

Abstract
While many antimicrobial peptides (AMPs) disrupt bacterial membranes, some translocate into bacteria and interfere with intracellular processes. Buforin II and DesHDAP1 are thought to kill bacteria by interacting with nucleic acids. Here, molecular modeling and experimental measurements are used to show that neither nucleic acid binding peptide selectively binds DNA sequences. Simulations and experiments also show that changing lysines to arginines enhances DNA binding, suggesting that including additional guanidinium groups is a potential strategy to engineer more potent AMPs. Moreover, the lack of binding specificity may make it more difficult for bacteria to evolve resistance to these and other similar AMPs.
Keywords: antimicrobial peptide, nucleic acid, molecular dynamics, electrostatics calculations
Introduction
Growing medical concern of drug resistance has prompted a need to develop alternatives to conventional antibiotics [1]. Antimicrobial peptides (AMPs), which are naturally occurring in numerous living organisms, are active against a wide range of bacteria and other pathogens and therefore show potential as a class of such alternatives [2–4]. While many AMPs kill bacteria through membrane permeabilization, a smaller subset of AMPs is believed to inhibit bacterial growth by interfering with essential intracellular functions [5, 6]. Buforin II (BF2) and DesHDAP1 are two AMPs that share this lesser studied mechanism, with both hypothesized to bind nucleic acid as part of their mechanism of action [7–9], although the peptides could potentially have other intracellular targets or cause more minor membrane damage that is not observed in common assays but still harms bacteria.
BF2 is a 21-amino acid long peptide derived from a naturally occurring peptide in Bufo bufo gargarizans [10], and DesHDAP1 [11] is a 20-amino acid long designed peptide based on the crystal structure of histone H2A. Previous studies have shown the activity of BF2 and DesHDAP1 against several bacterial strains [8, 10–14]. Because each peptide shares its entire sequence with a portion of a histone core subunit, both are histone-derived antimicrobial peptides (HDAPs). While BF2’s and DesHDAP1’s modes of action involve nucleic acid binding, this mechanism is certainly not unique to these two HDAPs [5, 6, 15–19]. For example, some puroindoline derived peptides were shown to inhibit macromolecular synthesis via binding DNA [20], whereas pseudin-2, an AMP derived from skin of the South American frog Pseudis paradoxa, did so via binding RNA [21]. In other studies, a cathelicidin derived peptide appeared to bind DNA and decrease replication of a plasmid containing an antimicrobial resistance gene [22], and piscidins can cause condensation of DNA in addition to their membrane effects [23]. With several peptides and peptidomimetics binding nucleic acids as part of their modes of antimicrobial activity, a better understanding of such peptide-nucleic acid binding could contribute to engineering of similar AMPs with improved activity.
While nucleic acid binding appears to play a role in the mechanism of these peptides, researchers have not considered whether peptides target particular base sequences or have less specificity in their binding. Past studies by our lab investigated nucleic acid binding of BF2 using a combination of experimental DNA binding assays and molecular dynamics (MD) simulations [9]. Subsequently, DesHDAP1, a peptide thought to share a similar mechanism of action as BF2, was designed and subjected to identical MD simulation modeling [11]. Although the structural models of BF2 and DesHDAP1 bound to DNA in these previous papers implied that the peptides likely had little nucleic acid binding specificity, only a single, relatively short (10 ns) MD simulation was reported for each peptide and any potential specificity was not considered experimentally.
Understanding any binding selectivity is central to design efforts aimed at developing more potent HDAPs. To this end, this current study characterizes the DNA binding of both BF2 and DesHDAP1 using multiple MD simulations, electrostatic analyses and experimental nucleic acid binding experiments with different repeating DNA sequences. Together, these results show a lack of sequence specificity in both of these peptides and emphasize the importance in peptide•phosphate interactions in DNA binding. Interestingly, our simulations of BF2 and DesHDAP1 also noted that arginine residues played a more significant role than lysine in mediating phosphate interactions. Thus, we also performed additional simulations and experimental studies to confirm that increased arginine composition does increase the DNA binding of these peptides. In fact, recent studies have shown that BF2 and DesHDAP1 variants containing increased arginine versus lysine composition have greater antimicrobial activity [24]. While this was attributed to enhanced membrane interactions, the data presented here implies that increased DNA interactions may also have played a role.
Materials and Methods
Molecular Modeling and MD Simulations
The initial models of BF2 and DesHDAP1 bound to DNA were extracted from the histone core particle crystal structure (1AOI) [25] as in previous work [9, 11]. These models incorporated the section of the protein with an identical sequence as the peptide along with an adjacent section of DNA (21 base pairs with BF2 and 20 base pairs with DesHDAP1). All modeling and experiments of BF2 included the F10W mutation. The BF2-DNA and DesHDAP1-DNA complexes were refined using MD simulations in Gromacs 4.5.5 [26] with the AMBER03 force field [27]. For all simulations, TIP4P-Ew waters were used for BF2 and DesHDAP1, with Na+ and Cl− added to neutralize overall charges and provide an additional salt concentration of 100 mM. Arginine and lysine residues and the N- and C- termini were ionized, while the single His side chain was left uncharged in the HID tautomer. Systems were subjected to 100 steps of steepest descents minimization and heated to 300 K over 20 ps. Each trajectory was extended to a total length of 50 ns at constant temperature (300 K) and isotropic constant pressure (1 bar). Simulations used a time step of 2 fs and long-range electrostatics longer than the 10 Å cutoff calculated using PME [28]. Bonds to hydrogens were constrained using LINCS [29]. For each peptide bound to DNA, five simulations with identical initial structures were run and analyzed. MD analyses were performed using tools in the Gromacs package.
Energetic analyses of simulation structures
Continuum electrostatic calculations
Continuum electrostatic calculations were performed by solving the linearized Poisson Boltzmann Equation with a single-grid red-black successive over-relaxation finite-difference solver (M. D. Altman and B. Tidor, unpublished; D. F. Green, E. Kangas, Z. S. Hendsch and B. Tidor, Technology Licensing Office, Massachusetts Institute of Technology) [30] for MD snapshots taken every ns from 40 to 50 ns. A dielectric constant of 4 was used for all peptide and DNA atoms, while the solvent was modeled using a dielectric constant of 80. Bondi radii [31], which have been shown to be reasonable for continuum electrostatics calculations [32, 33], were used for each atom along with AMBER03 charges. A probe radius of 1.4 Å was used for defining the surfaces of these dielectric boundaries. Potentials were solved on a 315×315×315 grid. A two-tiered focusing procedure was used, with the peptide-DNA complex occupying 23% and 92% of the grid, yielding a grid spacing of roughly 3.5 grids/Å at the higher focusing. Zero-radius and uncharged dummy atoms were placed at identical minimum and maximum points of every calculation for equal grid resolution. The ionic strength was set to 100 mM. Total electrostatic binding free energies were obtained by summing the desolvation penalties for each binding partner (in other words, desolvating the spatial cavities of the binding partners) and the solvent-screened interactions between the two partners. The penalty terms were obtained by multiplying one-half the potential differences due to charges on a given binding partner by its charges. Interaction terms were calculated multiplying bound-state potentials due to charges on a binding partner by charges on the other partner [34].
Component analyses
To quantify the contributions of selected portions of a binding partner on the overall electrostatic binding free energy, atomic charges of these portions were set to zero followed by a re-calculation of the binding free energy. The contribution of that portion, or the effect of zeroing out the charges of the portion, was reflected by the change in the electrostatic binding free energy, ΔΔG, obtained by subtracting the binding energy with zeroed atomic charges from the original binding energy with all initial charges.
Free energy of binding calculations
A free energy of binding (ΔGbind) for peptide•DNA systems was computed by considering electrostatics (ΔGelec), van der Waals interactions (ΔGvdw) and a cavitation penalty (ΔGSASA), as done in previous MM-PBSA based approaches [35]. ΔGelec was calculated using the linearized Poisson Boltzmann equation using the software and approach described above. ΔGvdw was computed from the AMBER03 force field Lennard-Jones term with a cutoff of 1.0 nm. The cavitation penalty (ΔGSASA), which is implemented to quantify the hydrophobic effect and favorable van der Waals interactions with water, was estimated using solvent accessible surface area. The probe radius used to compute the solvent accessible surface was 1.4 Å with a proportionality constant of 0.02267 kJ mol−1 Å−2 [36]. Structures from the last 10 ns of simulations were used for binding calculations. A rigid binding assumption was used where the “unbound” structures were also taken from the frames of the peptide•DNA simulation.
Experimental DNA binding measurements
Chemically synthesized wild type and mutant BF2 and DesHDAP1 (Table 1) at >95% purity were obtained from NeoScientific (Cambridge, MA) or GenScript (Piscataway, NJ). The dsDNA (double-stranded DNA) for this experiment was obtained from IDT (Coralville, IA) (Table 2). A fluorescent intercalator displacement (FID) assay was used to experimentally measure the relative DNA binding of each peptide [37, 38]. The FID assay, which has been used previously to measure relative DNA binding of HDAPs [9, 39], involved loading of thiazole orange (0.55 μM), an intercalator that fluoresces upon binding double-stranded nucleic acids, in STE buffer (10 mM Tris, 50 mM NaCl, 1 mM EDTA, pH 8.0) into a quartz cuvette. Fluorescence was measured with 509 nm excitation and 527 nm emission and normalized to 0% relative fluorescence. Double stranded DNA or RNA in STE buffer (1.1 μM) was then added. The solution was equilibrated for 5 minutes followed by measurement of fluorescence normalized to 100% relative fluorescence. Aliquots of a prepared peptide solution (7.8 × 10−5 M) were added periodically before mixing, with 5 minutes of incubation before subsequent measurements of fluorescence. Concentration of peptide required to displace sufficient thiazole orange to reduce fluorescence to half its initial value, or C50, was determined by a linear curve fit. All fits for data included in averaging had R2 > 0.88. The relative DNA binding constants were expressed as the reciprocal of C50, or 1/C50. In comparing dsDNA sequences, binding constants for both BF2 and DesHDAP1 were measured in at least seven independent experiments, and at least three independent measurements were performed for comparisons of peptide variants.
Table 1.
Sequences of peptides used in the study. Residues changed between parent peptides and variants are highlighted with boldface and underline. Note that the F10W variant of BF2 shown in the table was utilized for all models and experiments in this paper as in many previous studies.
| Peptide name | Sequence |
|---|---|
| BF2 | TRSSRAGLQWPVGRVHRLLRK |
| BF2R | TRSSRAGLQWPVGRVHRLLRR |
| BF2K | TKSSKAGLQWPVGKVHKLLKK |
| DesHDAP1 | ARDNKKTRIWPRHLQLAVRN |
| DesHDAP1R | ARDNRRTRIWPRHLQLAVRN |
| DesHDAP1K | AKDNKKTKIWPKHLQLAVKN |
Table 2.
dsDNA sequences used in experimental binding measurements. The H2A-15 sequence is identical to the DNA fragment bound by BF2 in the histone H2A structure used as a template for simulations [25] and was used in previous DNA binding measurements with BF2 [9].
| dsDNA name | Sequence |
|---|---|
| AC | ACA CAC ACA CAC ACA |
| CG | CGC GCG CGC GCG CGC |
| AG | AGA GAG AGA GAG AGA |
| AT | ATA TAT ATA TAT ATA |
| H2A-15 | AAA TAC ACT TTT GGT |
Results
Structural sampling in DNA binding simulations of buforin II and DesHDAP1
The initial models of BF2 and DesHDAP1 bound to DNA were based on the sections of histone H2A protein that were identical to the peptide sequences in a nucleosome core particle crystal structure. All modeling and experiments of BF2 included the F10W mutation to allow for quantification in experimental studies and more direct comparison to many previous studies using that variant [9, 10, 13, 24, 39, 40]. Clearly, the free peptides have significantly fewer structural constraints than the analogous region of the histone structure. Moreover, peptides are likely unstructured as they move through the cytosol, opening them up to assume different conformations upon nucleic acid binding in bacteria as observed in studies of indolicidin [41]. Thus, MD simulations were employed to refine these structures and explore potentially different binding orientations for the peptide•DNA complexes. To this end, we performed five independent 50 ns simulations starting with different initial velocities for each trajectory.
In order to quantify the change in binding conformation over the course of simulations, we measured the RMS deviation of the peptide from its initial conformation in two ways. First, we superimposed the peptide with its initial structure in order to determine how much the peptide changed from its initial conformation (Fig. 1A and 2A). However, this measurement does not give insight into any changes in the positioning of the peptide relative to its bound DNA. Thus, we also calculated the RMS deviation of the peptide from its initial position when superimposing structures throughout the trajectory to the initial position of the DNA backbone (Fig. 1B and 2B). This RMS deviation would reflect diffusion to different binding positions on the DNA and changes in the relative orientation of peptide and DNA in addition to changes in the peptide structure.
Figure 1.
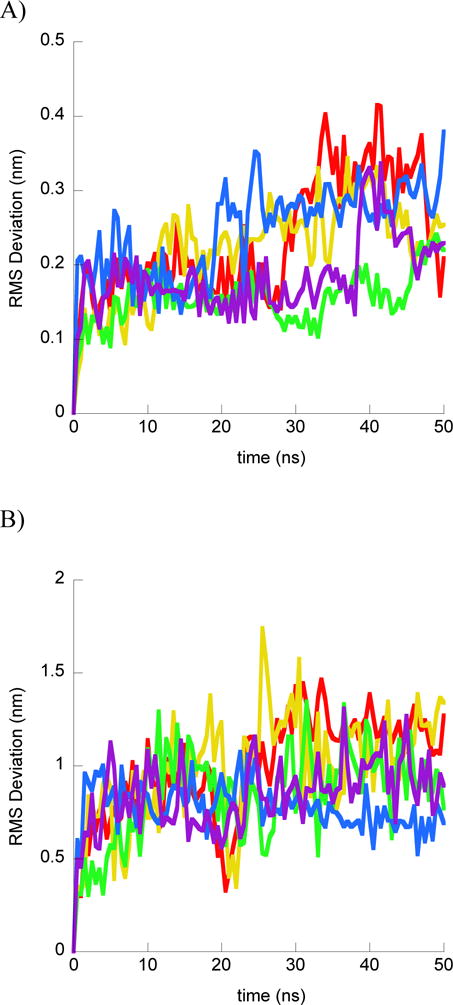
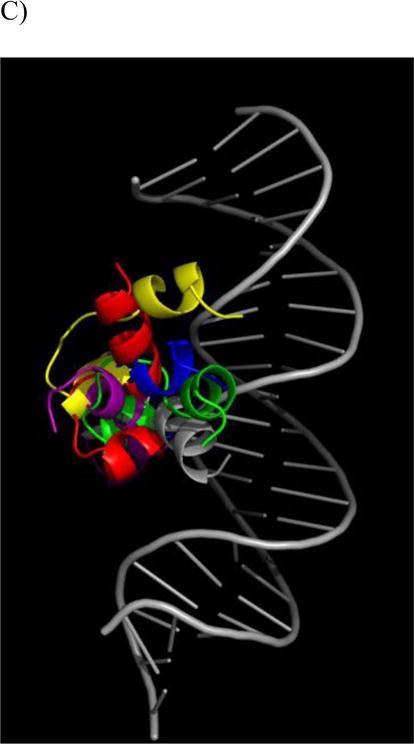
Structural results from BF2•DNA simulations. For each part simulations shown as different color: simulation 1 (red); simulation 2 (yellow); simulation 3 (green); simulation 4 (blue); simulation 5 (purple). A) RMS deviation of BF2 Cα from the initial peptide structure when structures at simulation frames were superimposed with the initial peptide structure. B) RMS deviation of BF2 Cα from the initial peptide structure when structures at simulation frames were superimposed with the initial DNA backbone structure. C) Final BF2 structures from each simulation and the initial BF2 structure. All structures superimposed on the DNA backbone. Peptide and DNA are shown as backbone ribbons with water and ions omitted for clarity.
Figure 2.
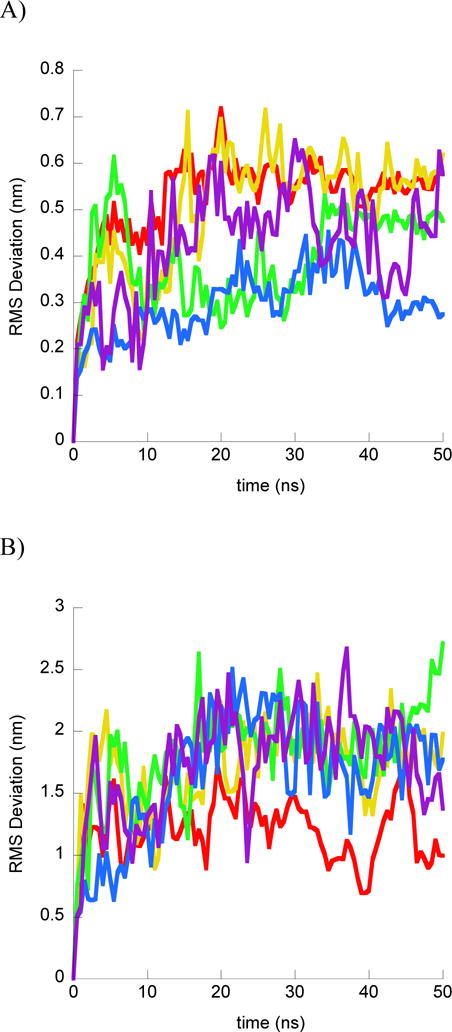
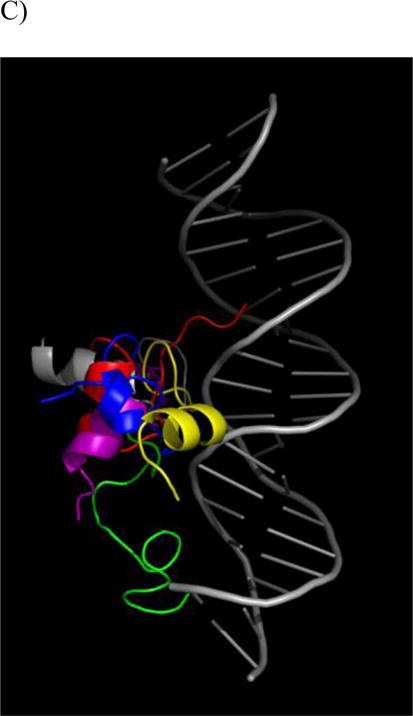
Structural results from DesHDAP1•DNA simulations. For each part simulations shown as different color: simulation 1 (red); simulation 2 (yellow); simulation 3 (green); simulation 4 (blue); simulation 5 (purple). A) RMS deviation of DesHDAP1 Cα from the initial peptide structure when structures at simulation frames were superimposed with the initial peptide structure. B) DesHDAP1 deviation of BF2 Cα from the initial peptide structure when structures at simulation frames were superimposed with the initial DNA backbone structure. C) Final DesHDAP1 structures from each simulation and the initial DesHDAP1 structure. All structures superimposed on the initial DNA backbone. Peptide and DNA are shown as backbone ribbons with water and ions omitted for clarity.
In simulations of BF2, all five simulations generally reached an equilibrated conformation of peptide with respect to DNA over the course of the 50 ns trajectories. Generally, the peptides maintained similar conformations in each simulation, with some α-helical residues distorted by the central Pro 11 residue (Fig. 1A and 1C). However, despite maintaining the same overall peptide conformations, the simulations did show different positions relative to the DNA (Fig 1B and 1C). These changes primarily increased the interactions between the helical region and the DNA backbone, although the helices assumed somewhat different positions in order to develop these increased interactions.
DesHDAP1 peptides also explored a range of peptide conformations relative to the bound DNA in simulations (Fig. 2). Notably, the terminal portions of DesHDAP1 were positioned relatively far from the DNA background in the histone-based initial structure, and all simulations adjusted the system to create additional peptide•DNA contacts. All simulations showed enhanced N-terminal region interactions, which was reasonable considering the concentration of positive charges in that region of the peptide. Notably, one of the simulations showed the peptide entering a groove region not explored in the other four trajectories. While the C-terminal helix region generally did move towards the peptide, this was less consistent between simulations.
Based on these results, it would be impossible to evaluate which of the conformations observed best reflect the “true” structure of the BF2•DNA and DesHDAP1•DNA complex. In fact, it is likely that the actual bound systems may assume a variety of different orientations, particularly considering the significant flexibility of a small peptide, and there are probably other potential conformations not sampled in our simulations. Although the 250 ns of simulations reported for each system were able to capture a range of conformations, more extended simulations may be able to uncover additional binding modes or dynamics that occur on a longer timescale. Previous circular dichroism measurements did not show a defined structure for BF2 bound to DNA [42]. This would be consistent with a peptide assuming a range of binding conformations, including some that are less helical than those observed in our simulations, although the overlap of peptide and DNA spectra can make determining peptide conformations from circular dichroism difficult in bound peptide•DNA systems [23].
Nonetheless, the different conformations observed in our simulations share several similarities in terms of the peptide•DNA interactions described below. These similarities give us increased confidence in the predictions based on our models as they are not biased by considering one particular conformation. To consider the effect of utilizing longer timescale simulations, we also performed three preliminary 100 ns simulations of each peptide with DNA, and these extended simulations did not cause any qualitative changes in averaged properties used for subsequent analyses of peptide•DNA interactions (data not shown). Thus, all results reported in the following sections are averaged over the last 10 ns of the five 50 ns simulations.
BF2 and DesHDAP1 do not show specific binding of DNA sequences
Previous work had predicted that BF2 and DesHDAP1 peptides might primarily interact with the phosphate groups of DNA [9, 11]. We tested whether this was true for our simulations of BF2 and DesHDAP1 by measuring the average percentage of hydrogen bonds formed between a peptide and DNA that involved the DNA phosphate group (Table 3). While the raw numbers of hydrogen bonds between a peptide and DNA fluctuated among the five simulations for each peptide, a very large percentage of the interactions involved phosphate groups in all simulations. For example, no simulation of BF2 had less than 88.0% of peptide•DNA H-bonds involving phosphates, and no DesHDAP1 simulation had less than 72.8% of peptide•DNA H-bonds with phosphate groups. Because these phosphate groups are identical for any nucleic acid sequence, these results imply that there is likely little to no selectivity for particular DNA sequences. Interestingly, there was no clear correlation between the extent of conformational change observed over the course of a simulation and significantly increased or decreased DNA interactions.
Table 3.
Interaction results from MD simulations of BF2 and DesHDAP1 with DNA. H-bonding data reports the average percentages of all peptide•DNA hydrogen bonds that involved the nucleic acid phosphate groups. ΔΔGphos:ΔΔGbases is the ratio of the electrostatic free energy contributions of the phosphate groups to the electrostatic free energy contributions of the nucleic acid bases, as determined from Poisson-Boltzmann electrostatics calculations. All values are averaged over structures taken from the last 10 ns of simulations.
| BF2 | DesHDAP1 | |||
|---|---|---|---|---|
| Simulations | % H-bonds to phosphate | ΔΔGphos:ΔΔGbases | % H-bonds to phosphate | ΔΔGphos:ΔΔGbases |
| 1 | 100.0 | 23.5 | 84.9 | 6.5 |
| 2 | 100.0 | 14.8 | 75.9 | 9.8 |
| 3 | 96.3 | 10.9 | 98.5 | 19.4 |
| 4 | 88.0 | 10.5 | 72.8 | 5.6 |
| 5 | 97.4 | 12.9 | 76.6 | 6.4 |
| average ± SE | 96.3±2.2 | 14.5±2.4 | 81.7±4.6 | 9.6±2.6 |
To provide a more direct energetic consideration of the peptide•DNA interactions, we employed electrostatic analysis to quantify average peptide•DNA interactions in frames taken over the last 10 ns of each BF2 and DesHDAP1 simulation. To consider the portion of overall interactions involving phosphate groups, we calculated the ratio ΔΔGphos:ΔΔGbases (Table 3). ΔΔGphos is essentially the contribution of peptide•phosphate group interaction to the overall electrostatics binding energy between the peptide and DNA. Similarly, ΔΔGbases is the contribution of peptide•base interaction. The fairly high ratios found for the simulations further support that the primary interactions between both peptides and DNA involve phosphate groups.
Our computational analyses suggested that little to no selectivity in base sequence should be expected for BF2 and DesHDAP1 binding of DNA, as both peptides interact primarily with the phosphate groups of nucleic acids. To experimentally test this observation, a fluorescent intercalator displacement (FID) assay was used to measure relative binding constants of BF2 and DesHDAP1 bound to one of four different double-stranded DNA sequences (Table 2). The 1/C50, which is proportional to the binding constant, was compared for the peptides with each DNA strand (Fig. 3). The uncertainty in 1/C50 measurements were overlapping for binding with different base sequences, and no significant differences arose in one-way ANOVA analyses of these results. Thus, these experiments appear to confirm the predicted lack of sequence specificity in the DNA binding for these two peptides.
Figure 3.
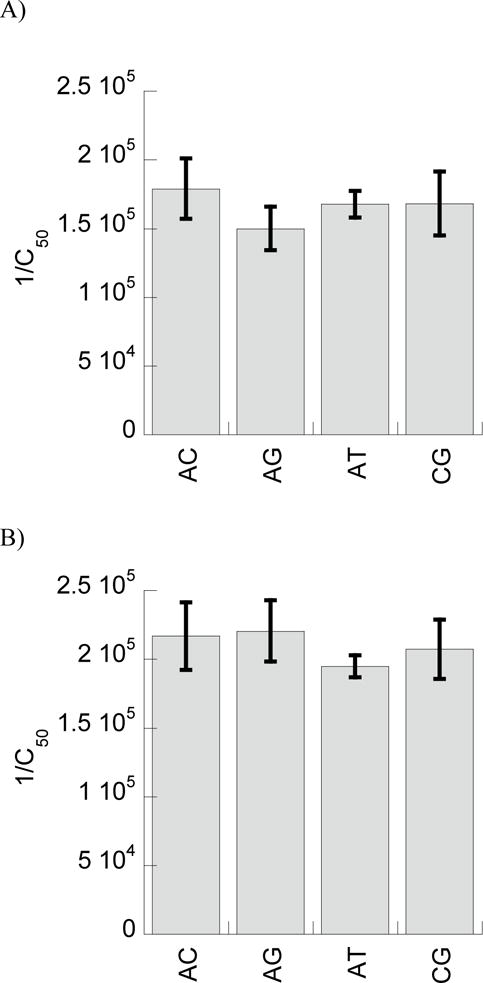
Experimentally measured relative binding constants, expressed as 1/C50, for BF2 (A) and DesHDAP1 (B) with different double-stranded DNA sequences, as given in Table 1. All data reported is averaged from at least seven independent binding experiments with error bars shown as standard error.
Relative arginine composition of basic residues increases DNA binding
In addition to the phosphate interactions, we also noted an interesting trend when considering which peptide residues were primarily involved in mediating interactions with DNA (Fig. 4). As one might expect, the cationic Arg and Lys residues formed the majority of H-bond interactions in each simulation. In fact, the only neutral residue that formed DNA interactions in all five simulations for either peptide was the Thr 7 residue of DesHDAP1, which is flanked on either side by cationic residues. However, within cationic residues, Arg residues form notably more DNA interactions than Lys, with Arg forming over 85% of the cationic residue•DNA H-bonds in both BF2 and DesHDAP1 simulations. While Arg residues are more prevalent in these peptides, they are nonetheless overrepresented relative to Lys in terms of direct interactions with DNA. This may be due to the ability of the guanidinium group in Arg sidechains to form bidentate interactions with the DNA phosphate groups, which cannot occur for the amine in Lys.
Figure 4.
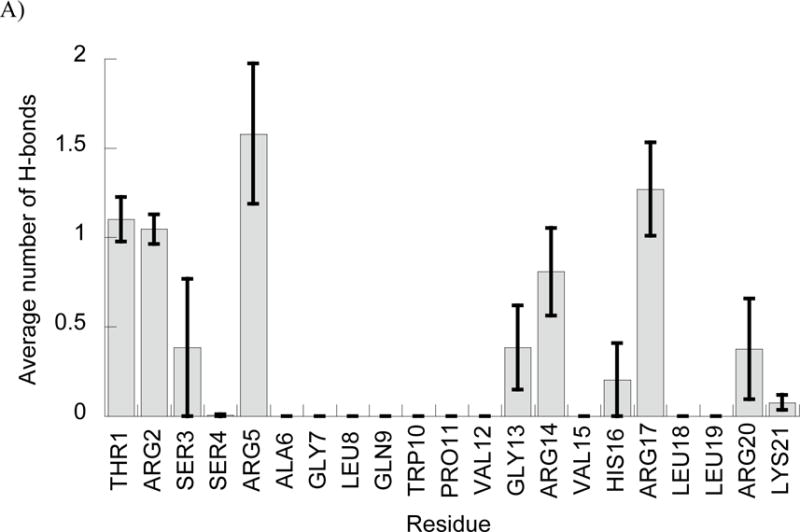
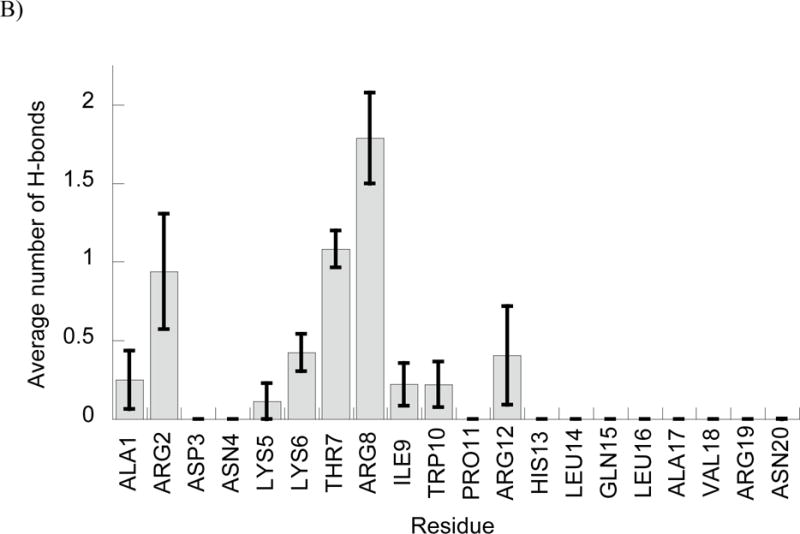
Average number of peptide•DNA H-bonds involving each peptide residue in simulations of BF2 (A) and DesHDAP1 (B). Averages for each simulation determined over the last 10 ns, with the error bars shown as the standard error from averaging values from the five simulations.
This potentially important role for arginine in the DNA interactions of these peptides is particularly intriguing in light of a recent study of BF2 and DesHDAP1 variants containing all lysine residues replaced by arginine (BF2R and DesHDAP1) or all arginine residues replaced by lysine (BF2K and DesHDAP1K) (Table 1). In this study, BF2R and DesHDAP1R showed increased activity relative to the wild type peptides while BF2K and DesHDAP1K showed decreased activity [24]. In order to test the relative importance of arginine composition on the DNA binding we performed additional sets of MD simulations of the all-arginine (BF2R and DesHDAP1) and all-lysine (BF2K and DesHDAP1K) peptide variants bound to DNA (Table 1). Based on the sampling observed for wild type simulations, we performed five replicate 50 ns simulations for each peptide variant.
In these simulations, increased arginine composition did lead to increased DNA interactions for both peptides. Simulations with BF2R and DesHDAP1R formed more peptide•DNA H-bonds than those for BF2K and DesHDAP1K, respectively (Fig. 5A). We also performed energetic analyses of our simulations in which we computed a ΔGbind for the different peptide variants with DNA including electrostatics, van der Waals interactions and a cavitation penalty. This ΔGbind showed an analogous trend to the H-bonding data, with arginine variants binding more strongly than those with increased lysine composition (Fig. 5B). While the quantitative ΔGbind values may be impacted through our rigid binding assumption, we believe they should appropriately capture the trends between arginine and lysine mutations due to the minor effect those mutations have on the unbound peptide structures [24]. The decreased interactions with lysine relative to arginine appeared to occur consistently regardless of the sequence position of a residue, as shown in average number of H-bonds per residue in the simulations (Fig. 5C and 5D).
Figure 5.
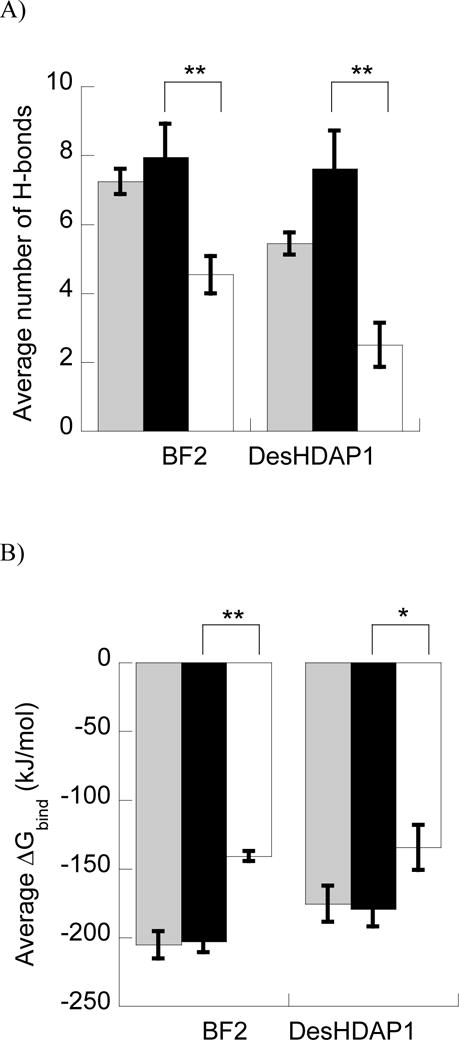
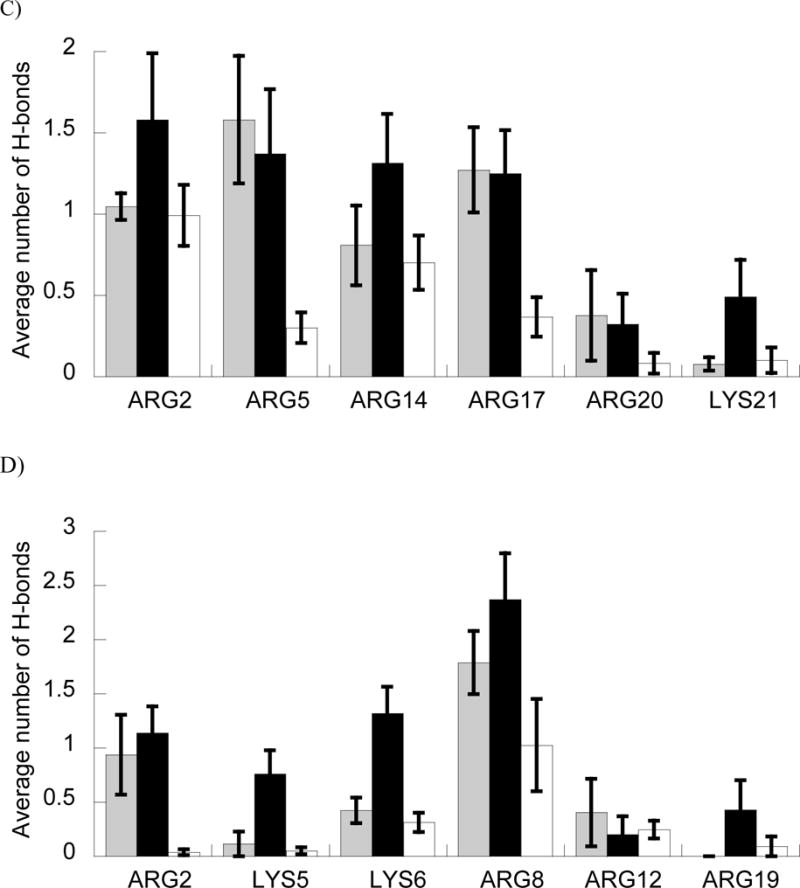
A and B) Average number of H-bonds (A) and ΔGbind (B) between BF2 and DesHDAP1 wild type (gray), arginine (black) and lysine (white) variants and DNA in MD simulations. * denotes p<0.05 and ** denotes p<0.01 for t-test comparisons of arginine and lysine variants of a peptide. C and D) Average number of peptide•DNA H-bonds for each cationic residue in simulations of wild type, arginine (black) and lysine (white) simulations of BF2 (C) and DesHDAP1 (D). Residue names correspond to the wild type peptides. Averages for each simulation determined over the last 10 ns, with the error bars shown as the standard error from averaging values from the five simulations.
In order to confirm these results experimentally, we measured the DNA binding of these peptides with the FID assay employed above. These results confirmed the increased DNA binding of BF2R and DesHDAP1R compared to BF2K and DesHDAP1K (Fig. 6). In both computational and experimental results, no significant difference was observed between the wt BF2 and BF2R peptides, which was consistent with the relatively modest change of a single residue between these peptides (Table 1).
Figure 6.
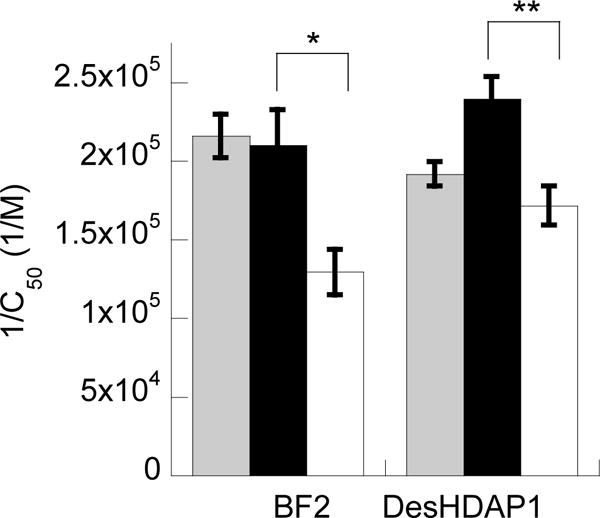
Experimentally measured relative binding constants, expressed as 1/C50, for BF2 and DesHDAP1 wild type (gray), arginine (black) and lysine (white) variants with the H2A-15 DNA sequence (Table 1). All data reported is averaged from at least three independent binding experiments with error bars shown as standard error. * denotes p<0.05 and ** denotes p<0.01 for t-test comparisons of arginine and lysine variants of a peptide.
Discussion
Past studies have noted that intracellular nucleic acids are a potential target for BF2 and other HDAPs, such as DesHDAP1, but have not considered whether these peptides might target specific nucleic acid sequences. Here, we have utilized MD simulations to explore potential DNA binding conformations for these peptides. Although simulations explored different binding conformations, all simulations consistently showed that peptide•DNA interactions primarily involved the nucleic acid phosphate groups. Because phosphates are identical for all DNA sequences, these simulations predicted little to no base sequence selectivity. This prediction was confirmed in peptide•DNA binding experiments. These results are consistent with the relatively broad spectrum behavior of BF2 and DesHDAP1, which are both active against a range of bacterial strains [8, 10, 12]. It may also be more difficult for bacteria to develop resistance mechanisms against these peptides, as they are relatively promiscuous in their DNA targeting. Moreover, these observations also imply that the differences in BF2 and DesHDAP1 activity that do occur between different strains must be due to factors other than nucleic acid differences between strains. Previous work showed that indolicidin does have at least some sequence specificity in its binding, although it is also hypothesized to primarily bind through phosphate groups [41]. Thus, it will be interesting for future studies to consider whether there are any general trends of binding specificity for other AMPs believed to interact with nucleic acids.
Our results also emphasized the particular importance of arginine sidechains in mediating peptide•DNA interactions. Both simulations and experimental data showed that both BF2 and DesHDAP1 variants with increased compositions of arginine residues bind DNA more strongly than those with increased lysine compositions. Recent work from our lab showed increased antimicrobial activity of the BF2 and DesHDAP1 variants (BF2R and DesHDAP1R) where all cationic residues were replaced with arginine [24]. Similar results have been observed for other systems, particularly in the design of antimicrobial peptidomimetics [43–48]. While arginine residues can also increase membrane interactions, our results here show that the enhanced activity of arginine containing peptides may also be due to their increased ability to target nucleic acids. This observation is particularly important for peptide design, as it emphasizes how increasing the presence of guanidinium groups may be a particular effective strategy since it has the potential to enhance multiple aspects of antimicrobial mechanisms.
Overall, these results support the importance of considering how AMPs interact with both the lipid membrane and potential intracellular components, as both interactions can play an important role in determining antimicrobial activity. For example, considering membrane translocation and DNA binding led to a more complete interpretation how proline mutants affect activity of BF2 [39]. Although AMPs are often characterized as primarily killing bacteria by either permeabilizing membranes or interacting with an intracellular component, several studies have proposed that at least some peptides may function through a combination of these different mechanisms [19, 23, 49]. Because of their well-characterized membrane and nucleic acid interactions, HDAPs may continue to provide a valuable model system for considering how different factors combine in AMP activity.
Acknowledgments
Research was supported by National Institute of Allergy and Infectious Diseases (NIH-NIAID) award R15AI079685, National Science Foundation (NSF) award MCB1615313 and Extreme Science and Engineering Discovery Environment (XSEDE) allocation MCB120100. S.S. was supported through the Arnold Beckman Scholars Program. B.N.B. was supported by National Science Foundation award CHE-1005032. D.E.E. is a Henry Dreyfus Teacher-Scholar.
Abbreviations
- AMP
antimicrobial peptide
- HDAP
histone-derived antimicrobial peptide
- BF2
buforin II
- MD
molecular dynamics
- LINCS
linear constraint solver
- FID
fluorescent intercalator displacement
References
- 1.Centers for Disease Control and Prevention. Antibiotic resistance threats in the United States. 2013:2013. [Google Scholar]
- 2.Hancock REW, Sahl HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 3.Toke O. Antimicrobial peptides: New candidates in the fight against bacterial infections. Biopolymers. 2005;80:717–735. doi: 10.1002/bip.20286. [DOI] [PubMed] [Google Scholar]
- 4.Yeaman MR, Yount NY. Mechanisms of antimicrobial peptide action and resistance. Pharmacol Rev. 2003;55:27–55. doi: 10.1124/pr.55.1.2. [DOI] [PubMed] [Google Scholar]
- 5.Hale JD, Hancock RE. Alternative mechanisms of action of cationic antimicrobial peptides on bacteria. Expert Rev Anti-Infe. 2007;5:951–959. doi: 10.1586/14787210.5.6.951. [DOI] [PubMed] [Google Scholar]
- 6.Nicolas P. Multifunctional host defense peptides: intracellular-targeting antimicrobial peptides. The FEBS journal. 2009;276:6483–96. doi: 10.1111/j.1742-4658.2009.07359.x. [DOI] [PubMed] [Google Scholar]
- 7.Park CB, Kim HS, Kim SC. Mechanism of action of the antimicrobial peptide buforin II: Buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem Bioph Res Co. 1998;244:253–257. doi: 10.1006/bbrc.1998.8159. [DOI] [PubMed] [Google Scholar]
- 8.Pavia KE, Spinella SA, Elmore DE. Novel histone-derived antimicrobial peptides use different antimicrobial mechanisms. Bba-Biomembranes. 2012;1818:869–876. doi: 10.1016/j.bbamem.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uyterhoeven ET, Butler CH, Ko D, Elmore DE. Investigating the nucleic acid interactions and antimicrobial mechanism of buforin II. FEBS Lett. 2008;582:1715–1718. doi: 10.1016/j.febslet.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 10.Cho JH, Sung BH, Kim SC. Buforins: Histone H2A-derived antimicrobial peptides from toad stomach. Bba-Biomembranes. 2009;1788:1564–1569. doi: 10.1016/j.bbamem.2008.10.025. [DOI] [PubMed] [Google Scholar]
- 11.Tsao HS, Spinella SA, Lee AT, Elmore DE. Design of novel histone-derived antimicrobial peptides. Peptides. 2009;30:2168–2173. doi: 10.1016/j.peptides.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 12.Birkemo GA, Mantzilas D, Luders T, Nes IF, Nissen-Meyer J. Identification and structural analysis of the antimicrobial domain in hipposin, a 51-mer antimicrobial peptide isolated from Atlantic halibut. Bba-Proteins Proteom. 2004;1699:221–227. doi: 10.1016/j.bbapap.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 13.Kobayashi S, Takeshima K, Park CB, Kim SC, Matsuzaki K. Interactions of the novel antimicrobial peptide buforin 2 with lipid bilayers: Proline as a translocation promoting factor. Biochemistry-Us. 2000;39:8648–8654. doi: 10.1021/bi0004549. [DOI] [PubMed] [Google Scholar]
- 14.Park CB, Yi KS, Matsuzaki K, Kim MS, Kim SC. Structure-activity analysis of buforin II, a histone H2A-derived antimicrobial peptide: The proline hinge is responsible for the cell-penetrating ability of buforin II. P Natl Acad Sci USA. 2000;97:8245–8250. doi: 10.1073/pnas.150518097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park KH, Nan YH, Park Y, Kim JI, Park IS, Hahm KS, Shin SY. Cell specificity, anti-inflammatory activity, and plausible bactericidal mechanism of designed Trp-rich model antimicrobial peptides. Biochim Biophys Acta. 2009;1788:1193–203. doi: 10.1016/j.bbamem.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 16.Song YM, Park Y, Lim SS, Yang ST, Woo ER, Park IS, Lee JS, Kim JI, Hahm KS, Kim Y, Shin SY. Cell selectivity and mechanism of action of antimicrobial model peptides containing peptoid residues. Biochemistry-Us. 2005;44:12094–106. doi: 10.1021/bi050765p. [DOI] [PubMed] [Google Scholar]
- 17.Scocchi M, Mardirossian M, Runti G, Benincasa M. Non-Membrane Permeabilizing Modes of Action of Antimicrobial Peptides on Bacteria. Current topics in medicinal chemistry. 2016;16:76–88. doi: 10.2174/1568026615666150703121009. [DOI] [PubMed] [Google Scholar]
- 18.Libardo MD, Cervantes JL, Salazar JC, Angeles-Boza AM. Improved bioactivity of antimicrobial peptides by addition of amino-terminal copper and nickel (ATCUN) binding motifs. ChemMedChem. 2014;9:1892–901. doi: 10.1002/cmdc.201402033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yi T, Huang Y, Chen Y. Production of an antimicrobial peptide AN5-1 in Escherichia coli and its dual mechanisms against bacteria. Chemical biology & drug design. 2015;85:598–607. doi: 10.1111/cbdd.12449. [DOI] [PubMed] [Google Scholar]
- 20.Haney EF, Petersen AP, Lau CK, Jing W, Storey DG, Vogel HJ. Mechanism of action of puroindoline derived tryptophan-rich antimicrobial peptides. Biochim Biophys Acta. 2013;1828:1802–13. doi: 10.1016/j.bbamem.2013.03.023. [DOI] [PubMed] [Google Scholar]
- 21.Park SC, Kim JY, Jeong C, Yoo S, Hahm KS, Park Y. A plausible mode of action of pseudin-2, an antimicrobial peptide from Pseudis paradoxa. Biochim Biophys Acta. 2011;1808:171–82. doi: 10.1016/j.bbamem.2010.08.023. [DOI] [PubMed] [Google Scholar]
- 22.Ma L, Wang Y, Wang M, Tian Y, Kang W, Liu H, Wang H, Dou J, Zhou C. Effective antimicrobial activity of Cbf-14, derived from a cathelin-like domain, against penicillin-resistant bacteria. Biomaterials. 2016;87:32–45. doi: 10.1016/j.biomaterials.2016.02.011. [DOI] [PubMed] [Google Scholar]
- 23.Hayden RM, Goldberg GK, Ferguson BM, Schoeneck MW, Libardo MD, Mayeux SE, Shrestha A, Bogardus KA, Hammer J, Pryshchep S, Lehman HK, McCormick ML, Blazyk J, Angeles-Boza AM, Fu R, Cotten ML. Complementary Effects of Host Defense Peptides Piscidin 1 and Piscidin 3 on DNA and Lipid Membranes: Biophysical Insights into Contrasting Biological Activities. The journal of physical chemistry B. 2015;119:15235–46. doi: 10.1021/acs.jpcb.5b09685. [DOI] [PubMed] [Google Scholar]
- 24.Cutrona KJ, Kaufman BA, Figueroa DM, Elmore DE. Role of arginine and lysine in the antimicrobial mechanism of histone-derived antimicrobial peptides. FEBS Lett. 2015;589:3915–20. doi: 10.1016/j.febslet.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 26.Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–54. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman P. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 28.Darden TD, York D, Pedersen L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 29.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: A linear constraint solver for molecular simulations. J Comp Chem. 1997;18:1463–1472. [Google Scholar]
- 30.Altman MD. Computational ligand design and analysis in protein complexes using inverse methods, combinatorial search, and accurate solvation modeling. Massachusetts Institute of Technology; Cambridge, MA: 2006. [Google Scholar]
- 31.Bondi A. van der Waals volumes and radii. J Phys Chem. 1964;68:441–451. [Google Scholar]
- 32.Su PC, Tsai CC, Mehboob S, Hevener KE, Johnson ME. Comparison of radii sets, entropy, QM methods, and sampling on MM-PBSA, MM-GBSA, and QM/MM-GBSA ligand binding energies of F. tularensis enoyl-ACP reductase (FabI) Journal of computational chemistry. 2015;36:1859–73. doi: 10.1002/jcc.24011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swanson JMJ, Adcock SA, McCammon JA. Optimized radii for Poisson-Boltzmann calculations with the AMBER force field. J Chem Theory Comput. 2005;1:484–493. doi: 10.1021/ct049834o. [DOI] [PubMed] [Google Scholar]
- 34.Radhakrishnan ML. Designing electrostatic interactions in biological systems via charge optimization or combinatorial approaches: insights and challenges with a continuum electrostatic framework. Theor Chem Acc. 2012;131:1252. [Google Scholar]
- 35.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE., 3rd Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000;33:889–97. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 36.Kumari R, Kumar R, Open Source Drug Discovery C. Lynn A. g_mmpbsa–a GROMACS tool for high-throughput MM-PBSA calculations. Journal of chemical information and modeling. 2014;54:1951–62. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 37.Jenkins TC. Optical absorbance and fluorescence techniques for measuring DNA-drug interactions. Methods Mol Biol. 1997;90:195–218. doi: 10.1385/0-89603-447-X:195. [DOI] [PubMed] [Google Scholar]
- 38.Tse WC, Boger DL. A fluorescent intercalator displacement assay for establishing DNA binding selectivity and affinity. Accounts Chem Res. 2004;37:61–69. doi: 10.1021/ar030113y. [DOI] [PubMed] [Google Scholar]
- 39.Xie Y, Fleming E, Chen JL, Elmore DE. Effect of proline position on the antimicrobial mechanism of buforin II. Peptides. 2011;32:677–682. doi: 10.1016/j.peptides.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kobayashi S, Chikushi A, Tougu S, Imura Y, Nishida M, Yano Y, Matsuzaki K. Membrane translocation mechanism of the antimicrobial peptide buforin 2. Biochemistry-Us. 2004;43:15610–15616. doi: 10.1021/bi048206q. [DOI] [PubMed] [Google Scholar]
- 41.Hsu CH, Chen C, Jou ML, Lee AY, Lin YC, Yu YP, Huang WT, Wu SH. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: evidence for multiple conformations involved in binding to membranes and DNA. Nucleic acids research. 2005;33:4053–64. doi: 10.1093/nar/gki725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lan Y, Ye Y, Kozlowska J, Lam JK, Drake AF, Mason AJ. Structural contributions to the intracellular targeting strategies of antimicrobial peptides. Biochim Biophys Acta. 2010;1798:1934–43. doi: 10.1016/j.bbamem.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andreev K, Bianchi C, Laursen JS, Citterio L, Hein-Kristensen L, Gram L, Kuzmenko I, Olsen CA, Gidalevitz D. Guanidino groups greatly enhance the action of antimicrobial peptidomimetics against bacterial cytoplasmic membranes. Biochim Biophys Acta. 2014;1838:2492–502. doi: 10.1016/j.bbamem.2014.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bahnsen JS, Franzyk H, Sandberg-Schaal A, Nielsen HM. Antimicrobial and cell-penetrating properties of penetratin analogs: effect of sequence and secondary structure. Biochim Biophys Acta. 2013;1828:223–32. doi: 10.1016/j.bbamem.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 45.Gabriel GJ, Madkour AE, Dabkowski JM, Nelson CF, Nusslein K, Tew GN. Synthetic mimic of antimicrobial peptide with nonmembrane-disrupting antibacterial properties. Biomacromolecules. 2008;9:2980–3. doi: 10.1021/bm800855t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Locock KE, Michl TD, Valentin JD, Vasilev K, Hayball JD, Qu Y, Traven A, Griesser HJ, Meagher L, Haeussler M. Guanylated polymethacrylates: a class of potent antimicrobial polymers with low hemolytic activity. Biomacromolecules. 2013;14:4021–31. doi: 10.1021/bm401128r. [DOI] [PubMed] [Google Scholar]
- 47.Svenson J, Karstad R, Flaten GE, Brandsdal BO, Brandl M, Svendsen JS. Altered Activity and Physicochemical Properties of Short Cationic Antimicrobial Peptides by Incorporation of Arginine Analogues. Mol Pharmaceut. 2009;6:996–1005. doi: 10.1021/mp900057k. [DOI] [PubMed] [Google Scholar]
- 48.Vedel L, Bonke G, Foged C, Ziegler H, Franzyk H, Jaroszewski JW, Olsen CA. Antiplasmodial and prehemolytic activities of alpha-peptide-beta-peptoid chimeras. Chembiochem. 2007;8:1781–4. doi: 10.1002/cbic.200700385. [DOI] [PubMed] [Google Scholar]
- 49.Wei L, LaBouyer M, Darling LE, Elmore DE. Bacterial Spheroplasts as a Model for Visualizing Membrane Translocation of Antimicrobial Peptides. Antimicrobial agents and chemotherapy. 2016;60:6350–6352. doi: 10.1128/AAC.01008-16. [DOI] [PMC free article] [PubMed] [Google Scholar]