

Summary
Eukaryotic mRNAs generally possess a 5′-end m7G cap that promotes their translation and stability. However, mammalian mRNAs can also carry a 5′-end nicotinamide adenine dinucleotide (NAD+) cap that, in contrast to the m7G cap, does not support translation but instead promotes mRNA decay. The mammalian and fungal noncanonical DXO/Rai1 decapping enzymes efficiently remove NAD+ caps and cocrystal structures of DXO/Rai1 with 3′-NADP+ illuminates the molecular mechanism for how the “deNADding” reaction produces NAD+ and 5′-phosphate RNA. Removal of DXO from cells increases NAD+-capped mRNA levels and enables detection of NAD+-capped intronic snoRNAs, suggesting NAD+ caps can be added to 5′-processed termini. Our findings establish NAD+ as an alternative mammalian RNA cap and DXO as a deNADding enzyme modulating cellular levels of NAD+-capped RNAs. Collectively, these data reveal mammalian RNAs can harbor a 5′-end modification distinct from the classical m7G cap that promotes, rather than inhibits, RNA decay.
Graphical abstract
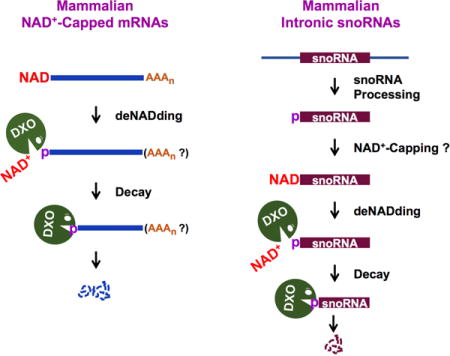
Introduction
The 5′ ends of eukaryotic mRNAs are cotranscriptionally modified by the addition of N7 methyl guanosine (m7G) linked to the first encoded nucleotide by a 5′-5′ linkage (Muthukrishnan et al., 1975; Shatkin, 1976; Wei et al., 1975b). The m7G cap fulfills multiple functions including nucleo-cytoplasmic transport, serving as an assembly platform for the cytoplasmic translation initiation complex to facilitate translation (Sonenberg et al., 1979) and protecting the 5′ end from 5′-3′ exonuclease decay (Furuichi et al., 1977; Hsu and Stevens, 1993; Sachs, 1993). Several derivatives of the canonical m7G cap have also been reported including a class of small U-rich noncoding RNAs that are further processed by the addition of two methyl moieties to generate a trimethylated, m2,2,7G cap (Mattaj, 1986). Modifications can also occur within the mRNA and constitute an epitranscriptomic level of gene regulation. For example, if the first nucleotide is an adenosine, it can be methylated at the N6 position to generate a cap with an N6 methyladenosine (m6A) (Linder et al., 2015; Wei et al., 1975a), which impact stability by curtailing Dcp2 decapping activity (Mauer et al., 2017). Collectively, the canonical m7G caps along with the modified m7G cap derivatives confer a layer of regulatory information to the 5′ ends of eukaryotic RNAs.
Although it had long been presumed RNA capping occurs only in eukaryotes, it was recently shown that certain RNA products in bacteria harbor a 5′ end NAD+ modification (Cahova et al., 2015; Chen et al., 2009). The 5′ end NAD+ can protect the RNA from 5′ end decay by the bacterial RppH and RNaseE nucleases in vitro (Cahova et al., 2015). The prokaryotic Nudix family hydrolase protein, NudC, hydrolyzes the 5′ end NAD+ to release Np + pA-RNA (where Np is nicotinamide mononucleotide), indicating that NudC can selectively initiate the decay of NAD+ containing RNAs in bacteria.
In contrast to m7G caps, which are added to RNA 5′ ends cotranscriptionally when the RNA reaches a length of ~25-nt, NAD+ caps can be added to RNA 5′ ends in bacteria during transcription initiation, through the use of NAD+ in place of ATP as an initiating nucleotide by bacterial RNA polymerase (Bird et al., 2016). Furthermore, it has been shown that eukaryotic RNAP II can also use NAD+ as an initiating nucleotide in vitro (Bird et al., 2016), raising the possibility that NAD+ caps are added to RNA 5′ ends in mammalian cells. In addition, the presence of multiple eukaryotic hydrolases capable of removing the 5′ end cap from an RNA (Grudzien-Nogalska and Kiledjian, 2016), suggests eukaryotic cells may harbor an enzyme that can remove a 5′ end NAD+ cap from an RNA.
In this report, we demonstrate that HEK293T cells contain mRNAs and small noncoding RNAs capped with a 5′-end NAD+ and that NAD+-capping of RNA plays a functional role in mammalian cells. We further establish that the DXO protein, previously implicated in the removal of incomplete 5′ cap structures (Grudzien-Nogalska and Kiledjian, 2016), plays a critical role in the turnover of NAD+-capped RNAs in mammalian cells.
Results
Mammalian Cells Contain NAD+-Capped mRNA
To determine whether mammalian cells contain NAD+-capped mRNAs, we utilized the NAD-CaptureSeq approach which uses click chemistry to selectively covalently link biotin to a 5′ NAD+ in the presence of adenosine diphosphate-ribosylcyclase (ADPRC) (Cahova et al., 2015). Total RNA from HEK293T cells was subjected to NAD+ capture in the presence or absence of ADPRC and biotinylated RNAs were isolated by streptavidin beads. The selectivity of this approach for NAD+-capped RNAs is shown in Figure 1A with the inclusion of 32P-labeled NAD+-capped or m7G-capped RNAs spiked into the reaction. The identity of the isolated cellular NAD+-capped RNAs was determined by high throughput sequencing of the generated double stranded cDNA.
Figure 1. Mammalian cells contain NAD+-capped mRNA.
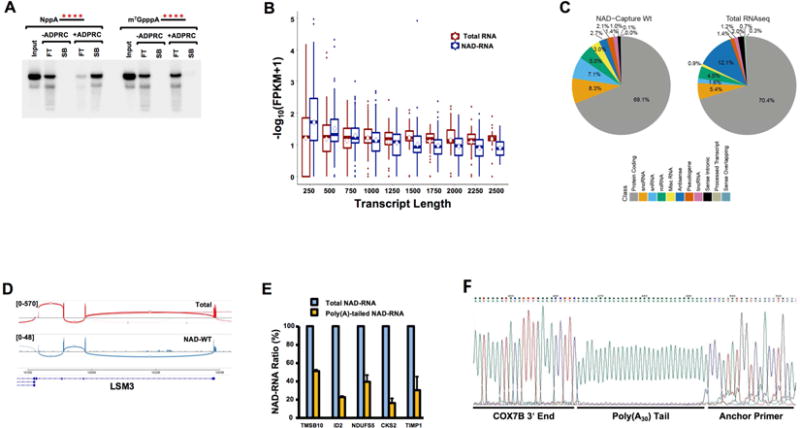
(A) Selectivity of NAD-capture approach. NAD-RNA capture was carried out with 100μg HEK293T total RNA in the presence of 32P-uniform-labeled 20pm NAD+-capped or m7GpppA-capped RNA with or without ADPRC treatment as indicated. RNA bound to magnet streptavidin beads (SB) or in the flow through (FT) were isolated resolved on 8% Denaturing Urea PAGE. The asterisks represent the positions of the 32P-labeling. Quantitation of the spiked RNA from two independent experiments revealed 1% of input NAD+-capped RNA was retained on the affinity matrix under –ADPRC conditions while 89% with ADPRC. For m7G capped RNA, 1% was retained with –ADPRC while 2% was detected with ADPRC. (B) Analysis of NAD-capture RNA-Seq FPKM distribution of assembled transcript lengths by sample. Calculated transcript length was obtained from Cufflinks output. FPKM values were summarized up to the length indicated using box plots, with the center line indicating median, the top and bottom of the box indicating 75th and 25th percentiles, and the mean by the diamond-shaped symbol. Total RNA: Ribosome-minus HEK293 RNA sample processed for RNA-seq by standard methods; NAD-RNA: NAD-capture RNA prepared from HEK293 cells. (C) Sequencing reads were intersected with a general genome map (GenCode, hg19 genome). The number of features overlapping by at least 50% of length is displayed by RNA class (color key) for RNAP II transcripts. (D) Aligned sequencing reads were visualized as a Sashimi plot using the Integrative Genomics Viewer (Broad Institute) where the density of reads are denoted above LSM3 gene. Boxes on the bottom represent exons and the curved lines represent adjoining exon-exon reads. (E) The percentage of the indicated NAD+-capped mRNAs that harbor a poly(A) tail relative to its corresponding total NAD+ capped mRNA is shown. Total NAD+ capped mRNA was designated as 100. Error bars represent ±SD. (F) Sequence read of COX7B 3′ end containing 30 adenosine poly(A) tail is shown. Sequencing of 5 indiviual clones revealed a range of poly(A) tails spanning from 18 to 51 adenosines.
Enriched NAD+-capped transcripts were compared with standard, ribosomal-depleted total RNA-Seq samples (labeled as “Total RNA”) prepared from the same cell type. To evaluate transcripts assembled de novo from NAD+-capture RNA-Seq (NAD-CaptureSeq) reads, Tophat-aligned reads merged from all three replicates were assembled using Cufflinks without a reference gene transfer format (GTF) transcript map. The resulting transcript maps from the NAD+-capture samples were then merged with a similar map created from traditional RNA-Seq using cuffMerge. Cufflinks was repeated to assign normalized expression levels in FPKM (read fragments per kilobase of transcript length per million reads in each sample) to each assembled transcript. Analysis of the sequencing data revealed a pattern of NAD+-capped transcripts that overlap capped transcripts. Starting with the map of Cufflinks-assembled transcripts, NAD+-capped transcripts had overall comparable distribution of RNAs with a slightly higher relative FPKM level than Total RNA at smaller transcript lengths and reduced FPKM levels than Total RNA at longer transcript lengths (Figure 1B). The reduced density reads towards the 3′ end are expected due to the intrinsic 5′ bias of the NAD-CaptureSeq approach which purifies RNAs through an intact 5′ end irrespective of the 3′ end.
Comparison of the overall NAD+-capped reads to that of Total reads, primarily consisting of m7G-capped RNAs, revealed extensive overlap between the two populations. Aligned reads were fit to a reference transcript model (UCSC hg19) to more appropriately interpret overlapping gene identity and predicted function. Many reads overlapped various classes of noncoding RNAs and these tended to be over-represented in FPKM due to their short lengths. Analyzing only protein-coding reference transcripts with a detection cut-off of 1 FPKM 19,451 transcripts were identified out of 41,426 present in the reference transcript map (47.0%), compared with 16,018 transcripts detected in the same cell type by traditional RNA-Seq (38.73%). A large proportion of these overlap with Total RNA (p<0.001, Fisher’s Exact Test). The higher number of NAD+ capped RNAs detected is likely influenced by the greater quantity of RNA required for the NAD-Capture approach and consequent enrichment of low abundance RNAs. We conclude that a large number of cellular transcripts are enriched by NAD+-Capture.
To assess distinct populations of mRNAs that are NAD+ capped, aligned sequencing reads were classified according to overlap with GenCode annotations (v.19), focusing on Pol II-specific transcripts (Figure 1C). A similar proportion overlapped traditional protein-coding sequences (69% for NAD-Capture RNA; 70.4% for Total RNA), but there were substantial differences in other classifications. For example, both small nuclear RNAs (snRNAs) and small nucleolar RNAs (snoRNAs) were proportionally increased in NAD-Capture RNA. Conversely, the fraction of antisense RNA was diminished in NAD-Capture compared with Total RNA. Collectively, these data demonstrate the presence of NAD+ capped RNAs in mammalian cells and suggest that specific transcripts are preferentially NAD+-capped.
The role of the m7G cap in mRNA splicing (Edery and Sonenberg, 1985; Konarska et al., 1984) and polyadenylation (Cooke and Alwine, 1996; Gilmartin et al., 1988; Hart et al., 1985), prompted us to analyze whether NAD+ capped RNAs are spliced and polyadenylated. Surprisingly NAD+-capped transcripts with multiple exons were detected. Alignment of raw sequencing read fitting of representative mRNAs are depicted in a standard Sashimi plot (Figure 1D and S1A) where sequencing reads that span exon-exon junctions are indicated by a curved line connecting the exon plots. Observed splicing patterns were similar between Total and NAD+-capped RNAs. To determine whether NAD+ capped RNAs can be polyadenylated, RNA were reverse transcribed with oligo(dT) and the 3′ ends amplified with gene specific primers and presented relative to the corresponding total level of each respective RNA. All oligo(dT) reverse transcribed RNAs tested, demonstrated the presence of a poly(A) tail and consistent with the 5′ polarity of NAD+-capped mRNA reads, 20% to 50% of the NAD+-capped mRNAs contain a polyadenylated tail (Figure 1E). Cloning and sequencing of the 3′ ends of one representative mRNA using anchored oligo(dT) amplification, confirmed the presence of a poly(A) tail on this NAD+-capped mRNA and its addition at the endogenous polyadenylation addition site (Figure 1F and S1B). We conclude, NAD+-capped mRNAs can be spliced and polyadenylated.
Mammalian DXO deNADs NAD+-capped RNA in vitro
In bacteria, the 5′ end NAD+ can protect the RNA from 5′ end decay by RppH and RNaseE nucleases in vitro (Cahova et al., 2015). The prokaryotic Nudix family hydrolase protein, NudC, selectively initiates the decay of NAD+-capped RNAs in bacteria by hydrolyzing the 5′ end NAD+ to release Np + pA-RNA (Bird et al., 2016; Cahova et al., 2015). The presence of NAD+-capped RNA in mammalian cells (Figure 1) suggests eukaryotic cells likely harbor an enzyme that can remove a NAD+ cap from an RNA.
Eukaryotes possess multiple hydrolases capable of removing 5′ end caps from an RNA (reviewed in (Grudzien-Nogalska and Kiledjian, 2016)). Among these hydrolases, the DXO family of proteins, which consist of fungal Rai1 and Dxo1 and mammalian DXO, hydrolyze incompletely capped mRNAs in a quality control mechanism to maintain 5′ end integrity (Grudzien-Nogalska and Kiledjian, 2016). All DXO family members characterized biochemically remove the entire cap structure by hydrolyzing the phosphodiester linkage between the first and second encoded nucleotides of an unmethylated capped RNA converting GpppN-RNA to GpppN + pRNA (Chang et al., 2012; Jiao et al., 2013; Jiao et al., 2010).
We reasoned that analogous to removal of the entire cap structure, DXO might also remove an NAD+ modification on the 5′ end of RNAs. To test this possibility, 32P-NAD+ cap-labeled RNA with the 32P as the second phosphate (Npp*A-RNA) was generated by in vitro transcription. Mouse DXO was incubated with m7G-capped RNA and NAD+-capped RNA, and the reaction products resolved by thin layer chromatography (TLC). As expected, DXO hydrolyzed the m7G capped RNA releasing the cap structure product (Figure 2A and (Jiao et al., 2013)). Surprisingly DXO exhibited robust preferential activity on NAD+-capped RNA with an approximate 6-fold greater efficiency releasing NAD+ (NppA) and pRNA (Figure 2A and 2B with quantitation in 2C and product confirmation in Figure S2). The lack of a 3′ phosphate on the NppA also indicates that the resulting RNA contains a 5′ monophosphate whereby NppA-RNA is hydrolyzed to NppA + pRNA. This activity was compromised by the E234A or K255Q catalytic site mutant (Figure 2D) demonstrating that the observed hydrolysis was from DXO and utilizes the same active site for both activities. The unexpected preferential activity of DXO to remove NAD+ from the 5′ end of an RNA relative to its previously characterized role on capped RNA indicates an additional function for DXO in the removal of NAD+ in a process we will refer to as “deNADding”.
Figure 2. Mammalian DXO possesses robust deNADding activity.
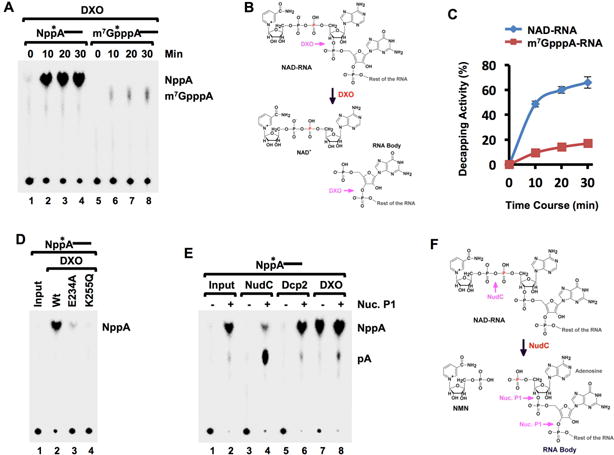
(A) In vitro decapping assays with 10nM mouse recombinant DXO protein and indicated 32P-cap-labeled RNA substrates. Reaction products were resolved by polyethyleneimine (PEI)-cellulose thin-layer chromatography (TLC) developed in 0.45 M (NH4)2SO4. (B) Schematic of NAD+-capped RNA and sites of DXO cleavage. The red “P” denotes the position of the 32P. (C) Quantitation from (A) carried out with ImageQuant software and plotted from three independent experiments with the error bars representing +/−SD. (D) Catalytically inactive mutants E234A and K256Q abolish DXO deNADding activity. (E) Mammalian Dcp2 does not possess intrinsic deNADding activity. Recombinant (50nM) human Dcp2, mouse DXO or bacteria NudC proteins were incubated with 32P-cap-labeled NAD-RNA at 37°C for 30 minutes. The reaction products were either untreated (−) or treated with 1U Nuclease P1 (+), resolved by PEI-TLC developed in 0.45 M (NH4)2SO4. Product standards were denoted on the right. The asterisks represent the position of the 32P-labeling. (F) Similar to B, except sites of NudC and Nuclease P1 (Nuc. P1) hydrolysis are denoted.
deNADding is not an activity shared by all decapping enzymes and NAD+-capped RNA was refractory to the Dcp2 decapping enzyme. A comparable NAD+ product resulting from hydrolysis of the phosphodiester linkage to the first encoded nucleotide was not observed with Dcp2 (Figure 2E). However, since the second phosphate within the NAD+ (Npp*A-RNA) is labeled, potential deNADding activity of Dcp2 within the diphosphate of NAD+ would release unlabeled nicotinamide mononucleotide (NMN; Np) and not be detected with this substrate (Figure 2F). To determine whether Dcp2 cleaved within the NAD+ diphosphate linkage, the reaction products were treated with nuclease P1, which cleaves all phosphodiester bonds within an RNA and would release the labeled p*A (Figure 2F). As expected, bacterial NudC treatment of Npp*A-RNA did not generate a detectable product in the absence of nuclease P1, but did generate adenosine monophosphate (p*A) in its presence (Figure 2E). However, Dcp2 failed to generate detectable products with either reaction condition demonstrating that the Dcp2 decapping enzyme is specific for m7G-capped RNA and does not function on NAD+-capped RNA (note that NppA is expected to be produced by nuclease P1 digestion even in the absence of Dcp2). Collectively, our data demonstrate that mouse DXO possess deNADding activity in vitro capable of removing 5′ end NAD+ from a NAD+-capped RNA to generate a 5′ end monophosphorylated mRNA susceptible to 5′-3′ exonucleolytic decay. Since DXO also contains 5′-3′ exonucleolytic activity (Jiao et al., 2013), it is plausible that DXO degrades the RNA body following deNADding.
Mammalian DXO deNADs NAD+-capped RNA in vivo
To test the deNADding role of DXO in mammalian cells, we asked whether NAD+-capped RNAs are more stable in the absence of DXO. RNA capped at its 5′ end with 32P-NAD+ and containing 16 consecutive guanosine nucleosides (G16) at the 3′ end to minimize 3′ end decay (Wang and Kiledjian, 2001) was used. The 32P-NAD+-capped RNA was transfected into either wild type (WT) cells, a pool of three different monoclonal HEK293T CRISPR directed homozygous DXO gene disrupted cells (DXO-KO; Figure S3A), or a pool of three different Dcp2 disrupted homozygous monoclonal lines (Dcp2-KO; (Mauer et al., 2017)). As shown in Figure 3A, transfected 32P-NAD+-capped RNA was more stable in the DXO-KO cells compared to WT cells with a half-life 6.3 hours and 2.8 hours respectively. Similarly, the NAD+-capped RNA was more stable in the DXO-KO cells when uniformly labeled RNA was used (Figure 3B). Consistent with the inability of Dcp2 to hydrolyze NAD+-capped RNA in vitro (Figure 2E), altered stability of NAD+-capped RNA was not observed in Dcp2-KO cells and half-lives in these cells were indistinguishable from control cells (Figures 3A and 3B). In contrast to results obtained with NAD+-capped RNA, m7G-capped RNA exhibited comparable stabilities in WT cells, DXO-KO cells, and Dcp2-KO cells. The results indicate that DXO modulates the stability of NAD+-capped RNA but not m7G-capped RNA in HEK293T cells.
Figure 3. DXO is a deNADding enzyme in cells and NAD-capped RNAs do not support translation.
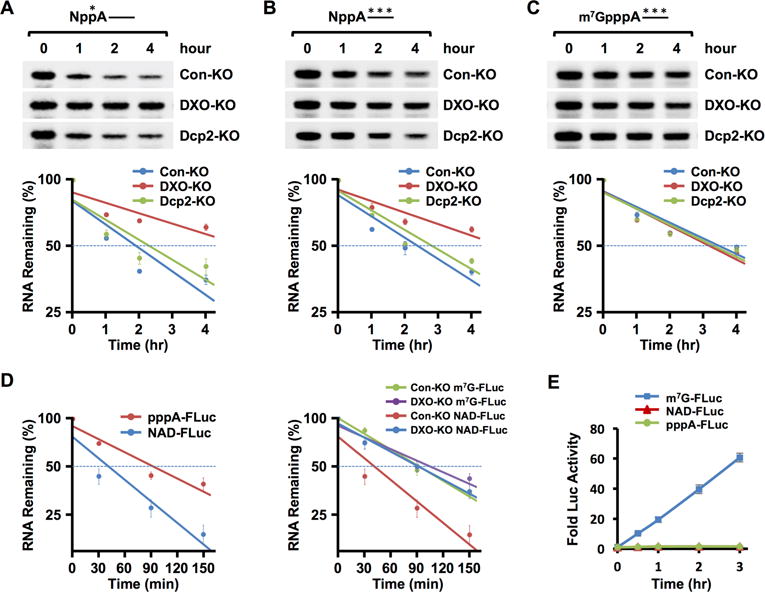
(A) 32P-NAD+ cap-labeled RNA was transfected into HEK293T control knock-out (Con-KO), DXO knock-out (DXO-KO) or Dcp2 knock-out (Dcp2-KO) cell lines with Lipofectamine 3000. Untransfected RNAs were degraded with micrococcal nuclease (MN) and total RNA was isolated at the indicated time points following MN treatment and resolved on 8% Denaturing PAGE and exposed to a PhosphorImager. RNA remaining was quantitated and plotted from three independent experiments with ±SD denoted by the error bars. Stabilities: Con-KO t1/2=2.8 hr [95%CI: 2.0 – 4.5]; DXO-KO t1/2=6.3 hr [95%CI: 4.2 – 11.9] (ANOVA, p=6.10 × 10−4); Dcp2-KO t1/2=3.2 hr, [95%CI: 2.3 – 5.8] (ANOVA, p=0.38). (B) Same as A except the NAD+-capped RNA was uniformly labeled 32P. Stabilities: Con-KO t1/2=3.0 hr [95%CI: 2.4 – 4.3]; DXO-KO t1/2=5.7 hr [95%CI: 4.1 – 8.9] (ANOVA, p=7.88 × 10−4); Dcp2-KO t1/2=3.3 hr, [95%CI: 2.7 – 4.4] (ANOVA, p=0.17). (C) Same as A except the RNA contained an m7G cap and was uniformly labeled within the RNA body with 32P. Stabilities: Con-KO t1/2=4.1 hr [95%CI: 3.2 – 5.8]; DXO-KO t1/2=3.9 hr [95%CI: 3.0 – 5.4] (ANOVA, p=0.51); Dcp2-KO t1/2=4.1 hr, [95%CI: 3.1 – 5.9] (ANOVA, p=0.64). (D). Luciferase RNAs with a 5′-end triphosphate lacking a cap, 5′-end NAD+ cap or m7G cap transfected into control or DXO-KO cells. Cell were treated with micrococcal nuclease to degrade untransfected RNA, harvested at the indicated times and remaining RNA levels quantitated from three independent experiments. The left panel shows RNAs transfections into control HEK293T (Con-KO) cells and the right panel presents RNAs introduced into either Con-KO or DXO-KO cells as indicated. Error bars denoted ±SD. Con-KO m7G-FLuc t1/2= 92.6 min (95% CI: 82.2 to 107.8 min, R2=0.96, p=2.10 × 10−8); DXO-KO m7G-FLuc t1/2=125 min (95% CI: 92.2 to 174 min, R2=0.84, p=1.47 × 10−5); Con-KO NAD-FLuc t1/2= 67.4 min (95% CI: 51.4 to 97.9 min, R2=0.82, p=3.05 × 10−5); DXO-KO NAD-FLuc t1/2= 103.6 min (95% CI: 85.2 to 132.3 min, R2=0.91, p=1.22 × 10−6); Con-KO pppA-RNA=110.7 min (95% CI: 88 to 149.2 min, R2=0.870, p=5.99 × 10−5). (E). Firefly luciferase mRNAs containing either a 5′-end NAD+ cap, m7G cap or no cap and 3′ poly(A)60 tail were co-transfected with m7G-capped Renilla Luciferase RNA into HEK293T cells. Cells were harvested and assayed at the indicated time points. Firefly Luciferase activity was plotted normalized to Renilla Luciferase and data from three independent experiments are presented with error bars representing +/−SD.
To directly test the stability of a NAD+-capped protein-coding mRNA, in vitro transcribed Firefly Luciferase mRNA containing a 5′ NAD+ cap, a 5′ m7G cap or 5′ triphosphate RNA lacking a cap, with 3′ end poly(A60) tails were individually transfected into WT or DXO-KO cells and mRNA isolated at increasing time points. Two important observations can be ascertained from these experiments. First, NAD+-capped luciferase mRNA is less stable than the same uncapped mRNA (Figure 3D, left panel; t1/2 =1.12 hr, 1.85 hr, respectively; p=5.06 × 10−4, ANOVA comparison) indicating the NAD+ cap promotes RNA decay. Second, the NAD+-capped luciferase mRNA was more stable in DXO-KO cells relative to control cells (Figure 3D, right panel; t1/2 =1.72 hours versus 1.12 hours respectively; p=2.8 × 10−4, ANOVA comparison) and this stability was comparable to that of the m7G-capped luciferase mRNA (t1/2 =2.08 hours). Importantly, a significant difference was not detected with m7G-capped RNA in control and DXO-KO cells (p=0.69). Collectively, these data demonstrate a NAD+ cap facilitates mRNA instability in WT cells and DXO is necessary for this rapid decay in cells.
NAD+-capped RNA does not support mRNA translation
The m7G-cap at the 5′ end of mRNAs is a major determinant of the translatability of an mRNA (Filipowicz, 1978). We next tested the capacity for an NAD+ cap to support mRNA translation. 5′ NAD+ capped or 5′ m7G capped Firefly luciferase mRNAs from above were individually cotransfected with a control m7G-capped Renilla luciferase into HEK293T cells. Extract was isolated at increasing time points and dual luciferase activities were determined as a function of the Renilla luciferase internal control. As expected an increase of Firefly luciferase over the 3-hour time course of the assay was detected from the m7G-capped RNA (Figure 3E). However, background levels of luciferase activity were detected from the NAD+-capped RNA throughout the duration of the experiment that were indistinguishable from that of uncapped Firefly luciferase mRNA (Figure 3E). Importantly, luciferase activity was also not detected from transfected NAD+ capped mRNA in DXO-KO cells (Figure S3B) where the stability of the NAD+ capped and m7G capped RNAs were comparable (Figure 3D), indicating the lack of detected firefly luciferase activity was not due to differential mRNA stability. We conclude that NAD+-capped mRNA are inefficiently, if at all, translated in cells.
DXO preferentially targets a subset of mRNAs for deNADding
To directly determine whether DXO contributes to the expression of NAD+-capped RNA, NAD-CaptureSeq was carried out with RNA from DXO-KO or WT HEK293T cells (n=3/group). Selecting transcripts with at least a 2-fold increase in DXO-KO compared with WT, at least 1 FPKM expression level in DXO-KO and ≤5% FDR revealed 539 NAD+-capped RNAs that were selectively enriched in the DXO-KO cells (Fig 4A, black dots; Table S1) suggesting these are targets of DXO deNADding. To assess the role of DXO on NAD+-capped mRNA in cells, a subset of mRNA were directly tested by quantitative RT (qRT)-PCR. Analysis of 12 randomly picked mRNAs that were predicted to either increase or not change, revealed two important findings. First, eight of the 10 NAD+ capped mRNAs identified from the NAD-CaptureSeq, were confirmed by qRT-PCR as significantly elevated in the NAD+-capped population of WT cells relative to the negative control (Figure 4B), further supporting the presence of endogenous NAD+-capped mRNAs in mammalian cells and validating the NAD-Capture results of Figure 1. Second, all 11 mRNAs predicted to be elevated in DXO-KO cells by NAD-CaptureSeq, were confirmed to be significantly higher in the DXO-KO cells (Figure 4B). In contrast, MRPL13 mRNA which was not expected to be NAD+ capped in either WT of DXO-KO cells, was indistinguishable from background controls. These data are consistent with a subset of mRNAs being NAD+ capped and subjected to either direct or indirect DXO-mediated decay in mammalian cells. Interestingly, the presence of a NAD+ 5′ end did not perturb RNA localization. Individual NAD+ capped RNAs tested contained a similar nuclear and cytoplasmic distribution as total cellular RNA (Figure S4).
Figure 4. DXO is a deNADding enzyme in cells.
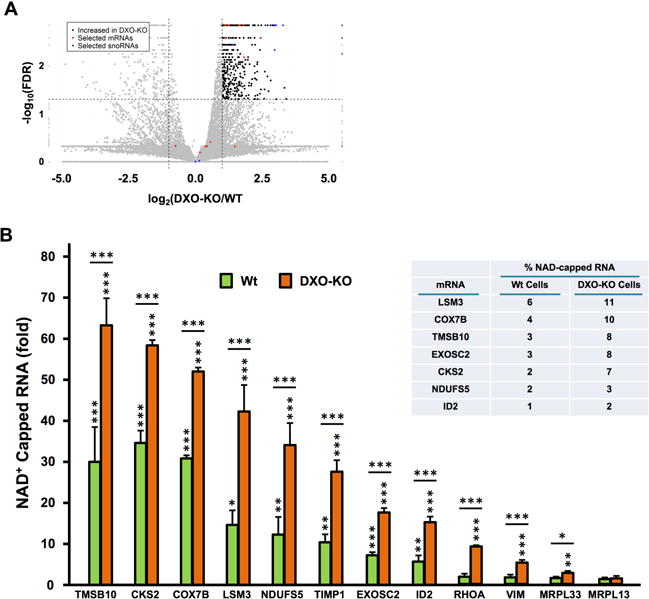
(A) Volcano plot of NAD-Capture transcripts comparing WT with DXO-KO samples. The log2 fold change (DXO-KO/WT) is plotted vs. the –log10 FDR (False Discovery Rate; corrected for multiple measurements). Each transcript is indicated as a dot on the plot. Dots above the horizontal dashed line are ≤ 5% FDR. The vertical dashed lines indicate ±2-fold differences, so dots to the right of the +2-fold line are colored black (only if they are detected with at least 1 FPKM). Red dots indicate the mRNAs assessed by qPCR in panel B and blue dots indicate snoRNAs assessed by qPCR in Fig. 5A. (B) qRT-PCR validation of NAD+ capped mRNAs in DXO-KO cells. Randomly selected NAD+-capped RNAs from the NAD-CaptureSeq were eluted from the beads, reverse transcribed and detected with gene specific primers. Data are presented relative to the -ADPRC negative control set to 1. MRPL13, which is not responsive to DXO in the NAD CaptureSeq, was included as a negative control. The inserted table represents levels of NAD+ capped RNAs relative to their respective total mRNA in the indicated cells. Error bars represent ±SD. P values are denoted by asterisks; (*) P < 0.05, (**) P < 0.01, (***) P < 0.001 (Student’s t-test).
To deduce the percentage of a given mRNA population that can be NAD+-capped, levels of NAD+-capped mRNAs retained by NAD-Capture relative to their total mRNA levels was quantitated. A range from 1% to 6% of a given mRNA was NAD+-capped in WT cells. As expected, the range of levels approximately doubled (2% to 11%) in DXO-KO cells (Figure 4B; insert). These data demonstrate DXO can influence the levels of a subset of NAD+ capped mRNAs in cells and the percentage of any given mRNA with an NAD+ cap can range at least up to 11% in cells.
DXO preferentially targets a subset of snoRNAs and scaRNAs for deNADding
In addition to mRNAs, a significantly disproportionate abundance of NAD+-capped small nucleolar RNAs (snoRNAs) and small Cajal body RNAs (scaRNAs) were elevated in the DXO-KO cells. Out of 113 H/ACA box snoRNAs, 16 are significantly increased in DXO-KO (p=9.02 × 10−20, hypergeometric distribution). Similarly, of the 24 scaRNAs, 9 increased in DXO-KO (p=7.65 × 10−7). These noncoding RNAs constituted 11 out of the top 17 most differentially elevated transcripts in DXO-KO cells (Figure 4A, blue dots), indicating they are DXO targets that are cleared in WT cells. SnoRNAs and scaRNAs function as guide RNAs in the pseudouridylation and methylation of rRNAs and U-rich small nuclear RNAs, respectively (Dieci et al., 2009; Filipowicz and Pogacic, 2002). Nine randomly chosen sno/scaRNAs indicated to be NAD+ capped (Figure 4A) were confirmed to be elevated in DXO-KO cells by qRT-PCR while two that lack an NAD+ cap were not (Figure 5A). The significant increase of a subset of NAD+-capped sno/scaRNAs in the absence of DXO, indicates that similar to NAD+ capped mRNAs, the presence of an NAD+ cap on a sno/scaRNA promotes its decay by a DXO dependent mechanism.
Figure 5. Small nucleolar and small Cajal body RNAs can be NAD+ capped and their levels are dependent on DXO in cells.
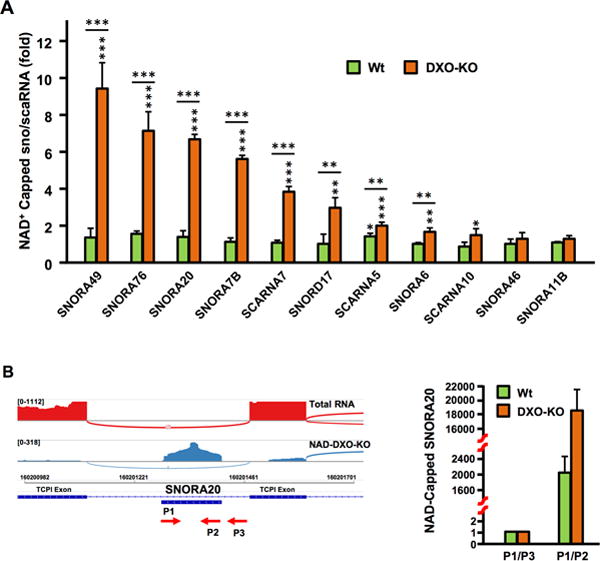
(A) A subset of NAD+ capped sno/scaRNAs were validated by qRT-PCR. NAD+-capped RNAs isolated by the NAD-Capture approach were eluted from the beads, reverse transcribed and the indicated sno/scaRNAs detected with gene specific primers. Data are presented relative to the -ADPRC negative control set to 1. SNORA46 and SNORA11B, which were not responsive to DXO in the NAD CaptureSeq, were also tested. Error bars represent ±SD. P values are denoted by asterisks; (*) P < 0.05, (**) P < 0.01, (***) P < 0.001 (Student’s t-test). (B) Sashimi plot with SNORA20 within the TCP1 intron 9 is shown. Labeling is as in Figure 1E. Detected SNORA20 reads are within the mature snoRNA. The P1 – P3 primers used to amplify SNORA20 or the SNORA20-intron junction are shown and qRT-PCR results graphed on the right.
Unlike, Archaea, yeast and plants, mammalian sno/scaRNAs are predominantly intronic and generated as exonucleolytic-resistant ribonucleoprotein complexes not known to possess a modification at their 5′ end (Dieci et al., 2009; Filipowicz and Pogacic, 2002; Kawaji et al., 2008). Although an NAD+-capped mRNA can be generated during transcription initiation by the use of NAD+ rather than ATP as an initiating nucleotide (Bird et al., 2016), addition of a NAD+ cap to an exonucleolytically processed sno/scaRNAs would require NAD+ capping to occur at a stage after transcription initiation. Furthermore, addition of the NAD+ cap onto these 5′-end monophosphate RNAs (Kawaji et al., 2008), would require a NAD+-capping mechanism. Shashimi plots of representative snoRNAs demonstrated the resulting reads were contained within the snoRNA genes and suggest they were not derived from a cryptic internal promoter or from stable spliced intronic sequences (Figure 5B and Figure S5). Quantitative RT-PCR of NAD+-capped RNAs with primers exclusively within SNORA20 or spanning the intron junction revealed NAD+-capped SNORA20 was restricted to the mature RNA and lacked adjoining intronic sequences. Moreover, only 26 out of 431 sno/scaRNAs are significantly increased in DXO-KO by NAD-Capture (Figure 4A) supporting the selectivity of NAD+ capping. Importantly, the snoRNAs are not indirectly retained onto the NAD-capture matrix through association with their target rRNA. In contrast to the elevation of NAD+-capped snoRNAs from DXO-KO cells (Figure 5A), levels of their 18S rRNA target (Kiss et al., 2004; Schattner et al., 2006) are comparable to levels detected in control samples and did not increase in DXO-KO cells (Figure S5A). Collectively, we conclude: (1) a subset of sno/scaRNAs are NAD+ capped; (2) DXO functions as a deNADding enzyme that targets NAD+-capped sno/scaRNAs and (3) mammalian cells likely possess a NAD+-capping mechanism that functions after transcription initiation to add NAD+ caps to intronic sno/scaRNAs.
Crystal Structure of DXO with 3′-phosphate NAD+
To understand the molecular mechanism for how DXO can function as a deNADding enzyme, we determined the crystal structures of wild-type mouse DXO in complex with 3′–phosphate NAD+ (3′-NADP+) and a Ca2+ ion at 2.1 Å resolution (Figure 6A). The 3′-phosphate is equivalent to the first backbone phosphate of the RNA body and is the scissile group of the deNADding reaction. The structure has excellent agreement with the X-ray diffraction data and the expected bond lengths, bond angles and other geometric parameters (Table S2).
Figure 6. Crystal structure of mouse DXO in complex with 3′-NADP+.
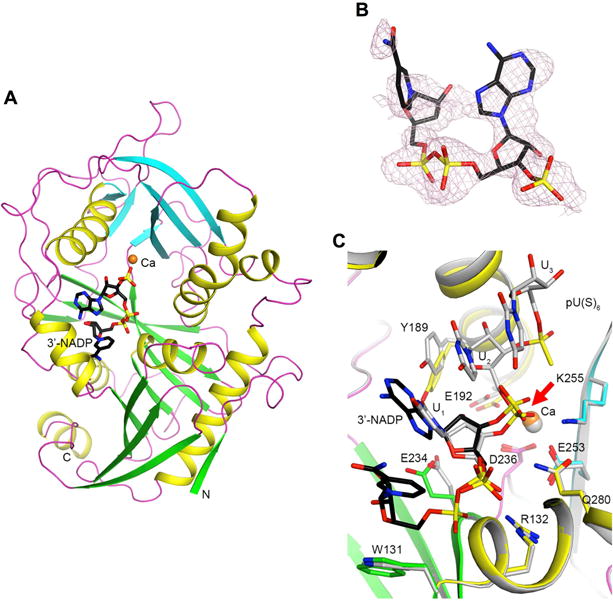
(A) Schematic drawing of the structure of DXO (in green and cyan for the large and small β-sheets, respectively, yellow for helices, and magenta for loops) in complex with 3′-NADP+ (in black for carbon atoms) and one Ca2+ ion (orange). (B). Omit Fo–Fc electron density at 2.1 Å resolution for 3′-NADP+, contoured at 2σ. (C). Overlay of the binding modes of 3′-NADP+ (black) and Ca2+ (orange) with those of the pU(S)6 substrate mimic and Ca2+ (gray) (Jiao et al., 2013). The scissile phosphate is denoted with a red arrow. All the structure figures were produced with PyMOL (www.pymol.org).
Good electron density was observed for the backbone of the 3′-NADP+ molecule, although the adenine base and especially the nicotinamide have weaker electron density (Figure 6B). Weak electron density was routinely observed for the bases with multiple analyzed X-ray diffraction data sets of crystals prepared for this complex. It is likely that they are partly disordered in the complex with DXO. There is one Ca2+ ion bound in the active site, similar to the observation on DXO in complex with Ca2+ and the substrate mimic pU(S)6 oligonucleotide (a hexanucleotide with the backbone phosphate groups of the first two nucleotides replaced with phosphorothioates) (Jiao et al., 2013).
Superposition of the 3′-NADP structure on that of DXO with the substrate mimic pU(S)6 shows the adenine of 3′-NADP overlaps well with the first nucleotide (U1) of the pU(S)6 substrate mimic and would likely stack with the next RNA base equivalent to U2 of pU(S)6 (Figure 6C). The 3′-phosphate is located at a similar position as the phosphate group of U2, the scissile phosphate of that substrate. The calcium ions in the two structures are located at essentially the same position, and most of the DXO residues in this binding site have similar conformations. Some variations are observed for the Tyr189 and Glu234 side chains, and the movement of Tyr189 is to avoid a steric clash with the larger adenine base of 3′-NADP+.
The observed binding mode of 3′-NADP+ illuminates the molecular mechanism for the deNADding activity of DXO. Combining the positions of 3′-NADP+ and pU(S)6 would produce the binding mode for an NAD+-capped RNA substrate. The 3′-phosphate is located at a similar position as the scissile phosphate of the pU(S)6 substrate mimic, and therefore it is also the scissile phosphate of the NAD+-capped RNA. Such a reaction would produce NAD+ and 5′–phosphate RNA, as observed in the biochemical assays.
Fungal Rai1 and Dxo1 Proteins are deNADding Enzymes
The comparable cap hydrolysis activity of the mammalian and fungal DXO family proteins (Grudzien-Nogalska and Kiledjian, 2016; Wang et al., 2015) prompted us to test whether Rai1 and Dxo1 proteins also possess deNADding activity. Schizosaccharomyces pombe Rai1 (SpRai1) was incubated with m7G-capped RNA, unmethylated G-capped RNA and NAD+-capped RNA and the reaction products resolved by TLC. As expected, SpRai1 inefficiently hydrolyzed m7G-capped RNA and had poor intrinsic activity to remove the cap structure from an unmethylated capped RNA with the low level of protein used but was stimulated by Rat1 (Figure 7A and (Jiao et al., 2010)). Importantly, SpRai1 exhibited robust preferential activity on NAD+-capped RNA that was saturated even at the 5-minute time point and released NAD+ analogous to DXO. This activity was compromised by the E199A, D201A catalytic site double mutant (Figure 7B) demonstrating that the detected activity was from SpRai1 and utilizes the same active site for both NAD+ cap deNADding and m7G cap decapping activities. Similarly, the Kluyveromyces lactis Dxo1 (KlDxo1) demonstrated more efficient deNADding activity releasing NAD+ than it did with m7G-capped RNA (Figure 7C). Catalytic site mutations that disrupt Dxo1 decapping (Chang et al., 2012) also disrupted its deNADding activity indicating both activities utilize the same active site. Our data reveal that the fungal DXO family proteins have robust deNADding activity in vitro.
Figure 7. SpRai1 and KlDxo1 are deNADding enzymes.
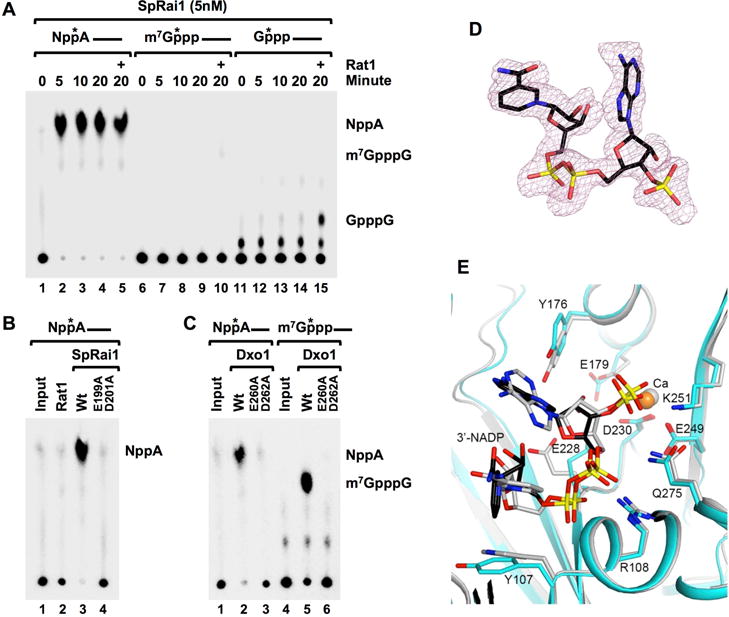
(A) SpRai1 is a robust deNADding enzyme. In vitro decapping assays were carried out with 5nM recombinant SpRai1 with 32P 5′ end-cap-labeled NAD+-capped, m7GpppA-capped or GpppA-capped RNAs and products detected as in legend to Figure 3. The (+) indicates the addition of 5nM recombinant Rat1. (B) SpRai1 catalytically inactive double mutant E199A/D201A and (C) catalytically inactive Dxo1 double mutant E260A/D262A, abolish deNADding or decapping activities. (D). Omit Fo–Fc electron density at 1.9 Å resolution for 3′-NADP+ in the complex with SsRai1, contoured at 2σ. (E). Overlay of the binding modes of 3′-NADP+ (black) and Ca2+ (orange) in the complex with SsRai1 (cyan) with those in the complex with DXO (gray).
We next determined the crystal structure of Scheffersomyces stipitis Rai1 (SsRai1) (Wang et al., 2015) in complex with 3′-NADP+ at 1.9 Å resolution (Table S2). Good quality electron density for 3′-NADP+ (Figure 7D) and a calcium ion was observed in the active site of one of the four SsRai1 molecules in the crystallographic asymmetric unit. The binding mode of 3′-NADP+ is similar to that in DXO (Figure 7E). Especially, the 3′ phosphate group is located in the same position in both structures, defining the molecular basis for the deNADding activity of Rai1. The analogous biochemical activity of SpRai1 and KlDxo1 to that of mammalian DXO indicates that Rai1 and Dxo1 also function as deNADding enzymes in yeast.
Discussion
A central premise in eukaryotic mRNA metabolism is the addition of an m7G cap at the 5′ end of the pre-mRNA. Our data demonstrate that mammalian mRNAs can also be capped with an NAD+ moiety at their 5′ end and in striking contrast to RNAs capped with m7G, RNAs capped with NAD+ are inefficiently translated and are less stable than capped mRNAs. Moreover, we identified a subset of sno/scaRNAs that also contain a NAD+ cap at their 5′ end, suggesting the presence of an NAD+ capping mechanism in mammalian cells that operates after transcription initiation. Furthermore, we identified the DXO family of proteins as potent deNADding enzymes that remove the intact NAD+ from the RNA. This was observed with the SpRai1 and KlDxo1 proteins in vitro as well as the mouse DXO both in vitro and in cells but not with the Dcp2 decapping enzyme demonstrating the removal of an NAD+ cap is a selective process.
Cotranscriptional addition of the m7G cap onto a nascent mammalian pre-mRNA is carried out by three enzymatic activities consisting of a 5′ triphosphatase, a guanylyl transferase and a methyl transferase (Shuman, 1995). Since its initial identification over four decades ago (Shatkin, 1976) all mRNAs produced by RNA polymerase II were thought to contain this modification at their 5′ end. This concept was modified following the demonstration that S. cerevisiae and mammalian mRNAs can possess incompletely capped mRNAs (Chang et al., 2012). Importantly, a quality control mechanism was identified whereby the DXO family of proteins can detect and remove the incomplete cap (Jiao et al., 2010). The ability of the DXO proteins to remove NAD+ caps from RNAs confers a second prominent function for these enzymes. The lack of Dcp2-directed deNADding on the same substrate RNAs supports the selectivity of DXO deNADding. Furthermore, based on the finding that the activity of DXO in removing NAD+ caps was more robust than the activity of DXO in removing guanosine caps in vitro, we propose that deNADding is the more prevalent activity of the DXO family of proteins in vivo. This deNADding activity further extends the known decapping, pyrophosphohydrolase and distributive 5′-3′ exoribonuclease activities of DXO, demonstrating the remarkable versatility of this enzyme in RNA metabolism.
The cocrystal structure of 3′ phosphate NAD+ provides exquisite insight into the deNADding reaction. The 3′ phosphate is at the same position as the scissile phosphate of an RNA substrate in a DXO-pU(S)6 RNA mimic we previously reported (Jiao et al., 2013), consistent with it being the scissile phosphate in the NAD+-capped RNA. The resulting products would be NAD+ and 5′ monophosphate RNA which would be a substrate for the 5′ to 3′ exonuclease activity of DXO (Jiao et al., 2013) and comparable to the reaction products observed biochemically (Figure 2 and 7).
In contrast to the m7G cap which is necessary for mRNA stability, our findings reveal NAD+ caps promote mRNA decay (Figure 3D). The presence of a NAD+ cap subjected the luciferase mRNA to rapid decay in the presence of DXO, yet conferred stability on to the same mRNA to levels comparable to an m7G cap in the absence of DXO. Similarly, our data are consistent with the NAD+ cap promoting DXO-directed decay of sno/scaRNAs. Whether the NAD+ cap functions as a tag to promote 5′ end decay analogous to uridylation of mRNA 3′ ends (Scheer et al., 2016; Scott and Norbury, 2013) remains to be determined. However, the intrinsic exoribonucleolytic property of DXO on 5′ monophosphorylated RNA (Chang et al., 2012; Jiao et al., 2013) supports a mechanism whereby recognition and deNADding of a 5′ NAD+ cap generates an exonucleolytic substrate for further DXO-mediated decay. The fact that the 5′ end of intronic snoRNA are generated by their resistance to exonucleolytic decay, may necessitate the requirement of a nongenomic encoded tag to trigger their decay.
NAD+ can serve as noncanonical transcription initiation nucleotide for bacterial RNA polymerase and eukaryotic RNA polymerase II (Bird et al., 2016) providing insight into a mechanism of NAD+ cap incorporation into mRNAs. Consistent with this mode of NAD+ cap formation, the LSM3 mRNA, which is transcribed with an adenosine as the first nucleotide, was among the NAD+ cap mRNAs confirmed in Figure 4B. Whether addition of an NAD+ cap is a stochastic event or a regulated process that is cleared by the DXO analogous to its quality control of incompletely capped mRNAs remains to be determined. However, the identification of NAD+ capped intronic sno/scaRNAs that normally contain a 5′ monophosphate, implies that mammalian cells possess a NAD+ capping mechanism distinct from transcription initiation with NAD+ for these noncoding RNAs. Whether mRNAs can also be capped with a NAD+ moiety or only utilize transcriptional incorporation of NAD+ cap is unknown.
Consistent with the functional significance of the m7G cap for mRNA translation, an NAD+-capped and polyadenylated luciferase mRNA did not support translation beyond levels attained with an uncapped mRNA (Figure 3E). The lack of translation was not a function of the NAD+ capped RNA being more labile, since a similar outcome was detected in DXO-KO cells where the half-lives of m7G and NAD+-capped luciferase mRNAs were indistinguishable (Figure 3D and S3). We conclude, unlike the m7G cap, a NAD+ cap does not facilitate translation. However, the possibility exists that endogenous NAD+-capped mRNAs can nevertheless undergo translation through a cap independent mechanism involving internal ribosome entry ((Martinez-Salas et al., 2012)) or 5′ UTR m6A directed translation (Meyer et al., 2015; Zhou et al., 2015).
The demonstration that human cells contain NAD+-capped mRNAs adds to the recent observation that bacterial mRNAs can possess an NAD+ modification at their 5′ end. Moreover, while this manuscript was under consideration, Saccharomyces cerevisiae was also reported to contain NAD+-capped RNA (Walters et al., 2017), indicating a wider prevalence of NAD+ capped RNAs in eukaryotic cells. Importantly, the deNADding activity of SpRai1 and KlDxo1 proteins (Figure 7) suggests the DXO family of proteins are general deNADding enzymes in eukaryotes.
The list of NAD+-capped RNAs reported here is likely an underestimation of the total cellular NAD+-capped RNAs. In particular, because not all NAD+-capped RNAs were responsive to DXO (Figure 4A), we anticipate mammalian cells contain enzymes in addition to DXO that can also hydrolyze NAD+-capped RNAs. Considering the bacterial NudC Nudix protein is a deNADding enzyme (Cahova et al., 2015) and the number of mammalian Nudix proteins that contain decapping capacity in vitro (Song et al., 2013), the presence of additional deNADding proteins in the human genome is a strong possibility. Furthermore, their identification and the analysis of cells lacking these enzymes may reveal additional classes of NAD+-capped RNAs that were not detected in this study.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESEOURCE SHARING
Requests for further information and requests for reagents can be directed to the lead contact, Megerditch Kiledjian (Kiledjian@biology.rutgers.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
Human embryonic kidney (HEK) 293T cells were obtained from ATCC. Cells were cultured in DMEM medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum, penicillin–streptomycin under 5% CO2 at 37°C.
METHOD DETAILS
Isolation and identification of NAD+-capped RNAs
Total RNAs from HEK 293T Con-KO and DXO-KO cell lines were isolated with TRIzol reagent (Thermo Fisher Scientific). NAD+-capped RNAs were isolated using the NAD-RNA Capture protocol (Cahova et al., 2015) with minor modifications as follows. NAD-Capture was carried out with 100μg total RNAs treated with 10μl 4-pentyn-1-ol (Sigma-Aldrich) and 1.5U Adenosine diphosphate-ribosylcyclase (ADPRC) in a 100μl reaction containing 50mM HEPES, 5mM MgCl2 (pH 7) and 40U RNasin® Ribonuclease Inhibitor (Promega) at 37°C 60 minutes. The reaction minus ADPRC was used as background control. The reaction was stopped with phenol/chloroform extraction and RNAs were precipitated with ethanol. The precipitated RNAs were subjected to copper-catalyzed azide-alkyne cycloaddition (CuAAC) reaction by incubating with 250μM biotin-PEG3-azide, freshly mixed 1mM CuSO4, 0.5mM THPTA, 2mM Sodium Ascorbate in 100μl reaction with 50mM HEPES, 5mM MgCl2 (pH7) and 40U RNasin® Ribonuclease Inhibitor (Promega) at 30°C for 30 minutes. Following extraction with phenol/chloroform and RNA precipitation, the NAD clicked RNAs were captured by binding to 20μl streptavidin magnet beads at room temperature with gentle rocking for 1 hour in 100μl binding buffer (1M NaCl, 10mM HEPES (pH7) and 5mM EDTA). The beads were washed three time with wash buffer (8mM Urea, 50mM Tris-HCl (pH7.4), 0.25% Triton-X100) and three washes with nuclease free H2O. Captured NAD-RNAs were reverse transcribed into cDNA and synthesized into dsDNA with the SMARTer® Universal Low Input RNA Kit for Sequencing (Clontech Laboratories, Inc.) as follows. Captured NAD-RNAs were initially converted to single-strand cDNA (ss-cDNA) with 1μl 3′ SMART N6 CDS Primer II A (24μM) and 2 μl SMARTScribe Reverse Transcriptase (100U/μl) at 42°C for 90 minutes in a 20μl reaction. The 5′ end adaptor was added to the 5′ end of the ss-cDNA with 1 μl SMARTer II A Oligonucleotide (24μM) by reverse transcriptase template switching. First-Strand cDNAs were purified with SPRI AMPure beads (Beckman Coulter, Part No. A63881) and double-stranded cDNAs (ds-cDNA) were generated and amplified by PCR with 2μl PCR Primer IIA (12μM) and 2μl 50X Advantage 2 Polymerase Mix for 12 cycles in a 50μl reaction (95°C 15 sec, 65°C 30 sec and 68°C 3 min). The amplified ds-cDNAs were purified with SPRI AMPure beads and digested with Rsa I to remove the adaptors. The digested ds-cDNAs were repurified with SPRI AMPure beads and used for sequencing.
RNA-Seq analysis
Double stranded DNA generated from the NAD-Capture was sequenced with Illumina high-throughput RNA sequencing technique (RUCDR, Rutgers University). RNA-Seq of HEK293T cell RNA was carried out using the Illumina TruSeq RNA Sample Preparation Kit v2. Sequencing was performed on an Illumina NextSeq instrument with paired ends of 75 nt each. RNA Seq reads were aligned to human hg19 genome (GRCh37) using Tophat (Trapnell et al., 2012) and the resulting BAM (binary sequence alignment/map format) files were sorted, and duplicates were removed using Picard (Broad Institute; http://broadinstitute.github.io/picard). The Cufflinks transcript assembly tool (Trapnell et al., 2012) was used to generate novel transcript assemblies from each sample. Overlap of raw sequencing reads with the GenCode comprehensive annotation (GRCh37.p13; http://www.gencodegenes.org) was completed using Bedtools package (Quinlan and Hall, 2010). The RNA class of each GenCode transcript intersecting a predicted transcript by at least 50% of the length (for both transcripts) was reported.
Plasmid Construction
The pcDNA3.1-Luciferase plasmid was generated by excising the firefly luciferase open reading frame from pGL3-Basic vector (Promega) with Xho I and Xba I and inserting into the same sites in pcDNA3.1 (Invitrogen). A 5′ leader ɸ2.5A-CA2 containing ɸ2.5 T7 promoter (Coleman et al., 2004) lacking adenosines other than the first transcribed nucleotide, was added to the luciferase open reading frame by PCR amplification of the ɸ2.5A-CA2 DNA fragment with primers containing restriction enzyme sites for Kpn I and Xho I at the 5′-end and 3′-ends, respectively (ɸ2.5A-Luc primers; Table S3). The amplified ɸ2.5A-CA2 promoter was cloned into 5′-end of the Firefly Luciferase gene in pcDNA3.1-Luciferase plasmid by Kpn I and Xho I. ɸ2.5A-CA2-Firefly Luciferase was amplified by PCR using a 3′ primer that incorporated 60 adenosines to the 3′ end (A60 tail) (F-Luc primers; Table S3). The pcDNA3.1-IRES-RF dual renilla and firefly luciferase plasmid was generated by PCR amplification of the two luciferase genes from the Renilla-hcvIRES-Firefly report plasmid using the RF-Forward and RF-Reverse Primers (Table S3). The primers were designed to insert Spe I and Hind III restrict enzyme sites on the 5′ and 3′ ends respectively and the resulting PCR product was inserted into the NheI and HindII sites of pcDNA3.1 (Invitrogen).
In vitro Transcription of NAD+-Capped RNAs
NAD+-capped RNAs were generated with the T7 ɸ2.5 promoter. ssDNA oligo ɸ2.5A-CA2 containing the T7 ɸ2.5 promoter at the 5′ end (Coleman et al., 2004) that retained the first adenosine at the transcription start site, but a uracil replacing all other adenosine was synthesized (ɸ2.5A-CA2; Table S3). The ssDNA oligo was amplified by PCR to generate dsDNA template for in vitro transcription and G16 track was added at the 3′-end (NAD-transcription primers; Table S3). In vitro transcription was carried out at 37°C for 90 minutes in 50μl reaction containing 400ng PCR-generated DNA template, 10μl 5× transcription buffer, 5μl DTT (10mM), 5μl BSA (10μg/μl), 5μl rNTP Mix (5mM), 2μl T7 RNA polymerase (Promega) and 1μl RNasin Ribonuclease Inhibitor (40U/μl, Promega). To generate 5′-end 32P-cap-labeled NAD-RNA, rATP was replaced with 32P-NAD+ or NAD+ in the reaction to generate 32P-NAD+-cap labeled or unlabeled NAD+-capped RNAs respectively. 32P-uniform-labeled 5′-end N7-methylated RNA was trancribed in with the inclusion of [α-32P]GTP and 0.5mM m7GpppA cap analog (New England BioLabs) in a reaction that omitted rATP. 5′-end N7-methylated 32P-cap-labelled RNA was generated as previously described (Wang et al., 1999) by capping 20 pmol 5′end triphosphate RNA in 15μl capping reaction containing 50 mM Tris-HCl (pH 7.9), 1.25 mM MgCl2, 6 mM KCl, 2.5 mM DTT, 20 U vaccinia virus capping enzyme (New England BioLabs), 1 mM S-adenosylmethionine (New England BioLabs), 20μCi [a-32P]GTP, 0.5μl RNasin Ribonuclease Inhibitor (40U/μl, Promega) and incubated at 37°C for 90 minutes.. The transcribed RNAs were purified by passing through MicroSpin G-50 Columns (GE Healthcare).
NAD+-capped, m7GpppA capped or uncapped Firefly Luciferase mRNA with an A60 tail was generated with T7 RNA polymerase in vitro. The transcription reactions were first incubated at 37°C for 15 minutes by adding unlabeled NAD+ or m7GpppA cap analog with rGTP, rUTP and rCTP in the absence of rATP to generate NAD+-capped, m7GpppA-capped or uncapped RNA oligos trapped at the first rATP addition site. The reaction was subsequently allowed to proceed at 37°C for 30 minutes in the presence of rATP and 200μg/ml heparin to inhibit transcription reinitiation (Ferrari et al., 2004). N7-methylguanosine-capped Renilla Luciferase was generated with T7 RNA polymerase from the template containing a T7 promoter amplified by PCR from pcDNA3.1-IRES-RF plasmid (R-Luc primers; Table S3). MicroSpin G-50 Columns (GE Healthcare) used to isolate the transcribed RNAs.
RNA in vitro Decapping and deNADding Assays
32P-5′-end labeled NAD+ capped, N7-methylated or non-methylated capped RNAs were incubated with 10 to 50nM recombinant proteins in 20μl decapping reaction containing 100mM KCl, 2mM MgCl2, 1mM MnCl2, 2mM DTT, 10mM Tris-HCl (pH 7.5) and incubated at 37°C for 30 minutes. Reactions were stopped with 30mM EDTA. Decapping products were resolved by PEI-cellulose TLC plates (Sigma-Aldrich) and developed in 0.45 M (NH4)2SO4 in a TLC chamber at room temperature (Liu et al., 2008). Reaction products were visualized and quantitated with a Molecular Dynamics PhosphorImager (Storm860) with ImageQuant-5 software.
NAD+ RNA poly(A) Tail Ratio and 3′ End Sequencing
NAD+-capped RNAs were captured from 100μg total RNA as descripted above (Isolation and identification of NAD+-capped RNAs) and eluted from the magnet streptavidin beads by heating at 95°C for 5 minutes with 100μl wash buffer and a second time with 100μl water and precipitated with Ethanol. The eluted RNA was aliquoted into two equal sections and reversed transcribed into cDNA with either Oligo d(T)18 or random hexanucleotides primers. The poly(A) tailed message NAD-capped RNAs were detected by real-time qPCR from Oligo d(T)18 reversed transcribed cDNA and normalized to total NAD+-capped RNA detected by the random hexanucleotides primer reversed transcribed cDNA. For NAD-capped mRNA 3′ end sequencing, the isolated NAD+-RNAs were reversed transcribed into cDNA with Anchor-Oligo d(T)18 primer and NAD+-capped mRNA 3′ ends were amplified with a gene specific forward primer and Anchor primer by PCR, cloned into pGEM-T vector and sequenced with T7 or SP6 primers.
NAD+-capped RNA Ratio Determination in Total RNA Population
NAD+-capped RNA was isolated from 10μg HEK293T WT or DXO-KO cell total RNA and eluted from the magnet streptavidin beads as descripted above. RNA in the unbound supernatant as well as the washes were also collected. Both pools of RNA were separated into two equivalent aliquots. The NAD+ capped RNA population was determined from one of the streptavidin bead bound RNA fraction while the second fraction was combined with one of the eluate fractions to designate the total RNA levels. RNAs were reverse transcribed into cDNA with random hexanucleotide primers and detected with gene specific primers by real-time PCR as descripted above.
Luciferase Assays and Luciferase RNA Stability Assays
NAD+-capped, m7G-capped or uncapped 5′ end triphosphate Firefly Luciferase RNAs containing an A60 tail were co-transfected with m7G-capped Renilla Luciferase RNA into HEK 293T WT or DXO-KO cells by electroporation in 0.4cm cuvettes at 220V, 25ms (Gene Pulser Xcell System, BIO-RAD). The transfected cells were equally aliquoted into different cell culture plates continually cultured at 37°C and harvested at 0, 0.5, 1, 2 and 3 hours. Firefly Luciferase and Renilla Luciferase expression levels were detected with the Dual-Luciferase® Reporter (DLR™) Assay System (Promega). For Firefly Luciferase RNA stability assays, the NAD+-capped, m7G-capped or uncapped Firefly Luciferase RNAs containing an A60 tail were electroporated as descripted above, except untransfected RNAs were degraded by Micrococcal Nuclease (New England Biolabs) in the presence of 5mM CaCl2 at 37°C for 15 minutes. Transfected cells were equally aliquoted into different cell culture plates and continually cultured at 37°C. Cells were harvested at the indicated time points and total RNA was isolated with TRIzol reagent (Thermo Fisher Scientific). Equal amounts of total RNA from each time point were reverse transcribed into cDNA with Firefly Luciferase RNA and GAPDH mRNA specific primers and RNA levels detected by real-time PCR with gene specific primers and normalized to endogenous GAPDH mRNA. Half-lives were determined by calculating the linear regression fit.
Protein Expression, Purification and Crystallization
SpRai1, SpRat1, SsRai1, KlDxo1 and mouse DXO His-tagged recombinant proteins were expressed by transforming E. coli BL21 (DE3) Rosetta cells with pET26b-SpRat1, pRT24d-SpRat1, pET28a-SsRail, pET28a-KlDxo1 and pET28a-mDom3Z (DXO) plasmids, respectively. Recombinant proteins were induced with 0.3 mM IPTG, the cells were allowed to grow at 20 °C for 14–16 h and purified by Ni-NTA Superflow (Qiagen) and gel filtration (Sephacryl S-300, GE Healthcare) chromatography. The proteins were concentrated to 25–30 mg/ml in a buffer containing 20 mM Tris (pH 7.5), 250 mM NaCl and 5% (v/v) glycerol, frozen in liquid nitrogen and stored at −80°C. DXO crystals were obtained using the hanging-drop vapor diffusion method at 20 °C with a reservoir solution containing 21–26% (w/v) PEG 3350. To obtain the DXO-3′–NADP-Ca2+ complex, free DXO crystals were soaked with 20 mM 3′-NADP+ and 10 mM CaCl2 overnight at 20 °C in a buffer containing 25% (w/v) PEG 3350 and 0.1 M Bis-Tris (pH 6.0). The crystals were cryoprotected with 25% (w/v) PEG 3350 and 25% (v/v) ethylene glycol before being flash frozen in liquid nitrogen for diffraction analysis and data collection at 100 K. SsRai1 crystals were obtained using the hanging-drop vapor diffusion method at 20 °C with a reservoir solution containing 15% (w/v) PEG 3350 and 0.2 M NH4Cl (Wang et al 2015).
Data Collection and Structure Determination
X-ray diffraction data were collected using the Pilatus-6M detector at the Advanced Photon Source (APS) beamline 24-IDC. The diffraction images were processed and scaled using the XDS program (Kabsch, 2010). The structure was solved with Phaser (McCoy et al., 2007) using the DXO or SsRai1 structure as a search model. Structure refinement was performed using PHENIX (Adams et al., 2002) and the atomic model was built with the Coot program (Emsley and Cowtan, 2004). The crystallographic information is summarized in Table S2.
Generation of Dcp2 and DXO CRISPR knockout cell lines
HEK293T cells were maintained in DMEM with 10% FBS and antibiotics (100 units/ml penicillin and 100 μg/ml of streptomycin) under standard tissue culture conditions. The three different monoclonal HEK293T cell lines harboring CRISPR-Cas9n double-nick generated distinct homozygous deletions within the DCP2 gene genomic region corresponding to its catalytic site have been reported elsewhere (Mauer et al., 2017). DXO knockout lines were similarly generated using CRISPR-Cas9n technology with two DXO-gRNAs (Table S3) designed to target genomic regions of DXO corresponding to the catalytic site in exon 4. The genomic modification was screened by PCR and confirmed by sequencing and a Western of the three individual lines is shown in Figure S3A.
RNA in vivo decay assay
32P-uniform-labeled RNAs were transfected into HEK293T control knock-out (Con-KO), DXO knock-out (DXO-KO) or Dcp2 knock-out (Dcp2-KO) cell lines with Lipofectamine 3000 (Thermo Fisher Scientific). 293T cell were grown in 60mm culture plates to 70% confluency. 0.5μg 32P- labeled RNAs were incubated with 10μl P3000 reagent in 500μl Opti-MEM medium at room temperature for 15 minutes and then mixed with 15μl Lipofectamine 3000 reagent diluted in 500μl Opti-MEM medium. The RNA-Lipofectamine 3000 complex was added to the cell culture plates and cells incubated for 30 minutes. Untransfected RNAs were degraded by Micrococcal Nuclease (New England Biolabs) 30 minutes post-transfection in the presence of 5mM CaCl2 for 15 minutes. The transfected cells were equally aliquoted into different cell culture plates continually cultured at 37°C. Cells were harvested at 0, 1, 2 and 4 hour time points after Micrococcal Nuclease treatment. Total RNAs were isolated with TRIzol reagent (Thermo Fisher Scientific) and equal amounts of total RNA from the different time points were fractionated by 8% polyacrylamide gel with 7M urea. The remaining 32P-labeled RNAs were analyzed by PhosphorImager.
Real-Time Quantitative Reverse Transcription and PCR
NAD Click-iT captured RNAs were isolated with MagVigen™ Streptavidin conjugates magnet beads (Nvigen Inc.) and reverse transcribed into cDNA in 20μl reaction containing 0.5μl random primers (Promega), 1μl M-MLV reverse transcriptase (Promega), 1μl dNTP (10mM), 4μl 5× reverse transcription buffer and 0.5μl RNasin Ribonuclease Inhibitor (40U/μl, Promega) incubated at 37°C 90 minutes. Real-time PCR was performed using iTaq Supermix (BioRad) on ABI Prism 7900HT sequence detection system (Jiao et al., 2006; Jiao et al., 2010) with transcript specific primers (Table S3). NAD+-RNA levels were computed by the comparative Ct method.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters for mRNA decay are reported in the Figure Legends. mRNA half-lives of reporter mRNAs were obtained by constructing linear models of the ln[remaining RNA] as a function of time, and then the half-life calculated as the ln[2]/slope (Chen et al., 2008). All data are presented ± the 95% confidence interval and p values were calculated from the linear model using R (Dudoit et al., 2003; Gentleman et al., 2004).
Following alignment with genome sequences, sequencing reads were assembled with the UCSC hg19 reference transcript map using Cufflinks, allowing a direct comparison of expression levels by Cuffdiff. Results were exported to the CummeRbund package (Trapnell et al., 2012) in R/Bioconductor (Dudoit et al., 2003; Gentleman et al., 2004). Only transcripts with expression levels of FPKM (fragments per kilobase of transcript length per million aligned reads) greater than 1.0 in at least one condition were considered to be expressed.
DATA AND SOFTWARE AVAILABILITY
Sequencing data have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE90884. The atomic coordinates have been deposited at the Protein Data Bank, with accession codes 5ULI and 5ULJ.
Supplementary Material
Figure S1 (related to Figure 1). (A) Sashimi plots for the indicated mRNAs is shown with the labeling as described in the legend to Figure 1E. (B) Sequence of COX7B 3′ end containing 30 adenosine poly(A) tail and the anchor sequence used in Figure 1G is shown. Primers used for the 3′-end amplification are highlighted.
Figure S2 (related to Figure 2). NAD+-capped RNA deNADding products. DXO deNADding product was resolved by polyethyleneimine-TLC developed in 0.5M LiCl with unlabeled NAD+, NADP, NADH or 32P-labeled NAD+. The localization of unlabeled NAD+, NADP and NADH were visualized under shortwave UV light and marked with radioactive pen.
Figure S3 (related to Figure 3). (A) Western blot of extract derived from Control knock out (Con-KO) and three individual monoclonal DXO knock out (DXO-KO) cell lines probed with DXO or GAPDH internal control is shown. (B) Firefly luciferase mRNAs containing either a 5′-end NAD+ cap or m7G cap with 3′ poly(A60) tail were co-transfected with m7G-capped Renilla Luciferase RNA into DXO-KO HEK293T cells. Cells were harvested and assayed at the indicated time points. Firefly Luciferase activity was plotted normalized to Renilla luciferase and data from three independent experiments are presented with error bars representing +/−SD.
Figure S4 (related to Figure 4). Distribution of NAD+ capped RNAs. HEK293T cells were fractionated to separate the nuclear and cytoplasmic compartments and indicated RNAs were tested with gene specific primers. Levels in each compartment are presented relative to the total that was set to 100. Data are derived from three independent samples, error bars representing +/−
Figure S5 (related to Figure 5). A. Lack of selective association of 18S rRNA to the NAD-Capture matrix. NAD+-capped RNAs isolated by the NAD-Capture approach were eluted from the beads, reverse transcribed and 18S rRNA detected with gene specific primers. Levels detected with the -ADPRC negative control was set to 1. Although a select number of NAD+-capped snoRNAs including, SNORA20 and SNORA76 are elevated in DXO-KO cells, their target 18S rRNA is not. B. Sashimi plots for the indicated snoRNAs and scaRNAs are shown. Labeling is as in the legend to Figure 1E.
Table S1. NAD-CaptureSeq (DXO-KO and WT HEK293T cells; Related to Figure 4A). The table includes a Genbank accession number as isoform_id, mean FPKM values for DXO-KO and Wt cells, the log2 fold-change (lfc), a test statistic, p-value, and q-value (essentially the false discovery rate) from the F-test, along with the associated gene symbol (gene_id) and genomic transcript region (locus).
Table S2. Summary of crystallographic information (Related to Figures 6 and 7)
Table S3. DNA Primer Sequences (Related to the STAR Methods section):
In Brief.
NAD+-caps destabilize eukaryotic mRNAs and snoRNAs.
Highlights.
-
-
Mammalian cells possess NAD+-capped mRNAs.
-
-
DXO family proteins are “deNADding” enzymes that remove the NAD+ cap from an mRNA.
-
-
A 5′ NAD+ cap promotes DXO-mediated RNA decay.
-
-
Intronic snoRNAs can be NAD+ capped in mammalian cells and degraded by DXO.
Acknowledgments
We thank members of the Kiledjian lab for helpful discussions. This work was supported by NIH grants GM118059 (B.E.N.) GM118093 (L.T.) and GM067005 (M.K.).
This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, funded by the NIH (P41 GM103403). The Pilatus 6M detector on 24-ID-C beam line is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and three tables and can be found with this article online at XXXX
AUTHOR CONTRIBUTIONS
M.K., X.J., and L.T. designed the experiments. X.J. carried out all experiments unless otherwise indicated. S.D. and L.T. carried out the structural analysis and interpretations. R.P.H. carried out all bioinformatics analyses. J.G.B. and B.E.N. provided NAD+-capped RNA for Figure 2 and NudC protein. M.K., X.J., L.T., R.P.H. and B.E.N. wrote the manuscript.
References
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Bird JG, Zhang Y, Tian Y, Panova N, Barvik I, Greene L, Liu M, Buckley B, Krasny L, Lee JK, et al. The mechanism of RNA 5′ capping with NAD+, NADH and desphospho-CoA. Nature. 2016;535:444–447. doi: 10.1038/nature18622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahova H, Winz ML, Hofer K, Nubel G, Jaschke A. NAD captureSeq indicates NAD as a bacterial cap for a subset of regulatory RNAs. Nature. 2015;519:374–377. doi: 10.1038/nature14020. [DOI] [PubMed] [Google Scholar]
- Chang JH, Jiao X, Chiba K, Oh C, Martin CE, Kiledjian M, Tong L. Dxo1 is a new type of eukaryotic enzyme with both decapping and 5′-3′ exoribonuclease activity. Nat Struct Mol Biol. 2012;19:1011–1017. doi: 10.1038/nsmb.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Ezzeddine N, Shyu AB. Messenger RNA half-life measurements in mammalian cells. Methods Enzymol. 2008;448:335–357. doi: 10.1016/S0076-6879(08)02617-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YG, Kowtoniuk WE, Agarwal I, Shen Y, Liu DR. LC/MS analysis of cellular RNA reveals NAD-linked RNA. Nat Chem Biol. 2009;5:879–881. doi: 10.1038/nchembio.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman TM, Wang G, Huang F. Superior 5′ homogeneity of RNA from ATP-initiated transcription under the T7 phi 2.5 promoter. Nucleic Acids Res. 2004;32:e14. doi: 10.1093/nar/gnh007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke C, Alwine JC. The cap and the 3′ splice site similarly affect polyadenylation efficiency. Mol Cell Biol. 1996;16:2579–2584. doi: 10.1128/mcb.16.6.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieci G, Preti M, Montanini B. Eukaryotic snoRNAs: a paradigm for gene expression flexibility. Genomics. 2009;94:83–88. doi: 10.1016/j.ygeno.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Dudoit S, Gentleman RC, Quackenbush J. Open source software for the analysis of microarray data. Biotechniques. 2003;(Suppl):45–51. [PubMed] [Google Scholar]
- Edery I, Sonenberg N. Cap-dependent RNA splicing in a HeLa nuclear extract. Proc Natl Acad Sci U S A. 1985;82:7590–7594. doi: 10.1073/pnas.82.22.7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Ferrari R, Rivetti C, Dieci G. Transcription reinitiation properties of bacteriophage T7 RNA polymerase. Biochem Biophys Res Commun. 2004;315:376–380. doi: 10.1016/j.bbrc.2004.01.071. [DOI] [PubMed] [Google Scholar]
- Filipowicz W. Functions of the 5,-terminal m7G cap in eukaryotic mRNA. FEBS Lett. 1978;96:1–11. doi: 10.1016/0014-5793(78)81049-7. [DOI] [PubMed] [Google Scholar]
- Filipowicz W, Pogacic V. Biogenesis of small nucleolar ribonucleoproteins. Curr Opin Cell Biol. 2002;14:319–327. doi: 10.1016/s0955-0674(02)00334-4. [DOI] [PubMed] [Google Scholar]
- Furuichi Y, LaFiandra A, Shatkin AJ. 5′-Terminal structure and mRNA stability. Nature. 1977;266:235–239. doi: 10.1038/266235a0. [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmartin GM, McDevitt MA, Nevins JR. Multiple factors are required for specific RNA cleavage at a poly(A) addition site. Genes Dev. 1988;2:578–587. doi: 10.1101/gad.2.5.578. [DOI] [PubMed] [Google Scholar]
- Grudzien-Nogalska E, Kiledjian M. New Insights into Decapping Enzymes and Selective mRNA Decay. Wiley Interdiscip Rev RNA. 2016 doi: 10.1002/wrna.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart RP, McDevitt MA, Nevins JR. Poly(A) site cleavage in a HeLa nuclear extract is dependent on downstream sequences. Cell. 1985;43:677–683. doi: 10.1016/0092-8674(85)90240-5. [DOI] [PubMed] [Google Scholar]
- Hsu CL, Stevens A. Yeast cells lacking 5′–>3′ exoribonuclease 1 contain mRNA species that are poly(A) deficient and partially lack the 5′ cap structure. Mol Cell Biol. 1993;13:4826–4835. doi: 10.1128/mcb.13.8.4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao X, Chang JH, Kilic T, Tong L, Kiledjian M. A mammalian pre-mRNA 5′ end capping quality control mechanism and an unexpected link of capping to pre-mRNA processing. Mol Cell. 2013;50:104–115. doi: 10.1016/j.molcel.2013.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao X, Wang Z, Kiledjian M. Identification of an mRNA-Decapping Regulator Implicated in X-Linked Mental Retardation. Mol Cell. 2006;24:713–722. doi: 10.1016/j.molcel.2006.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao X, Xiang S, Oh C, Martin CE, Tong L, Kiledjian M. Identification of a quality-control mechanism for mRNA 5′-end capping. Nature. 2010;467:608–611. doi: 10.1038/nature09338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr. 2010;66:133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaji H, Nakamura M, Takahashi Y, Sandelin A, Katayama S, Fukuda S, Daub CO, Kai C, Kawai J, Yasuda J, et al. Hidden layers of human small RNAs. BMC Genomics. 2008;9:157. doi: 10.1186/1471-2164-9-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss AM, Jady BE, Bertrand E, Kiss T. Human box H/ACA pseudouridylation guide RNA machinery. Mol Cell Biol. 2004;24:5797–5807. doi: 10.1128/MCB.24.13.5797-5807.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarska MM, Padgett RA, Sharp PA. Recognition of cap structure in splicing in vitro of mRNA precursors. Cell. 1984;38:731–736. doi: 10.1016/0092-8674(84)90268-x. [DOI] [PubMed] [Google Scholar]
- Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12:767–772. doi: 10.1038/nmeth.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SW, Jiao X, Welch S, Kiledjian M. Analysis of mRNA decapping. Methods Enzymol. 2008;448:3–21. doi: 10.1016/S0076-6879(08)02601-3. [DOI] [PubMed] [Google Scholar]
- Martinez-Salas E, Pineiro D, Fernandez N. Alternative Mechanisms to Initiate Translation in Eukaryotic mRNAs. Comp Funct Genomics. 2012;2012:391546. doi: 10.1155/2012/391546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattaj IW. Cap trimethylation of U snRNA is cytoplasmic and dependent on U snRNP protein binding. Cell. 1986;46:905–911. doi: 10.1016/0092-8674(86)90072-3. [DOI] [PubMed] [Google Scholar]
- Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, Linder B, Pickering BF, Vasseur JJ, Chen Q, et al. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature. 2017;541:371–375. doi: 10.1038/nature21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, Jaffrey SR. 5′ UTR m(6)A Promotes Cap-Independent Translation. Cell. 2015;163:999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthukrishnan S, Filipowicz W, Sierra JM, Both GW, Shatkin AJ, Ochoa S. mRNA methylation and protein synthesis in extracts from embryos of brine shrimp, Artemia salina. J Biol Chem. 1975;250:9336–9341. [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs AB. Messenger RNA degradation in eukaryotes. Cell. 1993;74:413–421. doi: 10.1016/0092-8674(93)80043-e. [DOI] [PubMed] [Google Scholar]
- Schattner P, Barberan-Soler S, Lowe TM. A computational screen for mammalian pseudouridylation guide H/ACA RNAs. RNA. 2006;12:15–25. doi: 10.1261/rna.2210406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheer H, Zuber H, De Almeida C, Gagliardi D. Uridylation Earmarks mRNAs for Degradation… and More. Trends in genetics: TIG. 2016;32:607–619. doi: 10.1016/j.tig.2016.08.003. [DOI] [PubMed] [Google Scholar]
- Scott DD, Norbury CJ. RNA decay via 3′ uridylation. Biochim Biophys Acta. 2013;1829:654–665. doi: 10.1016/j.bbagrm.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Shatkin AJ. Capping of eucaryotic mRNAs. Cell. 1976;9:645–653. doi: 10.1016/0092-8674(76)90128-8. [DOI] [PubMed] [Google Scholar]
- Shuman S. Capping enzyme in eukaryotic mRNA synthesis. Prog Nucleic Acid Res Mol Biol. 1995;50:101–129. doi: 10.1016/s0079-6603(08)60812-0. [DOI] [PubMed] [Google Scholar]
- Sonenberg N, Rupprecht KM, Hecht SM, Shatkin AJ. Eukaryotic mRNA cap binding protein: purification by affinity chromatography on sepharose-coupled m7GDP. Proc Natl Acad Sci U S A. 1979;76:4345–4349. doi: 10.1073/pnas.76.9.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MG, Bail S, Kiledjian M. Multiple Nudix family proteins possess mRNA decapping activity. RNA. 2013;19:390–399. doi: 10.1261/rna.037309.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters RW, Matheny T, Mizoue LS, Rao BS, Muhlrad D, Parker R. Identification of NAD+ capped mRNAs in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2017;114:480–485. doi: 10.1073/pnas.1619369114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang VY, Jiao X, Kiledjian M, Tong L. Structural and biochemical studies of the distinct activity profiles of Rai1 enzymes. Nucleic Acids Res. 2015;43:6596–6606. doi: 10.1093/nar/gkv620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Day N, Trifillis P, Kiledjian M. An mRNA stability complex functions with poly(A)-binding protein to stabilize mRNA in vitro. Mol Cell Biol. 1999;19:4552–4560. doi: 10.1128/mcb.19.7.4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Kiledjian M. Functional Link between the Mammalian Exosome and mRNA Decapping. Cell. 2001;107:751–762. doi: 10.1016/s0092-8674(01)00592-x. [DOI] [PubMed] [Google Scholar]
- Wei C, Gershowitz A, Moss B. N6, O2′-dimethyladenosine a novel methylated ribonucleoside next to the 5′ terminal of animal cell and virus mRNAs. Nature. 1975a;257:251–253. doi: 10.1038/257251a0. [DOI] [PubMed] [Google Scholar]
- Wei CM, Gershowitz A, Moss B. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell. 1975b;4:379–386. doi: 10.1016/0092-8674(75)90158-0. [DOI] [PubMed] [Google Scholar]
- Xiang S, Cooper-Morgan A, Jiao X, Kiledjian M, Manley JL, Tong L. Structure and function of the 5′–>3′ exoribonuclease Rat1 and its activating partner Rai1. Nature. 2009;458:784–788. doi: 10.1038/nature07731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian SB. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature. 2015;526:591–594. doi: 10.1038/nature15377. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (related to Figure 1). (A) Sashimi plots for the indicated mRNAs is shown with the labeling as described in the legend to Figure 1E. (B) Sequence of COX7B 3′ end containing 30 adenosine poly(A) tail and the anchor sequence used in Figure 1G is shown. Primers used for the 3′-end amplification are highlighted.
Figure S2 (related to Figure 2). NAD+-capped RNA deNADding products. DXO deNADding product was resolved by polyethyleneimine-TLC developed in 0.5M LiCl with unlabeled NAD+, NADP, NADH or 32P-labeled NAD+. The localization of unlabeled NAD+, NADP and NADH were visualized under shortwave UV light and marked with radioactive pen.
Figure S3 (related to Figure 3). (A) Western blot of extract derived from Control knock out (Con-KO) and three individual monoclonal DXO knock out (DXO-KO) cell lines probed with DXO or GAPDH internal control is shown. (B) Firefly luciferase mRNAs containing either a 5′-end NAD+ cap or m7G cap with 3′ poly(A60) tail were co-transfected with m7G-capped Renilla Luciferase RNA into DXO-KO HEK293T cells. Cells were harvested and assayed at the indicated time points. Firefly Luciferase activity was plotted normalized to Renilla luciferase and data from three independent experiments are presented with error bars representing +/−SD.
Figure S4 (related to Figure 4). Distribution of NAD+ capped RNAs. HEK293T cells were fractionated to separate the nuclear and cytoplasmic compartments and indicated RNAs were tested with gene specific primers. Levels in each compartment are presented relative to the total that was set to 100. Data are derived from three independent samples, error bars representing +/−
Figure S5 (related to Figure 5). A. Lack of selective association of 18S rRNA to the NAD-Capture matrix. NAD+-capped RNAs isolated by the NAD-Capture approach were eluted from the beads, reverse transcribed and 18S rRNA detected with gene specific primers. Levels detected with the -ADPRC negative control was set to 1. Although a select number of NAD+-capped snoRNAs including, SNORA20 and SNORA76 are elevated in DXO-KO cells, their target 18S rRNA is not. B. Sashimi plots for the indicated snoRNAs and scaRNAs are shown. Labeling is as in the legend to Figure 1E.
Table S1. NAD-CaptureSeq (DXO-KO and WT HEK293T cells; Related to Figure 4A). The table includes a Genbank accession number as isoform_id, mean FPKM values for DXO-KO and Wt cells, the log2 fold-change (lfc), a test statistic, p-value, and q-value (essentially the false discovery rate) from the F-test, along with the associated gene symbol (gene_id) and genomic transcript region (locus).
Table S2. Summary of crystallographic information (Related to Figures 6 and 7)
Table S3. DNA Primer Sequences (Related to the STAR Methods section):