the phosphorylation of MK2 substrates hnRNP A0, CREB, and HSP27 with and without treatment. Quantification of Western blots for phospho–hnRNP A0 (C), phospho-CREB (D), and phospho-HSP27 (E) in HSV. Data are means ± SEM (n = 3 separate biological replicates from three separate donors). P values determined by single-factor ANOVA. (F) Neointima were visualized using Verhoeff–van Gieson (VVG) staining of HSV samples that were treated for 2 hours and maintained in organ culture for 14 days. Red bars demarcate intimal thickness. Scale bars, 100 μm. (G) Intimal thickness in HSV samples was quantified after 14 days in organ culture. Data are means ± SEM (n ≥ 3 separate biological replicates from three separate donors). *P ≤ 0.01, **P ≤ 0.001 compared to no treatment (NT) controls; †P ≤ 0.05; single-factor ANOVA.
MK2i-NP bioactivity and duration of efficacy
Mechanistic evaluation of MK2i-NP bioactivity in both human VSMCs and ECs was performed to elucidate the impact of MK2 in hibition on inflammatory cytokine production and migration, both of which are integral processes in the pathogenesis of IH. MK2i-NPs were not cytotoxic and significantly increased the ability of the MK2i peptide to inhibit tumor necrosis factor–α (TNFα) and interleukin-6 (IL-6) inflammatory cytokine production in VSMCs stimulated with the proinflammatory peptide hormone angiotensin II or TNFα, respectively (Fig. 5, A and B, and fig. S6). MK2i-NPs were also more potent at inhibiting VSMC and EC migration compared to the free MK2i peptide (Fig. 5, C and D, and fig. S4, C and D). These results were found to be independent of cellular proliferation (fig. S7).
Fig. 5. NP formulation enhances MK2i bioactivity and duration of action in vitro.
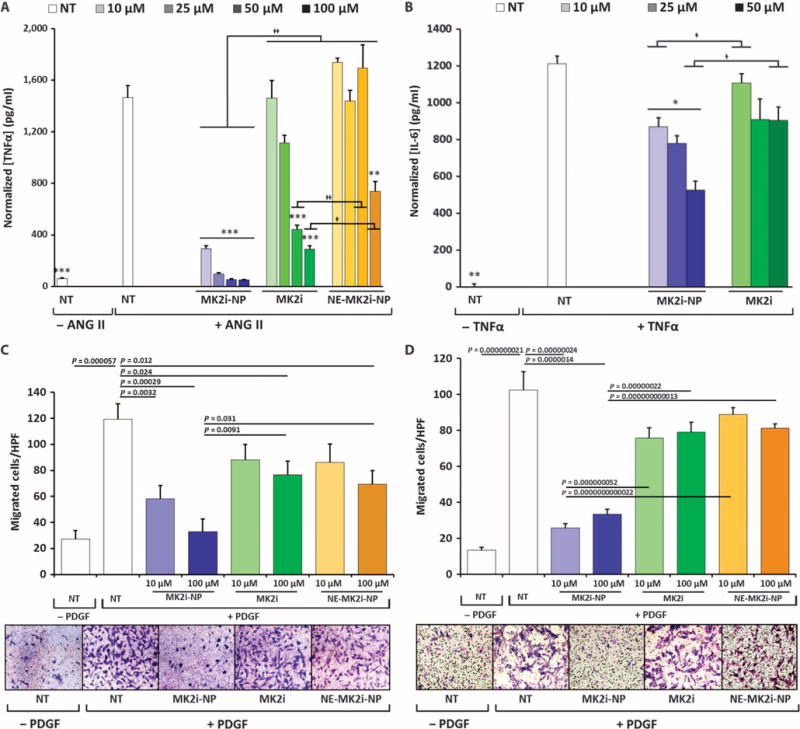
(A) TNFα production in human VSMCs stimulated with 10 μM angiotensin II (ANG II) for 4 hours, treated with MK2i formulations for 2 hours, and incubated in fresh medium for 24 hours. (B) IL-6 production in VSMCs stimulated with TNFα (20 ng/ml) for 4 hours, treated with MK2i formulations for 2 hours, and incubated in fresh medium for 24 hours. Data in (A) and (B) are means ± SEM (n = 4 technical replicates); *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 compared to NT, †P ≤ 0.05, ††P ≤ 0.001; single-factor ANOVA. (C and D) Quantification and representative images of VSMC migration immediately after (C) and 5 days after (D) treatment removal using a Boyden Transwell migration assay. Data are means ± SEM (n = 3 technical replicates from two separate experiments). P values determined by single-factor ANOVA.
In vitro bioactivity assays were carried out 5 days after treatment with MK2i to assess the impact of the NP formulation on longevity of peptide therapeutic action. MK2i-NPs sustained inhibitory activity against migration of VSMCs, whereas free MK2i or NE-MK2i-NPs showed minimal effect under these conditions (Fig. 5D). Similarly, the ability of the free MK2i peptide to inhibit the production of monocyte chemoattractant protein-1 (MCP-1), which is up-regulated through both hnRNP A0 and TNFα and implicated in vein graft IH, decreased in VSMCs and ECs at day 5 after treatment; conversely, MK2i-NPs demonstrated sustained inhibitory bioactivity (fig. S8). The decrease in antimigratory and anti-inflammatory activity at 5 days after treatment corresponds well with the shorter intracellular half-life of the free MK2i peptide relative to MK2i delivered via NPs (fig. S2B).
In vivo bioactivity in a rabbit vein graft interposition model
The therapeutic benefit of MK2i-NP–treated grafts in vivo was assessed in a rabbit bilateral jugular vein graft interpositional transplant model that uses a polymeric cuff method to induce turbulent blood flow and accelerate graft IH. Rabbit vein graft transplants were treated for 30 min with MK2i or MK2i-NPs. Twenty-eight days posttransplant, MK2i-NPs significantly inhibited neointima formation in grafts compared to tissues receiving no treatment or treatment with free MK2i peptide; conversely, free MK2i peptide was not statistically different from untreated grafts (Fig. 6A). Intimal proliferating cell nuclear antigen (PCNA) was ~17-fold lower in jugular vein grafts treated with MK2i-NPs compared with both free MK2i and untreated grafts, indicating that MK2i-NP treatment significantly decreased neointimal cell proliferation (Fig. 6B). Intimal expression of the contractile VSMC marker α-SMA was increased in MK2i-NP–treated grafts relative to free MK2i–treated grafts (Fig. 6C). Converse to contractile marker expression, intimal expression of the synthetic (that is, pathological) VSMC marker vimentin was decreased in MK2i-NP–treated grafts but not in grafts treated with free MK2i peptide (Fig. 6D).
Fig. 6. Intraoperative treatment with MK2i-NPs reduces neointima formation and promotes a contractile smooth muscle cell phenotype in vivo in transplanted rabbit vein grafts.
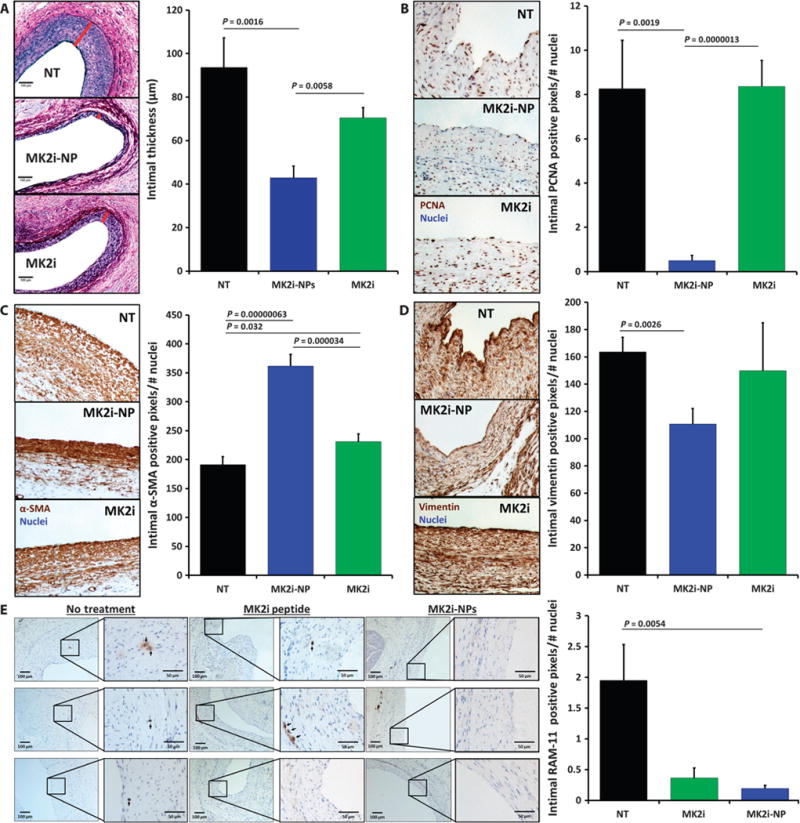
Rabbit jugular vein explants were treated ex vivo for 30 min with MK2i or MK2i-NP (30 μM peptide) before transplant into the carotid artery (n ≥ 7 grafts per treatment group). Histological analyses were done on graft tissues harvested 28 days later. (A) Neointima was visualized using VVG staining of vein grafts. Red bars demarcate intimal thickness on the representative images. Scale bars, 100 μm. (B) Proliferation of intimal cells quantified using PCNA immunohistochemistry. (C) Intimal expression of the contractile marker α-SMA. (D) Intimal expression of the synthetic vascular smooth muscle phenotypic marker vimentin. (E) RAM-11+ macrophages in jugular vein graft sections. Left column scale bar, 100 μm; right column zoomed view scale bar, 50 μm. Data in (B) to (E) are means normalized to total number of cells in intima (that is, number of nuclei) ± SEM. All P values determined by single-factor ANOVA.
There were fewer intimal macrophages in both MK2i- and MK2i-NP–treated grafts compared with untreated grafts (Fig. 6E), suggesting that MK2i blunts local macrophage recruitment and/or persistence. This mechanism is potentially mediated through decreased secretion of macrophage inflammatory protein 2 (MIP-2; also known as CXCL2) and/or MCP-1, both of which attract inflammatory cells and are up-regulated either directly or indirectly through hnRNP A0. Our in vitro results support this mechanism, demonstrating that MK2i-NPs inhibited MCP-1 production in both VSMCs and ECs (fig. S8).
DISCUSSION
A simple, translational approach was developed for the formulation of nanoparticles that enhance cellular uptake and retention of a highly specific peptide inhibitor of MK2 that has a broad mechanism of action relevant to IH. NP formulation enhanced the potency and longevity of action of the MK2i peptide, and the clinical translatability of this delivery technology was demonstrated in human tissue ex vivo and in a preclinical rabbit model of IH in vivo. These results also validate and provide mechanistic insight into the key role of MK2 in VSMC behavior and phenotype as well as vascular graft IH.
Fusion of therapeutic peptide sequences with CPPs is a common approach to increase peptide uptake and bioactivity. However, the choice of CPP sequence significantly influences peptide potency, and CPPs can cause nonspecific effects independent of the therapeutic sequence. The original MK2i peptide sequence was discovered and optimized as a substrate peptide based on the sequence surrounding the Ser86 phosphorylation site of HSP27 (13). Testing various MK2 inhibitory peptides revealed that fusion with some CPP sequences caused nonspecific kinase inhibitory activity that was associated with cytotoxicity in vitro (12). These findings led to the discovery that the CPP sequence used in our current study produces optimal specificity against MK2 and motivated our goal of developing a translational delivery technology to enhance the MK2i peptide’s therapeutic potential.
Formulation of the positively charged, CPP-based MK2i peptide with the anionic, endosomolytic polymer PPAA was conceptualized as a method to enhance peptide endolysosomal escape and therapeutic potency. This approach was inspired by the convention for nonviral gene therapy, which is based on electrostatic formation of polyplexes between anionic nucleic acids and positively charged CPP sequences, lipids, or polymeric transfection agents to enhance uptake and endosomal escape (19–21). MK2i-NP formulation yielded net negatively charged particles; serendipitously, the NP formulation significantly increased peptide uptake by VSMCs relative to the free MK2i peptide. Notably, the in vitro comparisons of MK2i-NPs and NE-MK2i-NPs suggest that the high level of MK2i-NP cell internalization was dependent on the specific composition of PPAA, rather than purely dictated by NP morphology and surface charge. The α-alkyl substitution of the propyl moiety makes PPAA more lipophilic/hydrophobic relative to acrylic acid, suggesting that the observed differences in uptake may be the result of increased hydrophobic interactions of MK2i-NPs with the cell membrane. Hydrophobic interactions may nonspecifically trigger MK2i-NP cell internalization, or MK2i-NP internalization could be mediated through a more specific interaction; for example, by VSMC scavenger receptors that are up-regulated in settings of vascular stress and that internalize negatively charged, hydrophobic particles (22).
In addition to initial cell internalization, intracellular persistence and longevity of action are important for a peptide-based vein graft therapeutic applied as a single, intraoperative treatment. Previous studies characterizing the time course of IH pathogenesis in rabbit and canine models have demonstrated an initial burst in cellular proliferation during the first week, followed by continued graft adaptation that reaches steady state by week 12 (23, 24). Our study suggests that peptide bioactivity exceeding 1 week would be ideal for reducing the impact of the acute inflammatory phase and accelerating resolution of the initial graft adaptation stage. A recent study on mouse vein grafts showed that, although the same free MK2i peptide prevented early intimal thickening, the rate of intimal thickening during weeks 1 to 4 posttransplant was similar between the treated and untreated control grafts (25). These data indicate that the therapeutic impact of MK2i on reducing graft IH may be improved by extending its longevity of action.
Here, we hypothesized that PPPA would extend intracellular MK2i peptide half-life by avoiding endolysosomal degradation and extracellular recycling, processes known to decrease cytosolically active peptide function (26). Indeed, avoiding endosomal entrapment was associated with increased intracellular peptide retention (half-life was increased 14-fold by incorporation into NPs) as well as prolonged bioactivity (migration and cytokine inhibition) for 5 days after treatment in both VSMCs and ECs in vitro. A sustained effect was also seen in intact HSVs ex vivo, with MK2i-NPs completely abrogating neointimal growth over 2 weeks, whereas the shorter-lived, free MK2i peptide delivered at an equivalent dose had modest effects. Furthermore, a sustained benefit was seen 28 days posttransplant in MK2i-NP–treated grafts in rabbits: minimal cell proliferation, residual inflammatory cell persistence, and intimal thickening were detected at day 28 in MK2i-NP–treated grafts. These data suggest that extended MK2 inhibition during the initial posttransplant period enabled the grafts to reach a steady state more rapidly and to avoid continued neointimal growth and consequent degeneration of graft function.
We further sought to clarify the effects of MK2i-NPs on the multifarious processes that characterize IH. The current studies validate the broad effects of MK2i-NPs on inflammation, cell migration, and VSMC phenotype and confirm the use of targeting the p38-MK2 pathway to inhibit multiple factors underlying IH pathogenesis. In human tissue, MK2i-NPs modulated proinflammatory mediators activated downstream of MK2, such as hnRNP A0 (7) and CREB (27, 28), which led to a decrease in production of the proinflammatory cytokines TNFα, IL-6, and MCP-1 in vitro and reduced inflammatory cell presence in vivo 28 days posttransplant. MK2i-NPs were also shown to modulate migration-related pathways in human tissue, as demonstrated by reduced phosphorylation of HSP27, which triggers VSMC transition to a migratory and fibrotic myofibroblast phenotype (10) and causes vein graft vasoconstriction (29). Inhibition of both HSP27 and CREB activation in HSVs correlated with reduced VSMC migration in vitro and may have been linked to maintaining a contractile VSMC phenotype in the MK2i-NP–treated grafts in vivo. Together, these results suggest that MK2i-NPs have pleiotropic effects that have desirable impacts on both inflammation- and migration-related pathways in vein grafts.
In sum, our data demonstrate that inhibition of MK2 is a promising approach for improving long-term graft patency and motivate further studies to establish clinical feasibility. In the rabbit graft transplant studies, MK2i-NP treatment provided a long-term (28-day) reduction in IH that correlated with reduced neointimal cell proliferation and macrophage persistence. Future in vivo studies focusing on a broader time scale (especially earlier time points) and larger animal models will provide in vivo MK2i-NP pharmacokinetic and pharmacodynamic information necessary to proceed toward clinical translation. MK2i-NP treatment was also found to prevent transition from a contractile to a synthetic VSMC phenotype in vivo; however, the underlying mechanisms of this observation remain unclear, and further studies will be necessary to elucidate the role that MK2 plays in VSMC phenotypic modulation in vein grafts.
Last, although VSMCs are considered the primary target cell for therapeutically blocking IH, MK2i-NPs were shown to also be internalized by ECs ex vivo and have effects on vascular ECs in vitro. These results not only highlight the need to optimize MK2i-NP delivery to more homogeneously penetrate the medial region of the vessel wall but also raise questions about the functional importance of MK2i concentration at the luminal surface of the graft where ECs are present. Focal disruption of the endothelium can occur during vein graft harvest (30), raising the potential concern that MK2i-NP therapy may have deleterious effects on vein graft reendothelialization by reducing EC migration. However, previous evidence suggests that vein graft reendothelialization predominantly occurs through ECs derived from the surrounding artery (31). This finding further justifies ex vivo, intraoperative treatment of grafts with MK2i-NPs as the ideal therapeutic strategy to enhance delivery to the target tissue and avoid potential off-target effects. Furthermore, others have suggested that inhibition of the p38 MAPK and MK2 pathways may have beneficial effects on ECs in vascular grafts. For example, the p38 MAPK pathway has been implicated in transforming growth factor β–mediated endothelial-to-mesenchymal transdifferentiation (5), which is believed to be a key mechanism underlying IH (32). MK2 deficiency may also promote endothelial healing by decreasing inflammatory chemokine production and associated inflammatory cell recruitment (33), which is in agreement with our results that MK2 inhibition decreased endothelial production of inflammatory cytokines. These combined findings by our group and others prompt further investigation into the relative importance of MK2 inhibition in EC migration and inflammatory signaling in vein bypass grafts.
Our overall findings provide new insights into the molecular mechanisms underlying the initiation and progression of human IH and highlights new therapeutic targets that modulate VSMC phenotype and improve vein graft patency. The collective anti-inflammatory, antimigratory, and phenotype-modulating actions of MK2i-NPs comprehensively address the multifactorial pathophysiology of IH and may overcome the pitfalls of previous therapeutic candidates with narrow mechanisms of action (for example, E2F decoys). The current findings also firmly establish the potential use of nanotechnology to enhance drug delivery into cells and tissue. These findings also raise new questions about optimizing NP penetration into the vascular wall, understanding the impacts of MK2i-NPs on different cell types, and elucidating MK2i-NP in vivo pharmacokinetics and pharmacodynamics. These aspects motivate our ongoing studies to translate MK2i-NPs as an intraoperative, prophylactic therapy to improve long-term vein graft patency.
MATERIALS AND METHODS
Study design
We hypothesized that a single treatment with MK2i-NPs would prevent IH in vein graft tissue. Intimal thickening was used as a measure of IH in HSV explants ex vivo and in rabbit jugular vein bypass grafts in vivo. For all ex vivo HSV histomorphometric measurements, the researcher was blinded to the treatment group. For in vivo studies in rabbits, an a priori power analysis based on pilot data determined that a sample size of eight grafts per treatment group would provide 95% power to detect significant difference in our primary endpoints with α = 0.05. Endpoints and peptide dosage were selected on the basis of existing precedents for the evaluation of IH. Graft treatments in each rabbit were randomized, and samples were blinded before histological analyses. Partially and completely thrombosed grafts were excluded. Grafts in which the contralateral graft failed were also excluded owing to concerns with compensatory changes in blood flow affecting tissue remodeling. One rabbit was euthanized at day 14 owing to thrombosis of both grafts. The incidences of graft failure were as follows: NT—10 grafts harvested, 2 failed due to thrombosis, 1 excluded due to contralateral thrombosis, 7 patent; MK2i—16 grafts harvested, 8 grafts failed due to thrombosis, 0 excluded due to contralateral thrombosis, 8 patent; and MK2i-NP—14 grafts harvested, 2 failed due to thrombosis, 3 excluded due to contralateral thrombosis, 9 patent. Altogether, n ≥ 7 patent grafts without contralateral occlusion per treatment group were included in the reported data.
MK2i-NP synthesis and characterization
PPAA was dissolved in 1 M NaOH and diluted into a phosphate buffer (pH 8) to obtain a stock solution. MK2i was dissolved in phosphate buffer (pH 8). The MK2i peptide and PPAA polymer were mixed at a range of CRs from [NH3+]:[COO−] = 10:1 to 1:10 to form MK2i-NPs. The resulting polyplexes were syringe-filtered through 0.45-μm PTFE (polytetrafluoroethylene) filter, and the hydrodynamic diameter and ζ-potential were characterized on a Malvern Zetasizer Nano ZS (table S1 and fig. S2, A and B). A CR of 1:3 was used in subsequent studies. NPs formulated at the same CR with the nonendosomolytic polymer PAA (that is, NE-MK2i-NPs) were analyzed by DLS and used as a vehicle control in all subsequent studies. MK2i-NPs were also visualized through transmission electron microscopy (TEM) imaging. TEM samples were prepared by inverting carbon film–backed copper grids onto a droplet of aqueous polyplex suspensions (1 mg/ml) and blotted dry. All samples were then counterstained with 3% uranyl acetate. Samples were desiccated in vacuo for 2 hours before TEM imaging (fig. S5C). The pH-dependent size changes of polyplexes at a CR of 1:3 were then quantified by DLS analysis at various pH values in phosphate-buffered saline.
Cell culture
Primary human coronary artery VSMCs and human umbilical vein endothelial cells (HUVECs) were obtained from Lonza. VSMCs were cultured in vascular cell basal medium (American Type Culture Collection) supplemented with a VSMC growth kit [5% fetal bovine serum (FBS), human basic fibroblast growth factor (5 ng/ml), human insulin (5 μg/ml), ascorbic acid (50 μg/ml), L-glutamine (10 mM), human epidermal growth factor (5 ng/ml), 1% penicillin-streptomycin and plasmocin (5 μg/ml)]. HUVECs were cultured in endothelial basal medium (Lonza) supplemented with an EGM-2 BulletKit, 1% penicillin-streptomycin, and plasmocin (5 μg/ml). All cultures were maintained in 75-cm2 polystyrene tissue culture flasks in a 37°C and 5% CO2 environment with cell culture medium refreshed every other day. Cells were grown to 80 to 90% confluence before being harvested and passaged. All cells were seeded at a density of 20,000 to 30,000 cells/cm2, as required for each specific experiment. Only cells from early passages (numbers 3 to 8) were used in experiments.
Inflammatory cytokine analysis
Two hundred microliters of cell suspension (at 10,000 cells per well) was seeded onto 96-well plates to yield an about 70% confluence per well. Cells were allowed to adhere to the plate overnight. Human TNFα, IL-6, and MCP-1 enzyme-linked immunosorbent assay (ELISA) development kits (PeproTech) were used to measure cytokine levels in supernatant collected from treated cells (Supplementary Methods) according to the manufacturer’s protocol. All ELISA data were then normalized to cell viability determined by a CytoTox-ONE Homogenous Membrane Integrity Assay (Promega) according to the manufacturer’s protocol.
Migration assays
Chemokinesis and chemotaxis were evaluated by scratch wound and Boyden chamber assays, respectively, as described in Supplementary Methods.
Intracellular uptake and trafficking
Cell uptake of the MK2i peptide formulations was analyzed by microscopy and flow cytometry, as described in Supplementary Methods.
Human saphenous vein
HSV tissue was procured, and tissue viability, peptide uptake, and Western blot analysis of protein phosphorylation were performed as described in Supplementary Methods.
HSV organ culture and ex vivo assay for IH
After vessel viability was verified with multiple KCl challenges, additional rings were cut and placed in a 24-well plate and maintained in RPMI 1640 medium supplemented with 30% FBS, 1% L-glutamine, and 1% penicillin-streptomycin for 14 days at 37°C in an atmosphere of 5% CO2 in air. The rings were treated with MK2i-NPs, NE-MK2i-NPs, MK2i peptide, or buffer alone for 2 hours, washed, and given fresh medium. The culture medium without treatments was replaced every 2 days for 14 days. After 14 days of organ culture, vein segments were fixed in 0.5 ml of 10% formalin at 37°C for 30 min and embedded in paraffin for sectioning. Beginning at the midportion of each ring, five transverse sections, spaced 5 μm apart, were cut from each specimen. Sections were then stained with VVG. Histology sections were imaged using a Nikon Eclipse Ti inverted fluorescence microscope, and six radially parallel measurements of intimal and medial thickness were randomly taken from each section using NIS Elements imaging software (total of 6 to 12 measurements per ring, n ≥ 3 rings per treatment group from separate donors). Intima was defined as tissue on the luminal side of the internal elastic lamina or the chaotic organization of the cells contained within it, whereas the medial layer was contained between the intimal layer and the external elastic lamina.
Rabbit bilateral jugular vein graft interposition model
This study was approved by the Vanderbilt Institutional Animal Care and Use Committee and conformed to the Guide for the Care and Use of Laboratory Animals. Male New Zealand White rabbits (3.0 to 3.5 kg) were anesthetized through an intramuscular injection with ketamine hydrochloride (1.4 mg/kg) and xylazine (0.2 mg/kg). Anesthesia was maintained with endotracheal intubation and inhaled isoflurane (2.0 to 5.0%). A high-dose intravenous heparin bolus (250 U/kg) was administered immediately before carotid cross clamp. The operative procedure was performed with aseptic technique under ×2.5 optical magnification.
Vein bypass grafts were constructed with an anastomotic cuff technique as previously described (34). Briefly, polymer cuffs consisting of a 2.0-mm body loop were fashioned from a 4-Fr introducer sheath. After ligation of smaller tributary vessels, the external jugular veins were harvested (3.0 to 4.0 cm in length) for creation of an interposition graft into the common carotid artery. Jugular vein ends were passed through a cuff, everted, and fixed with 6-0 silk. Vein grafts were subsequently treated for 30 min in 2 ml of either 30 μM MK2i-NP, 30 μM MK2i peptide, or heparin Plasma-Lyte (no treatment). Afterward, the carotid artery lumen was exposed with a 2.0-cm arteriotomy, and the cuffed, reversed vein ends were inserted. A 3-0 silk was used to secure the artery around the cuff. Finally, 1.0 cm of carotid artery back wall was cut away between the cuffs to permit vein graft extension.
Rabbits were euthanized at 28 days postoperatively, and vein grafts were perfusion-fixed in situ with 10% neutral buffered formalin under 50 mmHg pressure with a roller pump. Vein grafts were subsequently excised and sectioned into four segments avoiding the tissue overlying the cuff to allow for evaluation of morphological variation along the length of the graft. Histological sections were prepared, and separate sections were stained with VVG, PCNA (Abcam), α-SMA (Abcam), vimentin (Abcam), or RAM-11 (Dako). Intimal and medial thicknesses were quantified by taking three measurements from each quadrant of each VVG-stained vessel section (12 measurements per segment = 48 measurements per graft). PCNA-, α-SMA–, vimentin-, and RAM-11–positive staining in the intima was quantified by free-form selection of the intima in ImageJ and using a color deconvolution method as previously described (35). Positive staining was normalized to the number of intimal cell nuclei. Sixteen histological images from four different graft sections were analyzed for each treatment group.
Statistical analysis
Statistical analysis was performed with one-way ANOVA followed by Tukey’s post hoc test to validate statistical significance of mean differences between experimental groups. Analyses were done with OriginPro 8 software or Minitab 16 software. Statistical significance was accepted within a normal-based 95% confidence limit (α = 0.05). Results are presented as arithmetic means ± SEM graphically, and P values are included in the figures or in the figure legends.
Supplementary Material
Acknowledgments
We thank A. Panitch, J.C. Flynn, and P. Komalavilas for input on experimental study design.
Funding: Confocal imaging was performed in part through the use of the Vanderbilt University Medical Center Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, HD15052, DK59637, and Ey008126). DLS and TEM were conducted at the Vanderbilt Institute of Nanoscale Sciences and Engineering. Histological sectioning and immunostaining were performed in part by the Vanderbilt Translational Pathology Shared Resource. This work was supported by the American Heart Association (11SDG4890030), NIH (1R21HL110056), and a National Science Foundation Graduate Research Fellowship to B.C.E. (DGE-0909667).
Footnotes
Author contributions: C.L.D. and C.M.B. conceived and directed the project. B.C.E. synthesized, formulated, and characterized NPs and performed in vitro assays, flow cytometry, organ culture experiments, image analysis, histological analysis, and statistical analysis. K.M.H. performed smooth muscle physiology and Western blot experiments. K.V.K. performed fluorescence confocal microscopy. J.D. performed cell migration experiments. M.J.O and I.V. performed rabbit surgeries. B.C.E. wrote the manuscript and received feedback and final approval from all the other authors.
Competing interests: C.M.B. is the chief medical officer of and a stockholder in Moerae Matrix Inc., which is conducting a phase 1 clinical trial using MK2i. A patent application has been filed for the NP technology (PCT/US14/33873).
Data and materials availability: All data are in the manuscript, and all materials are available.
REFERENCES AND NOTES
- 1.Alexander JH, Hafley G, Harrington RA, Peterson ED, Ferguson TB, Jr, Lorenz TJ, Goyal A, Gibson M, Mack MJ, Gennevois D, Califf RM, Kouchoukos NT, PREVENT IV Investigators Efficacy and safety of edifoligide, an E2F transcription factor decoy, for prevention of vein graft failure following coronary artery bypass graft surgery: PREVENT IV: A randomized controlled trial. JAMA. 2005;294:2446–2454. doi: 10.1001/jama.294.19.2446. [DOI] [PubMed] [Google Scholar]
- 2.Kent KC, Liu B. Intimal hyperplasia—Still here after all these years! Ann Vasc Surg. 2004;18:135–137. doi: 10.1007/s10016-004-0019-4. [DOI] [PubMed] [Google Scholar]
- 3.Conte MS, Bandyk DF, Clowes AW, Moneta GL, Seely L, Lorenz TJ, Namini H, Hamdan AD, Roddy SP, Belkin M, Berceli SA, DeMasi RJ, Samson RH, Berman SS. Results of PREVENT III: A multicenter, randomized trial of edifoligide for the prevention of vein graft failure in lower extremity bypass surgery. J Vasc Surg. 2006;43:742–751. doi: 10.1016/j.jvs.2005.12.058. [DOI] [PubMed] [Google Scholar]
- 4.Saunders PC, Pintucci G, Bizekis CS, Sharony R, Hyman KM, Saponara F, Baumann FG, Grossi EA, Colvin SB, Mignatti P, Galloway AC. Vein graft arterialization causes differential activation of mitogen-activated protein kinases. J Thorac Cardiovasc Surg. 2004;127:1276–1284. doi: 10.1016/j.jtcvs.2003.07.017. [DOI] [PubMed] [Google Scholar]
- 5.Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL. p38 mitogen-activated protein kinase is required for TGFβ-mediated fibroblastic transdifferentiation and cell migration. J Cell Sci. 2002;115:3193–3206. doi: 10.1242/jcs.115.15.3193. [DOI] [PubMed] [Google Scholar]
- 6.Hedges JC, Dechert MA, Yamboliev IA, Martin JL, Hickey E, Weber LA, Gerthoffer WT. A role for p38(MAPK)/HSP27 pathway in smooth muscle cell migration. J Biol Chem. 1999;274:24211–24219. doi: 10.1074/jbc.274.34.24211. [DOI] [PubMed] [Google Scholar]
- 7.Rousseau S, Morrice N, Peggie M, Campbell DG, Gaestel M, Cohen P. Inhibition of SAPK2a/p38 prevents hnRNP A0 phosphorylation by MAPKAP-K2 and its interaction with cytokine mRNAs. EMBO J. 2002;21:6505–6514. doi: 10.1093/emboj/cdf639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dambach DM. Potential adverse effects associated with inhibition of p38α/β MAP kinases. Curr Top Med Chem. 2005;5:929–939. doi: 10.2174/1568026054985911. [DOI] [PubMed] [Google Scholar]
- 9.Engel K, Kotlyarov A, Gaestel M. Leptomycin B-sensitive nuclear export of MAPKAP kinase 2 is regulated by phosphorylation. EMBO J. 1998;17:3363–3371. doi: 10.1093/emboj/17.12.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen HF, Xie LD, Xu CS. Role of heat shock protein 27 phosphorylation in migration of vascular smooth muscle cells. Mol Cell Biochem. 2009;327:1–6. doi: 10.1007/s11010-009-0034-4. [DOI] [PubMed] [Google Scholar]
- 11.Molnar P, Perrault R, Louis S, Zahradka P. The cyclic AMP response element-binding protein (CREB) mediates smooth muscle cell proliferation in response to angiotensin II. J Cell Commun Signal. 2014;8:29–37. doi: 10.1007/s12079-013-0215-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ward B, Seal BL, Brophy CM, Panitch A. Design of a bioactive cell-penetrating peptide: When a transduction domain does more than transduce. J Pept Sci. 2009;15:668–674. doi: 10.1002/psc.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayess K, Benndorf R. Effect of protein kinase inhibitors on activity of mammalian small heat-shock protein (HSP25) kinase. Biochem Pharmacol. 1997;53:1239–1247. doi: 10.1016/s0006-2952(96)00877-5. [DOI] [PubMed] [Google Scholar]
- 14.Lopes LB, Brophy CM, Flynn CR, Yi Z, Bowen BP, Smoke C, Seal B, Panitch A, Komalavilas P. A novel cell permeant peptide inhibitor of MAPKAP kinase II inhibits intimal hyperplasia in a human saphenous vein organ culture model. J Vasc Surg. 2010;52:1596–1607. doi: 10.1016/j.jvs.2010.06.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flynn CR, Cheung-Flynn J, Smoke CC, Lowry D, Roberson R, Sheller MR, Brophy CM. Internalization and intracellular trafficking of a PTD-conjugated anti-fibrotic peptide, AZX100, in human dermal keloid fibroblasts. J Pharm Sci. 2010;99:3100–3121. doi: 10.1002/jps.22087. [DOI] [PubMed] [Google Scholar]
- 16.Murthy N, Robichaud JR, Tirrell DA, Stayton PS, Hoffman AS. The design and synthesis of polymers for eukaryotic membrane disruption. J Control Release. 1999;61:137–143. doi: 10.1016/s0168-3659(99)00114-5. [DOI] [PubMed] [Google Scholar]
- 17.Foster S, Duvall CL, Crownover EF, Hoffman AS, Stayton PS. Intracellular delivery of a protein antigen with an endosomal-releasing polymer enhances CD8 T-cell production and prophylactic vaccine efficacy. Bioconjug Chem. 2010;21:2205–2212. doi: 10.1021/bc100204m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruttekolk IR, Witsenburg JJ, Glauner H, Bovee-Geurts PHM, Ferro ES, Verdurmen WPR, Brock R. The intracellular pharmacokinetics of terminally capped peptides. Mol Pharm. 2012;9:1077–1086. doi: 10.1021/mp200331g. [DOI] [PubMed] [Google Scholar]
- 19.Mislick KA, Baldeschwieler JD. Evidence for the role of proteoglycans in cation-mediated gene transfer. Proc Natl Acad Sci USA. 1996;93:12349–12354. doi: 10.1073/pnas.93.22.12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richard JP, Melikov K, Brooks H, Prevot P, Lebleu B, Chernomordik LV. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J Biol Chem. 2005;280:15300–15306. doi: 10.1074/jbc.M401604200. [DOI] [PubMed] [Google Scholar]
- 21.Nelson CE, Kintzing JR, Hanna A, Shannon JM, Gupta MK, Duvall CL. Balancing cationic and hydrophobic content of pegylated sirna polyplexes enhances endosome escape, stability, blood circulation time, and bioactivity in vivo. ACS Nano. 2013;7:8870–8880. doi: 10.1021/nn403325f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mietus-Snyder M, Friera A, Glass CK, Pitas RE. Regulation of scavenger receptor expression in smooth muscle cells by protein kinase C: A role for oxidative stress. Arterioscler Thromb Vasc Biol. 1997;17:969–978. doi: 10.1161/01.atv.17.5.969. [DOI] [PubMed] [Google Scholar]
- 23.Kalra M, Miller VM. Early remodeling of saphenous vein grafts: Proliferation, migration and apoptosis of adventitial and medial cells occur simultaneously with changes in graft diameter and blood flow. J Vasc Res. 2000;37:576–584. doi: 10.1159/000054091. [DOI] [PubMed] [Google Scholar]
- 24.Zwolak RM, Adams MC, Clowes AW. Kinetics of vein graft hyperplasia: Association with tangential stress. J Vasc Surg. 1987;5:126–136. [PubMed] [Google Scholar]
- 25.Muto A, Panitch A, Kim N, Park K, Komalavilas P, Brophy CM, Dardik A. Inhibition of mitogen activated protein kinase activated protein kinase II with MMI-0100 reduces intimal hyperplasia ex vivo and in vivo. Vascul Pharmacol. 2012;56:47–55. doi: 10.1016/j.vph.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duvall CL, Convertine AJ, Benoit DS, Hoffman AS, Stayton PS. Intracellular delivery of a proapoptotic peptide via conjugation to a RAFT synthesized endosomolytic polymer. Mol Pharm. 2010;7:468–476. doi: 10.1021/mp9002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee GL, Chang YW, Wu JY, Wu ML, Wu KK, Yet SF, Kuo CC. TLR 2 induces vascular smooth muscle cell migration through cAMP response element-binding protein-mediated interleukin-6 production. Arterioscler Thromb Vasc Biol. 2012;32:2751–2760. doi: 10.1161/ATVBAHA.112.300302. [DOI] [PubMed] [Google Scholar]
- 28.Nakanishi K, Saito Y, Azuma N, Sasajima T. Cyclic adenosine monophosphate response-element binding protein activation by mitogen-activated protein kinase-activated protein kinase 3 and four-and-a-half LIM domains 5 plays a key role for vein graft intimal hyperplasia. J Vasc Surg. 2013;57:182–93. doi: 10.1016/j.jvs.2012.06.082. [DOI] [PubMed] [Google Scholar]
- 29.McLemore EC, Tessier DJ, Thresher J, Komalavilas P, Brophy CM. Role of the small heat shock proteins in regulating vascular smooth muscle tone. J Am Coll Surg. 2005;201:30–36. doi: 10.1016/j.jamcollsurg.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 30.Motwani JG, Topol EJ. Aortocoronary saphenous vein graft disease: Pathogenesis, predisposition, and prevention. Circulation. 1998;97:916–931. doi: 10.1161/01.cir.97.9.916. [DOI] [PubMed] [Google Scholar]
- 31.Zhang L, Freedman NJ, Brian L, Peppel K. Graft-extrinsic cells predominate in vein graft arterialization. Arterioscler Thromb Vasc Biol. 2004;24:470–476. doi: 10.1161/01.ATV.0000116865.98067.31. [DOI] [PubMed] [Google Scholar]
- 32.Cooley BC, Nevado J, Mellad J, Yang D, Hilaire CSt, Negro A, Fang F, Chen GB, San H, Walts AD, Schwartzbeck RL, Taylor B, Lanzer JD, Wragg A, Elagha A, Beltran LE, Berry C, Feil R, Virmani R, Ladich E, Kovacic JC, Boehm M. TGF-β signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci Transl Med. 2014;6:227ra34. doi: 10.1126/scitranslmed.3006927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kapopara PR, von Felden J, Soehnlein O, Wang Y, Napp LC, Sonnenschein K, Wollert KC, Schieffer B, Gaestel M, Bauersachs J, Bavendiek U. Deficiency of MAPK-activated protein kinase 2 (MK2) prevents adverse remodelling and promotes endothelial healing after arterial injury. Thromb Haemost. 2014;112:1264–1276. doi: 10.1160/TH14-02-0174. [DOI] [PubMed] [Google Scholar]
- 34.Jiang Z, Wu L, Miller BL, Goldman DR, Fernandez CM, Abouhamze ZS, Ozaki CK, Berceli SA. A novel vein graft model: Adaptation to differential flow environments. Am J Physiol Heart Circ Physiol. 2004;286:H240–H245. doi: 10.1152/ajpheart.00760.2003. [DOI] [PubMed] [Google Scholar]
- 35.Ruifrok AC, Johnston DA. Quantification of histochemical staining by color deconvolution. Anal Quant Cytol Histol. 2001;23:291–299. [PubMed] [Google Scholar]
- 36.Ferrito MS, Tirrell DA. Macromolecular Syntheses. Vol. 11. Wiley; New York: 1992. Poly(2-ethylacrylic acid) pp. 59–62. [Google Scholar]
- 37.Convertine AJ, Benoit DSW, Duvall CL, Hoffman AS, Stayton PS. Development of a novel endosomolytic diblock copolymer for siRNA delivery. J Control Release. 2009;133:221–229. doi: 10.1016/j.jconrel.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans BC, Nelson CE, Yu SS, Beavers KR, Kim AJ, Li H, Nelson HM, Giorgio TD, Duvall CL. Ex vivo red blood cell hemolysis assay for the evaluation of pH-responsive endosomolytic agents for cytosolic delivery of biomacromolecular drugs. J Vis Exp. 2013;73:e50166. doi: 10.3791/50166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.