

Abstract
Right ventricular (RV) remodeling represents a complex set of functional and structural adaptations in response to chronic pressure or volume overload due to various inborn defects or acquired diseases and is an important determinant of patient outcome. However, the underlying molecular mechanisms remain elusive. We investigated the time course of structural and functional changes in the RV in the murine model of pressure overload‐induced RV hypertrophy in C57Bl/6J mice. Using magnetic resonance imaging, we assessed the changes of RV structure and function at different time points for a period of 21 days. Pressure overload led to significant dilatation, cellular and chamber hypertrophy, myocardial fibrosis, and functional impairment of the RV. Progressive remodeling of the RV after pulmonary artery banding (PAB) in mice was associated with upregulation of myocardial gene markers of hypertrophy and fibrosis. Furthermore, remodeling of the RV was associated with accumulation and activation of mast cells in the RV tissue of PAB mice. Our data suggest possible involvement of mast cells in the RV remodeling process in response to pressure overload. Mast cells may thus represent an interesting target for the development of new therapeutic approaches directed specifically at the RV.
Keywords: Mast cell, pulmonary artery banding, right ventricular hypertrophy
Introduction
Right ventricular (RV) remodeling represents a complex set of functional and structural adaptations induced by chronic pressure or volume overload due to various inborn defects or acquired diseases. Although the response of the RV to chronic pressure overload is an important determinant of patient outcome, the underlying molecular mechanisms remain elusive (Guarracino et al. 2005). A better understanding of the mechanisms of the RV remodeling may help identify candidate targets for novel therapeutic strategies directed specifically at the RV and thus improve survival in these patients. The availability of a murine model of pulmonary artery banding (PAB) provides us with a valuable tool to explore the mechanisms involved in the RV remodeling following pressure overload (Rockman et al. 1994; Tarnavski et al. 2004; Tarnavski 2009). Recently, a serial echocardiographic assessment of RV function changes in FVB mice subjected to PAB has been reported (Urashima et al. 2008). However, a more detailed investigation of longitudinal changes in concomitant maladaptive processes, including cardiomyocyte hypertrophy, myocardial fibrosis, and capillary density alterations, was not performed. In addition, numerous studies have demonstrated that significant differences in cardiac structure and function exist between mouse strains (Shah et al. 2010; Moreth et al. 2014). Even when cardiovascular parameters may not be different between strains under physiological conditions, genetic background may affect the cardiovascular response to various insults, such as hypoxia, aortic constriction, ischemia‐reperfusion, or myocardial infarction (Campen et al. 2004; Gao et al. 2005; Barrick et al. 2007; Izikki et al. 2007; Tada et al. 2008; van den Borne et al. 2009; Barnabei et al. 2010). Moreover, strain‐specific differences may affect results of pharmacological treatments (Lucki et al. 2001). Therefore, a direct comparison of findings of studies in which different inbred strains have been used may be challenging or impossible.
As recently shown in the rat SU/Hypoxia model of pulmonary arterial hypertension, differences in background strain can also affect the ability of the right heart to cope with increased pulmonary artery pressure (Jiang et al. 2016). However, the interstrain differences in the response of the RV to pressure overload have not been investigated comprehensively yet. Interestingly, it has recently been shown that C57BL/6J, the most widely used strain in the biomedical research (Simon et al. 2013), and FVB mice have different cardiovascular phenotypes at baseline and in response to acute hypoxic challenge (Barnabei et al. 2010). As a detailed characterization of the time course of pressure overload‐induced changes in RV morphology and function in other commonly used mouse strains is still lacking, we sought to investigate serial changes in RV morphology and function in C57BL/6J mice subjected to sustained pressure overload.
Although mast cell functions have been related to immune responses, a rapidly growing body of evidence has implicated these cells in a large variety of pathophysiologic processes (Puxeddu et al. 2003). Mast cells are also present in the normal heart tissues in humans and animals (Marone et al. 2000) and an increasing number of studies suggest possible roles of cardiac mast cells in the pathogenesis of various cardiovascular diseases (Levick et al. 2011). In particular, enhanced accumulation of mast cells in hypertensive and failing left ventricles has been documented (Panizo et al. 1995; Shiota et al. 2003; Batlle et al. 2006). Recent studies have identified important roles for mast cells in left ventricular hypertrophy and failure (Hara et al. 2002; Kitaura‐Inenaga et al. 2003; Stewart et al. 2003). However, the role of mast cells in the development of RV hypertrophy is poorly understood. The goal of this study was therefore twofold. First, to better understand longitudinal changes in RV morphology and function in C57BL/6J mice subjected to sustained pressure overload. Second, to investigate the involvement of mast cells in the RV remodeling process.
Material and Methods
8–10‐week‐old male C57BL/6J mice (Charles River Laboratories, Sulzfeld, Germany) were maintained under appropriate barrier conditions in a 12‐h/12‐h light‐dark cycle and received standard laboratory food ad libitum and free access to water. All procedures involving animals were approved by the governmental Animal Ethics Committee (Regierungspräsidium Darmstadt).
Model of PAB
Pressure overload was induced by PAB (n = 10 mice for each time point) as described (Kreymborg et al. 2010). Briefly, buprenorphine hydrochlorid (0.05 mg/kg bw, Vetergesic, Braun) was administered s.c. as an analgesic prior to operation. Surgery was performed under general anesthesia using isoflurane (1.5–2.5% v/v). The animals were placed on a heating pad to maintain body temperature and were ventilated with a rodent ventilator (MiniVent Type 845, Hugo Sachs Elektronik KG, March, Germany). A lateral thoracotomy was performed to gain access to the pulmonary artery. The pulmonary artery was constricted to 350 μm using titanium clips (Hemoclip®, Weck, Germany) and a modified, adjustable clip applier (Hemoclip®). The chest and the skin incisions were closed by standard surgery techniques. Sham‐operated mice (n = 10 mice for each time point) served as controls and underwent an identical procedure except for the placement of the clip around the pulmonary artery. Carprofen (8 mg/kg per day, Rimadyl®, Pfizer) was administered via drinking water for 3 days post operation. Thereafter, animals were observed for various time periods after surgical procedures.
Magnetic resonance imaging
Cardiac magnetic resonance imaging (MRI) was performed at baseline (before surgery) and on postoperative days 3, 7, 14, and 21 (n = 10 mice per group for each time point). Mice were anesthetized by inhalation of isoflurane (2% v/v) in oxygen/medical air (0.5/0.5 L/min) mixture. Cardiac MR measurements were performed on a 7.0T Bruker PharmaScan, equipped with a 300 mT/m gradient system, using a custom‐built circularly polarized birdcage resonator and the IntraGate™ self‐gating tool (Bruker, Ettlingen, Germany). The measurement is based on the gradient echo method (repetition time = 6.2 msec; echo time = 1.6 msec; field of view = 2.20 × 2.20 cm; slice thickness = 1.0 mm; matrix = 128 × 128; repetitions = 100; resolution 0.0172 cm/pixel). The imaging plane was localized using scout images showing the sagittal and coronal views of the heart, followed by acquisition in axial view, orthogonally to the septum of both scout scans. Multiple (9–10) contiguous axial slices were acquired for complete coverage of the ventricles. MRI data were analyzed using MASS4Mice digital imaging software (Medis, Leiden, Netherlands). The RV and left ventricular end‐diastolic volume (EDV) and end‐systolic volume (ESV) were determined and based on these values stroke volume (SV = EDV‐ESV), ejection fraction (EF = (SV/EDV)*100%), and cardiac output (CO = SV × heart rate) were calculated automatically. The myocardial volume was determined as the area inside the epicardial border minus the endocardial area. The myocardial mass was then obtained by multiplying the myocardial volume and the density of myocardial tissue (1.05 g/mL). Left ventricular eccentricity index (LVEI) values were calculated for both end‐diastole and end‐systole in the axial plane at midpapillary level. It is calculated as the ratio of the length of the major axis of the left ventricle (L1), which runs parallel to the septum, to the length of the minor axis of left ventricle (L2), which runs perpendicular to the septum: LVEI = L1/L2.
Hemodynamic measurements
In vivo hemodynamic measurements were performed after completion of the protocols to induce RV hypertrophy (n = 10 mice per group for each time point). Mice were anesthetized using isoflurane (1.5% v/v) and placed on controlled heating and the core temperature, measured via rectal probe, was maintained at 37°C. The right jugular vein was used for catheterization of the RV to measure RV systolic pressure (RVSP), RV end‐diastolic pressure (RVEDP), and time constant of isovolumic pressure decay Tau. Systemic arterial pressure (SAP) was measured by catheterizing the right carotid artery. Hemodynamic measurements were performed using a Millar microtip catheter (SPR‐671, FMI, Foehr Medial Instruments GmbH, Seeheim/Ober‐Beerbach, Germany) and a PowerLab 8/30 System with the Chart 7.0 Software (AD Instruments GmbH, Spechbach, Germany).
Tissue processing and histology
Hearts were harvested immediately after animals were killed (n = 5 mice per group for each time point). The ventricles were dissected free of the great vessels and atria. The RV was isolated from the left ventricle (LV) + septum (LV + S) by dissection along the septal insertion. The RV and (LV + S) were patted dry and weighed. The RV hypertrophy was assessed using the ratio of the RV mass to tibia length and to body weight.
Freshly dissected RV tissue was fixed in 4% paraformaldehyde overnight, then dehydrated and embedded in paraffin and sectioned at a thickness of 3 μm. To detect collagen fibers, RV sections were stained with 0.1% sirius red (sirius red F3B, Niepoetter, Bürstadt, Germany) in picric acid (Fluka, Neu‐Ulm, Germany). For cardiomyocyte size determination transverse sections of the RV were stained with FITC conjugated wheat germ agglutinin (WGA‐FITC, Sigma‐Aldrich, Steinheim, Germany) for membrane staining. For quantification of the capillaries, sections were stained with IB4‐TRITC (Sigma Aldrich). Nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI, Invitrogen, Karlsruhe, Germany) and mounted in DAKO fluorescence mounting medium (DAKO, Hamburg Germany). Sections without WGA‐FITC, IB4‐TRITC, and DAPI staining were used as a negative control. Photomicrographs were quantified to determine the mean cross‐sectional area of cardiomyocytes and interstitial collagen fraction using Leica Qwin V3 computer‐assisted image analysis software (Leica Microsystem, Wetzlar, Germany). Photomicrographs were quantified to count the number of cardiomyocytes and capillaries using STEPanizer image analysis tool (University of Bern, Department of Anatomy, Bern, Switzerland). Myocardial capillary density was expressed as a number of capillaries per cardiomyocyte. Average data reflect results from at least four or five different hearts in each group (more than 100 cells for each heart).
Mast cell density and activity
To identify mast cells, toluidine blue staining was performed using standard protocol (Dahal et al. 2011). Briefly, paraffin‐embedded RV tissue sections were dewaxed, rehydrated and incubated with 0.05% w/v toluidine blue for 2–3 min. Mast cell density was quantified by counting the number of toluidine blue‐positive cells from each entire RV longitudinal sections (Leica QWin, 200X) in two random sections. The mast cell density was expressed as a number of mast cells per mm2. Degranulated mast cells are identified as cells in which granules are substantially reduced by 70–90% (Kanellakis et al. 2012). The results were expressed as percentage of degranulated mast cell to total amount of mast cells.
Quantitative real‐time PCR
RV tissue samples were harvested immediately after mice were killed, snap‐frozen in liquid nitrogen and stored at −80°C until further analysis (n = 5 mice per group for each time point). Total mRNA was extracted from snap‐frozen mouse RV tissue samples using RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. The primers used for real‐time quantitative PCR are presented in the Table 1. All results were normalized to the relative expression of the constitutively expressed gene porphobilinogen deaminase (PBGD). The relative transcript abundance of the target gene is expressed in ΔCt values (ΔCt = Ct reference–Ct target).
Table 1.
Sequence of the primers used in quantitative real‐time PCR reactions
| Gene | Primer sequence |
|---|---|
| ANP |
Forward Primer: TCTGCCCTCTTGAAAAGCAA Reverse Primer: TTCGGTACCGGAAGCTGTT |
| BNP |
Forward Primer: GAACGTGCTGTCCCAGATGA Reverse Primer: TCCAGGAGCTTCTGCATCTT |
| Col1α |
Forward Primer: GACGGGAGGGCGAGTGCTGT Reverse Primer: ACGGGTCCCCTTGGGCCTTG |
| Col3α |
Forward Primer: AAAGGGTGAAATGGGTCCCAG Reverse Primer: TCACCTGAAGGACCTCGAGT |
| TGF‐β |
Forward Primer: AGAAGGCAAGCCGGAGGGCA Reverse Primer: TGGGCGGGATGGCATTTTCGG |
| PAI‐1 |
Forward Primer: TGGCGTCTTCCTCCACAGCCTT Reverse Primer: GTCGGGTTGTGCCGAACCACA |
| cKit |
Forward Primer: TCTTCCGGCACAACGGCACG Reverse Primer: GTGGGCCTGGATTTGCTCTTTGTTGT |
| mMCP‐1 |
Forward Primer: GAGCTGGAGCTGAGGAGATTA Reverse Primer: CTCAGAACCTCTGTCCGT |
| mMCP‐2 |
Forward Primer: GCACTTCTTTTGCCTTCTGG Reverse Primer: TAAGGACGGGAGTGTGGTTT |
| mMCP‐4 |
Forward Primer: TCTGGGGCTGGAGCTGAGGAGA Reverse Primer: GCAGCAGTCAACACAAATTGGCGG |
| mMCP‐5 |
Forward Primer: ATCTGCTGCTCCTTCTCCTG Reverse Primer: ACTCCGTGCCTCCAATGA |
| mMCP‐6 |
Forward Primer: GGCAGGTGAGCCTGAGATTT Reverse Primer: GGAAGAGCTGTGGGCTTTTG |
| CPA3 |
Forward Primer: CAAGGATTAAAATTGGATCAACTG Reverse Primer: GATAGCCTTTCTTTCTCCATCTTT |
| IL6 |
Forward Primer: CCTCTCTGCAGGAGACTTCCATCCA Reverse Primer: AGCCTCCGACTTGTGAGGTGGT |
| TNF‐α |
Forward Primer: TACTGAACTTCGGGGTGATTGGTCC Reverse Primer: CAGCCTTGTCCCTTGAAGAGAACC |
| PBGD |
Forward Primer: AGAAGAGCCTGTTTACCAAGGAG Reverse Primer: TTTCTCTGTAGCTGAGCCACTCT |
Statistical analysis
Data are expressed as mean ± standard error the mean (SEM). All statistics were performed using GraphPad Prism software version 5.00 for Windows (GraphPad Software Inc., San Diego, CA). Differences between parameters of control and corresponding PAB mice at each time point were compared by an unpaired two‐sided Student's t‐test or one‐way ANOVA followed by Dunnett's post hoc test. Differences in the time course of MRI parameters between sham and PAB mice were assessed using repeated measures ANOVA with Bonferroni post hoc test for multiple comparisons. Differences were considered statistically significant when P < 0.05.
Results
In order to characterize the time course of morphological and functional changes in the RV in response to pressure overload, mice were randomly assigned to either sham or PAB group. Then a MRI analysis of the RV structure and function was performed at baseline before surgery and subsequently at 3, 7, 14, and 21 days after surgery.
Noninvasive MRI demonstrated that PAB led to severe dilatation, hypertrophy, and functional impairment of the RV (Fig. 1A–E). Pressure overload resulted in increased end‐systolic and end‐diastolic RV volumes on day 3 after PAB (Fig. 1A and B). The RV chamber dilatation was attenuated on postoperative day 7 but significantly increased afterwards. The RV dilatation was accompanied by impaired contractile function as reflected by decreased ejection fraction in PAB animals (Fig. 1C). In PAB mice, abnormal bulging of the interventricular septum toward the left ventricle due to compression exerted by dilated and hypertrophied RV, as reflected by a rapid progressive increase in the LVEI both in systole and diastole (Fig. 1F and G), led to significant reduction in left ventricular end‐systolic and end‐diastolic volumes at all time points after surgery (Fig. 1H and I). The impaired ability of the left ventricle to properly dilate and fill resulted in reduced left ventricular stroke volume and cardiac output, whereas the left ventricular ejection fraction basically remained unchanged (Fig. 1J–L).
Figure 1.
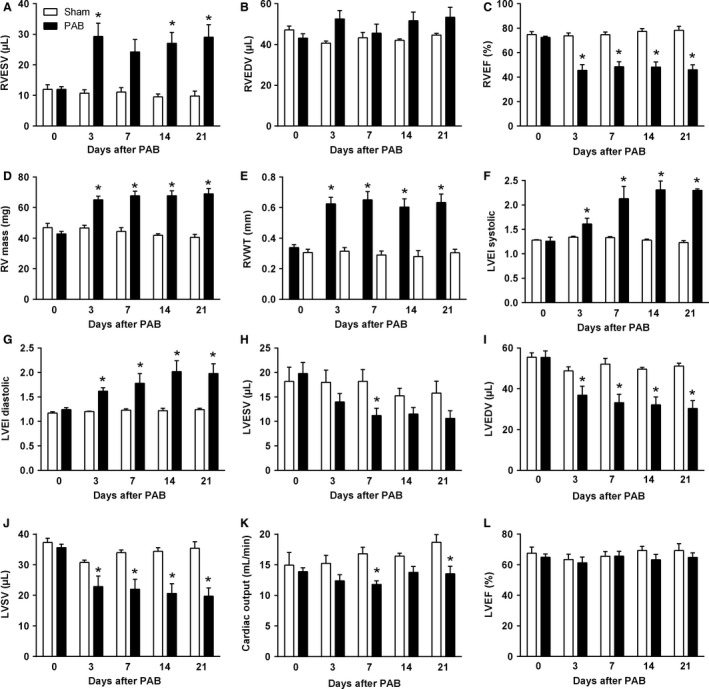
Characterization of right and left ventricular structure and function and interventricular interaction. (A) Right ventricular end‐systolic volume. (B) Right ventricular end‐diastolic volume. (C) Right ventricular ejection fraction. (D) Right ventricular mass. (E) Right ventricular wall thickness. (F) Left ventricular eccentricity index during end‐systole. (G) Left ventricular eccentricity index during end‐diastole. (H) Left ventricular end‐systolic volume. (I) Left ventricular end‐diastolic volume. (J) Left ventricular stroke volume. (K) Cardiac output. (L) Left ventricular ejection fraction. Values are means ± SEM. *P < 0.05 versus sham at corresponding time points, n = 10 mice per group.
The RVSP in PAB mice increased with time coinciding with development of RV hypertrophy (Fig. 2A). The diastolic parameter Tau was elevated in PAB mice at all time points, whereas RVEDP started increasing from day 14 (Fig. 2B and C). The systemic blood pressure decreased slightly but significantly in PAB mice (Fig. 2D). The invasively determined RV hypertrophy developed rapidly after induction of pressure overload. The RV mass significantly increased in PAB mice as early as 3 days after surgery and continued to increase thereafter until day 21 (Fig. 2E and F). On a cellular level, significant cardiomyocyte hypertrophy was evident on day 7 after PAB and gradually increased thereafter (Fig. 2G and J). Significant interstitial collagen deposition in PAB mice occurred at day 7 after surgery and progressively increased over time (Fig. 2H and J). The number of microvessels per cardiomyocyte increased significantly by day 7 and remained elevated thereafter (Fig. 2I and J).
Figure 2.
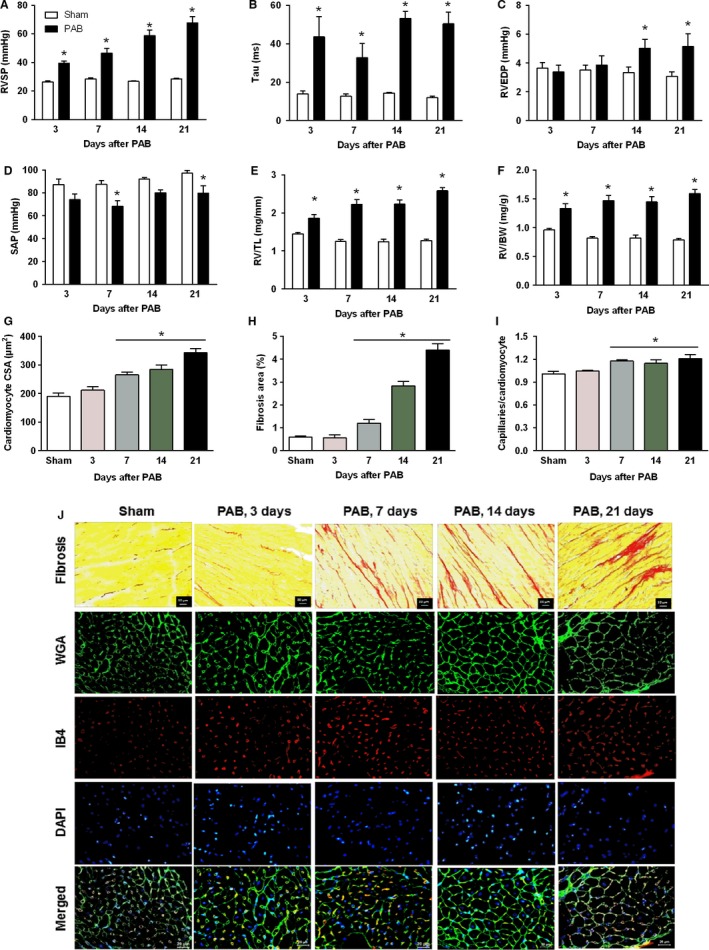
Characterization of hemodynamics and right ventricular remodeling. (A) Right ventricular systolic pressure. (B) Time constant of isovolumic relaxation of the right ventricle Tau. (C) Right ventricular end‐diastolic pressure. (D) Systemic arterial pressure. (E) Post‐mortem measured right ventricular weight values normalized to tibia length (RV/TL). (F) Post‐mortem measured right ventricular weight values normalized to body weight (RV/BW). (G) Bar graphs summarizing quantification of mean right ventricular cardiomyocyte cross‐sectional area. (H) Bar graphs summarizing quantification of interstitial fibrosis. (I) Bar graphs summarizing quantification of angiogenesis. (J) Representative images of picro‐sirius red staining of right ventricles cut in cross‐section and stained with wheat germ agglutinin‐FITC conjugate (WGA), isolectin B4‐TRITC conjugate (IB4), DAPI and merged. WGA marks cell boundaries (green), isolectin B4 (red) marks endothelial cells and DAPI marks nuclei (blue). Values are means ± SEM. *P < 0.05 vs. sham at corresponding time points, n = 10 mice per group for hemodynamic parameters and n = 5 mice per group for histological parameters.
The RV remodeling process was associated with significantly increased mRNA expression levels of hypertrophic and profibrotic (ANP, BNP, collagen 1, collagen 3, Tgf‐β, and PAI1) markers in RV tissue from PAB mice compared with those in the sham‐operated control group. Expression of ANP and BNP mRNA increased early and was maintained at high levels up to 21 days after PAB (Fig. 3A and B). Interestingly, mRNA expression of profibrotic genes started increasing as early as 3 days after surgery, reached a maximum by day 7 and then gradually declined (Fig. 3C–F).
Figure 3.
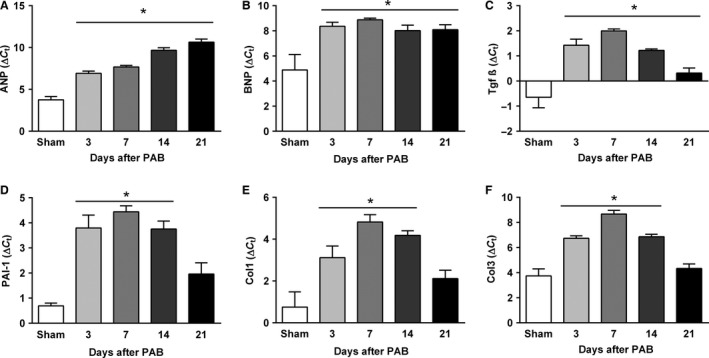
Time course of the right ventricular mRNA gene expression of hypertrophic (A and B) and profibrotic markers (C–F) in sham and PAB mice. (A) Atrial natriuretic peptide. (B) B‐type natriuretic peptide. (C) Transforming growth factor 1. (D) Plasminogen activator inhibitor‐1. (E) Collagen 1 (Col1). (F) Collagen 3 (Col 3). Values are means ± SEM. *P < 0.05 versus sham at corresponding time points, n = 5 mice per group. SEM, standard error the mean.
A time‐dependent increase in total number of mast cells and the proportion of degranulated mast cells was detected in the remodeled RV tissue from PAB mice (Fig. 4A–C). The proportion of degranulated mast cells started increasing from day 3 after PAB and reached a plateau by day 7. Remarkably, the mast cell density in PAB mice started increasing after 14 days and reached maximal values 21 days post surgery. The increased number of mast cells was accompanied by significantly increased mRNA expression of the mast cell marker c‐Kit and mast cell‐specific genes encoding murine mast cell proteases (mMCP)‐2, 4, 5, and 6 in the RV at 7 and 14 days post surgery (Fig. 5A–F). Expression of the carboxypeptidase A3 (CPA3) increased early after PAB and remained elevated for the entire period (Fig. 5G). Similarly, augmented expression of the cytokine IL‐6 was observed in RV tissue of PAB mice at all time points (Fig. 5H). Significantly elevated RV expression of TNF‐alpha in PAB mice was noted at 7 and 14 days after PAB which further increased by day 21 (Fig. 5I).
Figure 4.
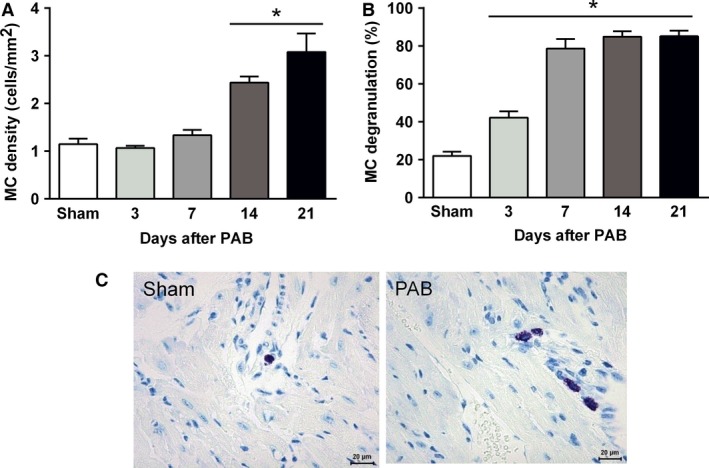
Accumulation and activation of mast cells (MC) in the process of the RV remodeling. (A) Mast cell density. (B) Mast cell activity. (C) Representative images of mast cells in RV tissue from sham and PAB mice, n = 5 mice per group.
Figure 5.
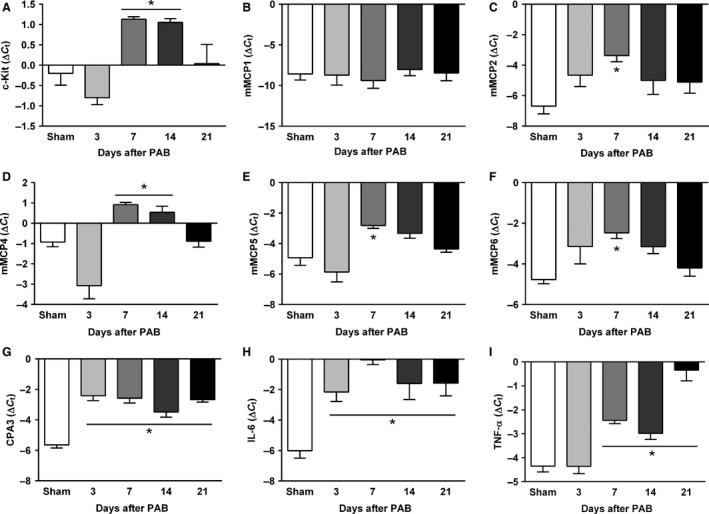
Time course of the right ventricular mRNA gene expression analysis of genes related to mast cells (A–G) and inflammation (H and I) in sham and PAB mice. (A) c‐Kit. (B) Murine mast cell protease (mMCP)‐1. (C) mMCP2. (D) mMCP4. (E) mMCP5. (F) mMCP6. (G) carboxypeptidase‐3 (CPA3). (H) IL‐6. (I) TNF‐α. Values are means ± SEM. *P < 0.05 versus sham at corresponding time points, n = 5 mice per group. PAB, pulmonary artery banding.
Discussion
In this study, we characterized the longitudinal changes in RV morphology and function in response to pressure overload in C57BL/6J mice. We demonstrated that PAB led to significant dilatation, cellular and chamber hypertrophy, and functional impairment of the RV. Compression of the left ventricle exerted by dilated and hypertrophied RV caused flattening of the interventricular septum and was reflected by the increased LVEI. The latter has been shown to increase in various RV pressure and volume overload states (Ryan et al. 1985; Ascah et al. 1990; Louie et al. 1992). Due to deleterious interventricular interaction, compression of the left ventricle led to significant reduction in left ventricular end‐systolic and end‐diastolic volumes. The impaired ability of the left ventricle to properly dilate and fill resulted in reduced left ventricular stroke volume and cardiac output, whereas its contractile function remained unchanged. Progressive remodeling of the RV in PAB mice was associated with upregulation of myocardial gene markers of hypertrophy and fibrosis. Pressure overload of the RV may also lead to reduction in capillary density (Hudlicka et al. 1992), which may contribute to ventricular dysfunction. In contrast, our study demonstrated that number of capillaries per cardiomyocyte actually slightly increased in PAB mice as compared with sham mice. However, taking into account significant increase in cardiomyocyte size in PAB mice, blood supply to individual cardiomyocytes may remain reduced.
Although, pulmonary artery banding has recently become an important model of pressure overload induced RV hypertrophy and several research groups including our own have reported maladaptive alterations (Kreymborg et al. 2010; Kapur et al. 2013; Rajagopalan et al. 2013; Egemnazarov et al. 2015; West et al. 2016), serial assessment of RV morphology and function in mice subjected to pressure overload of various degrees was performed only in one study, which utilized FVB mice (Urashima et al. 2008). In that study, severe pulmonary artery stenosis was associated with significant RV cardiomyocyte and chamber hypertrophy, RV dilatation and dysfunction, reduced cardiac output, and increased mortality. Although, we have not performed a direct comparison of FVB and C57Bl/6J mice, we think that the degree of pressure overload, RV hypertrophy, and dysfunction in C57Bl/6J mice was comparable with that of FVB mice with severe pulmonary artery constriction. Our findings suggest that compared with the FVB strain, C57Bl/6J mice subjected to severe RV pressure overload are not prone to increased mortality during the first 3 weeks. The cause for increased mortality in FVB mice is not clear as RV dysfunction did not decline with time but may be related to development of malignant arrhythmias. Unfortunately, a detailed characterization of longitudinal changes in concomitant maladaptive processes, including myocardial fibrosis and capillary density alterations, is lacking for FVB mice. Nevertheless, our findings suggest that interstrain differences can affect the ability of the RV to cope with sustained pressure overload.
Inflammatory activation contributes to the pathogenesis of both left ventricular (Sun et al. 2007; Coggins and Rosenzweig 2012) and RV adverse remodeling and dysfunction (Campian et al. 2010; Rondelet et al. 2012; Dewachter et al. 2015). Previous reports have shown that cardiac mast cells are a component of the inflammatory response in various models of adverse left ventricular remodeling (Levick et al. 2008, 2009; Zhang et al. 2011). Mast cells are cells of hematopoietic lineage residing in the tissues. In normal human and rodent hearts, the number of mast cells ranges between 1.5 and 5.3 cells/mm2 (Rakusan et al. 1990; Patella et al. 1998). Enhanced accumulation of mast cells in left ventricular tissue from late‐stage heart failure patients (194 ± 19.6 cells/cm2 vs. 131 ± 22.2 cells/cm2 in control ventricles) has been documented (Batlle et al. 2006). Interestingly, an increase in cardiac mast cell density of similar magnitude is reported in various animal models of left ventricular hypertrophy and failure. In the rat model of left ventricular hypertrophy induced by transverse aortic constriction, the number of cardiac mast cells increased from about 2 cells/mm2 in sham animals to 3 cells/mm2 5 weeks post surgery (Li et al. 2016). Comparable cardiac mast cell densities are reported in spontaneously hypertensive rats (Shiota et al. 2003). The temporal responses in the density of myocardial mast cells in the model of biventricular volume overload was similar for the left ventricle and RV (Brower et al. 2002). The number of mast cells in the RV at 21 days post fistula was 3.11 ± 2.01 versus 2.34 ± 1.00 cells/mm2 in the control. The issue of whether degranulation of this small number of cells is sufficient to cause ventricular remodeling has specifically been addressed in a study using isolated hearts, which findings clearly demonstrated that degranulation of mast cells present in normal hearts is sufficient to induce extracellular matrix degradation via activation of MMPs and produce alteration in ventricular function (Chancey et al. 2002). Importance of this small number of mast cells for cardiac remodeling is further supported by studies utilizing mast cell stabilizers, inhibitors of mast cell proteases and mast cell‐deficient mice (Hara et al. 2002; Matsumoto et al. 2003; Levick et al. 2009; Liu et al. 2012). Given the role of mast cells in the hypertensive and failing left ventricle, we were interested, if mast cells are also involved in the RV remodeling. Earlier studies in rats have shown that prolonged duration (200 days) of pressure overload on the RV induced by PAB is associated with more than threefold increase in the mast cell density (Olivetti et al. 1989). In contrast, cardiac mast cells density was not affected in 3‐month‐old rats born at high altitude despite development of significant RV hypertrophy (Rakusan et al. 1990). However, it remains unclear, if this discrepancy is due to the differences in the duration of the overload or if it is model‐dependent. In addition, temporal responses to pressure overload in the density of cardiac mast cells has not been addressed yet. We investigated the number and activity of mast cells in RV tissue at different time points after PAB. We found an early activation of mast cells evidenced by an increase in the proportion of the degranulated mast cells in the RV tissue. In contrast, mast cell density started increasing after 2 weeks and reached maximal values 3 weeks post surgery. Interestingly, increased expression of c‐Kit and soluble stem cell factor and enhanced cardiac mast cells density were observed in the prehypertensive spontaneously hypertensive rats even before any signs of cardiac hypertrophy or fibrosis (Shiota et al. 2003). An early elevation in mast cell density in both left and right ventricles was observed in a rat model of biventricular volume overload (Brower et al. 2002; Forman et al. 2006). In this model, the number of mast cells increased within 24 h and remained elevated for the first 5 days postfistula before returning to normal values by day 14.
Increase in mast cell density can result from proliferation and maturation of immature resident mast cells (Forman et al. 2006; Li et al. 2012) as well as recruitment of mast cell progenitors from the bone marrow or white adipose tissue and further proliferation and maturation in the tissue (Frangogiannis et al. 1998; Ngkelo et al. 2016). Increased chymase activity secondary to mast cell activation may increase tissue levels of stem cell factor by cleaving membrane‐bound stem cell factor to release a soluble form (Longley et al. 1997). Higher levels of the stem cell factor then rapidly elevate mast cell numbers by stimulating maturation of immature resident mast cells in the heart (Li et al. 2012). However, the mast cell density returns to normal values at 5–7 days post fistula (Brower et al. 2002; Forman et al. 2006). Thus, the increase in cardiac mast cell density in response to volume overload is due to maturation and differentiation, but not proliferation of a resident population of immature cardiac mast cells. Rather late increase in mast cell density in our experiments may be due to different underlying mechanisms. Indeed, recruitment of mast cell precursors and further maturation in the tissue requires a considerable amount of time (Jamur et al. 2010). In addition, interspecies differences (Halapas et al. 2008) and type of overload stress (Bartelds et al. 2011; Borgdorff et al. 2013) may account for this discrepancy. Nevertheless, this issue deserves further investigation.
Depending on the magnitude of the stimulus, activation mode and selective activation of specific signaling pathways, mast cells release unique arrays of preformed and newly synthesized biologically active substances (Rao and Brown 2008; Gilfillan et al. 2011). Potential mechanisms of the involvement of mast cells in the cardiac remodeling include stimulation of collagen synthesis leading to myocardial fibrosis or activation of matrix metalloproteinases resulting in collagen degradation and ventricular dilatation. Mast cells serve as a source of a number of cytokines, which are mitogenic and chemotactic for fibroblasts and stimulate production of extracellular matrix by fibroblasts. It has been demonstrated that cardiac mast cells participate in the induction of cardiac hypertrophy and cardiac fibrosis by synthesizing and secreting prohypertrophic cytokines and profibrotic growth factors TGF‐β and bFGF (Shiota et al. 2003). Moreover, cardiac mast cells can promote tissue fibrosis by stimulating cell proliferation and collagen expression in cardiac fibroblasts through PDGF‐A (Liao et al. 2010). Mast cells also release both preformed and newly synthesized cytokines TNF‐α and IL‐6 (Gordon and Galli 1990, 1991; Gagari et al. 1997) which have previously been shown to mediate cardiac fibrosis and hypertrophy in various animal models (Sun et al. 2007; Melendez et al. 2010; Gonzalez et al. 2015; Sriramula and Francis 2015). Similar to the findings from the left ventricular hypertrophy studies, we found significantly elevated mRNA levels of TNF‐α and IL‐6 in RV tissues from PAB mice compared with those from sham mice suggesting their potential contribution to RV remodeling process. Besides cytokines, mast cell granules contain very high levels of a number of proteases and mast cell degranulation leads to release of active mast cell proteases (Stevens and Adachi 2007). Mouse mast cell consists of various proteases including chymases (mMCP‐1, ‐2, ‐4, ‐5, ‐9), tryptases (mMCP‐6, ‐7, membrane‐bound tryptase and a number of other trypsin‐like serine proteases), and the exopeptidase CPA3 (Pejler et al. 2010). Mast cell chymases and tryptases are potent activators of fibroblast proliferation and inducers of matrix protein synthesis (Cairns and Walls 1997; Akers et al. 2000; Zhao et al. 2008; McLarty et al. 2011). Mast cells proteases can promote extracellular matrix accumulation through formation of angiotensin II (Urata et al. 1990), activation of TGF‐β (Lindstedt et al. 2001), or by acting as mitogens for fibroblasts (Ruoss et al. 1991). Previous studies have demonstrated that expression and activity of mast cell proteases tryptase and chymase are elevated in the hypertensive left ventricles (Shiota et al. 1997; Li et al. 2002; Levick et al. 2009; Li et al. 2016). We performed analysis of gene expression in RV tissues from PAB mice and found a significant upregulation of several mouse mast cell proteases including mMcp‐2, 4, 5, 6, and CPA3. Inhibition of mast cell chymase and tryptase has been shown to prevent cardiac fibrosis and improve left ventricular dysfunction in various left heart disease models (Matsumoto et al. 2003; Kanemitsu et al. 2006, 2008; Li et al. 2016). Interestingly, inhibition of mast cell chymase increases the cardiac efficacy of angiotensin I‐converting enzyme inhibitor therapy and improves survival after myocardial infarction in hamsters (Jin et al. 2003; Wei et al. 2010). These data demonstrate that selective inhibition of mast cell proteases might represent an important strategy for management of cardiac dysfunction.
In summary, our work provided a detailed description of the longitudinal changes in RV morphology and function in response to pressure overload in C57BL/6J mice. We also demonstrated accumulation and activation of mast cells in the RV tissue of PAB mice. Our data suggest possible involvement of mast cells in the RV remodeling process in response to pressure overload. Mast cells may thus represent an interesting target for the development of new therapeutic approaches directed specifically at the RV.
Conflicts of Interest
No conflicts of interest, financial or otherwise, are declared by the author(s).
Acknowledgments
We thank Ewa Bieniek and Christina Vroom for expert technical assistance.
Luitel H. , Sydykov A. , Schymura Y. , Mamazhakypov A. , Janssen W. , Pradhan K. , Wietelmann A. , Kosanovic D. , Dahal B. K. , Weissmann N. , Seeger W. , Grimminger F. , Ghofrani H. A. , Schermuly R. T.. Pressure overload leads to an increased accumulation and activity of mast cells in the right ventricle, Physiol Rep, 5 (6), 2017, e13146, doi: 10.14814/phy2.13146
Funding Information
This work was supported by the DFG (German Research Foundation) grant SCHE 691/6‐1, the CRC1213 (Collaborative Research Center 1213), the ECCPS (Excellence Cluster Cardiopulmonary System), Giessen and UGMLC (Universities of Giessen and Marburg Lung Center).
References
- Akers, I. A. , Parsons M., Hill M. R., Hollenberg M. D., Sanjar S., Laurent G. J., et al. 2000. Mast cell tryptase stimulates human lung fibroblast proliferation via protease‐activated receptor‐2. Am. J. Physiol. Lung Cell. Mol. Physiol. 278:L193–L201. [DOI] [PubMed] [Google Scholar]
- Ascah, K. J. , King M. E., Gillam L. D., and Weyman A. E.. 1990. The effects of right ventricular hemodynamics on left ventricular configuration. Can. J. Cardiol. 6:99–106. [PubMed] [Google Scholar]
- Barnabei, M. S. , Palpant N. J., and Metzger J. M.. 2010. Influence of genetic background on ex vivo and in vivo cardiac function in several commonly used inbred mouse strains. Physiol. Genomics 42A:103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick, C. J. , Rojas M., Schoonhoven R., Smyth S. S., and Threadgill D. W.. 2007. Cardiac response to pressure overload in 129S1/SvImJ and C57BL/6J mice: temporal‐ and background‐dependent development of concentric left ventricular hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 292:H2119–H2130. [DOI] [PubMed] [Google Scholar]
- Bartelds, B. , Borgdorff M. A., Smit‐van Oosten A., Takens J., Boersma B., Nederhoff M. G., et al. 2011. Differential responses of the right ventricle to abnormal loading conditions in mice: pressure vs. volume load. Eur. J. Heart Fail. 13:1275–1282. [DOI] [PubMed] [Google Scholar]
- Batlle, M. , Roig E., Perez‐Villa F., Lario S., Cejudo‐Martin P., Garcia‐Pras E., et al. 2006. Increased expression of the renin‐angiotensin system and mast cell density but not of angiotensin‐converting enzyme II in late stages of human heart failure. J. Heart Lung Transplant. 25:1117–1125. [DOI] [PubMed] [Google Scholar]
- Borgdorff, M. A. , Bartelds B., Dickinson M. G., Steendijk P., de Vroomen M., and Berger R. M.. 2013. Distinct loading conditions reveal various patterns of right ventricular adaptation. Am. J. Physiol. Heart Circ. Physiol. 305:H354–H364. [DOI] [PubMed] [Google Scholar]
- van den Borne, S. W. , van de Schans V. A., Strzelecka A. E., Vervoort‐Peters H. T., Lijnen P. M., Cleutjens J. P., et al. 2009. Mouse strain determines the outcome of wound healing after myocardial infarction. Cardiovasc. Res. 84:273–282. [DOI] [PubMed] [Google Scholar]
- Brower, G. L. , Chancey A. L., Thanigaraj S., Matsubara B. B., and Janicki J. S.. 2002. Cause and effect relationship between myocardial mast cell number and matrix metalloproteinase activity. Am. J. Physiol. Heart Circ. Physiol. 283:H518–H525. [DOI] [PubMed] [Google Scholar]
- Cairns, J. A. , and Walls A. F.. 1997. Mast cell tryptase stimulates the synthesis of type I collagen in human lung fibroblasts. J. Clin. Invest. 99:1313–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campen, M. J. , Tagaito Y., Li J., Balbir A., Tankersley C. G., Smith P., et al. 2004. Phenotypic variation in cardiovascular responses to acute hypoxic and hypercapnic exposure in mice. Physiol. Genomics 20:15–20. [DOI] [PubMed] [Google Scholar]
- Campian, M. E. , Hardziyenka M., de Bruin K., van Eck‐Smit B. L., de Bakker J. M., Verberne H. J., et al. 2010. Early inflammatory response during the development of right ventricular heart failure in a rat model. Eur. J. Heart Fail. 12:653–658. [DOI] [PubMed] [Google Scholar]
- Chancey, A. L. , Brower G. L., and Janicki J. S.. 2002. Cardiac mast cell‐mediated activation of gelatinase and alteration of ventricular diastolic function. Am. J. Physiol. Heart Circ. Physiol. 282:H2152–H2158. [DOI] [PubMed] [Google Scholar]
- Coggins, M. , and Rosenzweig A.. 2012. The fire within: cardiac inflammatory signaling in health and disease. Circ. Res. 110:116–125. [DOI] [PubMed] [Google Scholar]
- Dahal, B. K. , Kosanovic D., Kaulen C., Cornitescu T., Savai R., Hoffmann J., et al. 2011. Involvement of mast cells in monocrotaline‐induced pulmonary hypertension in rats. Respir. Res. 12:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewachter, C. , Belhaj A., Rondelet B., Vercruyssen M., Schraufnagel D. P., Remmelink M., et al. 2015. Myocardial inflammation in experimental acute right ventricular failure: Effects of prostacyclin therapy. J. Heart Lung Transplant. 34:1334–1345. [DOI] [PubMed] [Google Scholar]
- Egemnazarov, B. , Schmidt A., Crnkovic S., Sydykov A., Nagy B. M., Kovacs G., et al. 2015. Pressure overload creates right ventricular diastolic dysfunction in a mouse model: assessment by echocardiography. J. Am. Soc. Echocardiogr. 28:828–843. [DOI] [PubMed] [Google Scholar]
- Forman, M. F. , Brower G. L., and Janicki J. S.. 2006. Rat cardiac mast cell maturation and differentiation following acute ventricular volume overload. Inflamm. Res. 55:408–415. [DOI] [PubMed] [Google Scholar]
- Frangogiannis, N. G. , Perrard J. L., Mendoza L. H., Burns A. R., Lindsey M. L., Ballantyne C. M., et al. 1998. Stem cell factor induction is associated with mast cell accumulation after canine myocardial ischemia and reperfusion. Circulation 98:687–698. [DOI] [PubMed] [Google Scholar]
- Gagari, E. , Tsai M., Lantz C. S., Fox L. G., and Galli S. J.. 1997. Differential release of mast cell interleukin‐6 via c‐kit. Blood 89:2654–2663. [PubMed] [Google Scholar]
- Gao, X. M. , Xu Q., Kiriazis H., Dart A. M., and Du X. J.. 2005. Mouse model of post‐infarct ventricular rupture: time course, strain‐ and gender‐dependency, tensile strength, and histopathology. Cardiovasc. Res. 65:469–477. [DOI] [PubMed] [Google Scholar]
- Gilfillan, A. M. , Austin S. J., and Metcalfe D. D.. 2011. Mast cell biology: introduction and overview. Adv. Exp. Med. Biol. 716:2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez, G. E. , Rhaleb N. E., D'Ambrosio M. A., Nakagawa P., Liu Y., Leung P., et al. 2015. Deletion of interleukin‐6 prevents cardiac inflammation, fibrosis and dysfunction without affecting blood pressure in angiotensin II‐high salt‐induced hypertension. J. Hypertens. 33:144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, J. R. , and Galli S. J.. 1990. Mast cells as a source of both preformed and immunologically inducible TNF‐alpha/cachectin. Nature 346:274–276. [DOI] [PubMed] [Google Scholar]
- Gordon, J. R. , and Galli S. J.. 1991. Release of both preformed and newly synthesized tumor necrosis factor alpha (TNF‐alpha)/cachectin by mouse mast cells stimulated via the Fc epsilon RI. A mechanism for the sustained action of mast cell‐derived TNF‐alpha during IgE‐dependent biological responses. J. Exp. Med. 174:103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarracino, F. , Cariello C., Danella A., Doroni L., Lapolla F., Vullo C., et al. 2005. Right ventricular failure: physiology and assessment. Minerva Anestesiol. 71:307–312. [PubMed] [Google Scholar]
- Halapas, A. , Papalois A., Stauropoulou A., Philippou A., Pissimissis N., Chatzigeorgiou A., et al. 2008. In vivo models for heart failure research. In Vivo 22:767–780. [PubMed] [Google Scholar]
- Hara, M. , Ono K., Hwang M. W., Iwasaki A., Okada M., Nakatani K., et al. 2002. Evidence for a role of mast cells in the evolution to congestive heart failure. J. Exp. Med. 195:375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudlicka, O. , Brown M., and Egginton S.. 1992. Angiogenesis in skeletal and cardiac muscle. Physiol. Rev. 72:369–417. [DOI] [PubMed] [Google Scholar]
- Izikki, M. , Hanoun N., Marcos E., Savale L., Barlier‐Mur A. M., Saurini F., et al. 2007. Tryptophan hydroxylase 1 knockout and tryptophan hydroxylase 2 polymorphism: effects on hypoxic pulmonary hypertension in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 293:L1045–L1052. [DOI] [PubMed] [Google Scholar]
- Jamur, M. C. , Moreno A. N., Mello L. F., Souza Junior D. A., Campos M. R., Pastor M. V., et al. 2010. Mast cell repopulation of the peritoneal cavity: contribution of mast cell progenitors versus bone marrow derived committed mast cell precursors. BMC Immunol. 11:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, B. , Deng Y., Suen C., Taha M., Chaudhary K. R., Courtman D. W., et al. 2016. Marked strain‐specific differences in the SU5416 rat model of severe pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 54:461–468. [DOI] [PubMed] [Google Scholar]
- Jin, D. , Takai S., Yamada M., Sakaguchi M., Kamoshita K., Ishida K., et al. 2003. Impact of chymase inhibitor on cardiac function and survival after myocardial infarction. Cardiovasc. Res. 60:413–420. [DOI] [PubMed] [Google Scholar]
- Kanellakis, P. , Ditiatkovski M., Kostolias G., and Bobik A.. 2012. A pro‐fibrotic role for interleukin‐4 in cardiac pressure overload. Cardiovasc. Res. 95:77–85. [DOI] [PubMed] [Google Scholar]
- Kanemitsu, H. , Takai S., Tsuneyoshi H., Nishina T., Yoshikawa K., Miyazaki M., et al. 2006. Chymase inhibition prevents cardiac fibrosis and dysfunction after myocardial infarction in rats. Hypertens. Res. 29:57–64. [DOI] [PubMed] [Google Scholar]
- Kanemitsu, H. , Takai S., Tsuneyoshi H., Yoshikawa E., Nishina T., Miyazaki M., et al. 2008. Chronic chymase inhibition preserves cardiac function after left ventricular repair in rats. Eur. J. Cardiothorac. Surg. 33:25–31. [DOI] [PubMed] [Google Scholar]
- Kapur, N. K. , Paruchuri V., Aronovitz M. J., Qiao X., Mackey E. E., Daly G. H., et al. 2013. Biventricular remodeling in murine models of right ventricular pressure overload. PLoS ONE 8:e70802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitaura‐Inenaga, K. , Hara M., Higuchi K., Yamamoto K., Yamaki A., Ono K., et al. 2003. Gene expression of cardiac mast cell chymase and tryptase in a murine model of heart failure caused by viral myocarditis. Circ. J. 67:881–884. [DOI] [PubMed] [Google Scholar]
- Kreymborg, K. , Uchida S., Gellert P., Schneider A., Boettger T., Voswinckel R., et al. 2010. Identification of right heart‐enriched genes in a murine model of chronic outflow tract obstruction. J. Mol. Cell. Cardiol. 49:598–605. [DOI] [PubMed] [Google Scholar]
- Levick, S. P. , Gardner J. D., Holland M., Hauer‐Jensen M., Janicki J. S., and Brower G. L.. 2008. Protection from adverse myocardial remodeling secondary to chronic volume overload in mast cell deficient rats. J. Mol. Cell. Cardiol. 45:56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levick, S. P. , McLarty J. L., Murray D. B., Freeman R. M., Carver W. E., and Brower G. L.. 2009. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension 53:1041–1047. [DOI] [PubMed] [Google Scholar]
- Levick, S. P. , Melendez G. C., Plante E., McLarty J. L., Brower G. L., and Janicki J. S.. 2011. Cardiac mast cells: the centrepiece in adverse myocardial remodelling. Cardiovasc. Res. 89:12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, P. , Chen P. M., Wang S. W., and Chen L. Y.. 2002. Time‐dependent expression of chymase and angiotensin converting enzyme in the hamster heart under pressure overload. Hypertens. Res. 25:757–762. [DOI] [PubMed] [Google Scholar]
- Li, J. , Lu H., Plante E., Melendez G. C., Levick S. P., and Janicki J. S.. 2012. Stem cell factor is responsible for the rapid response in mature mast cell density in the acutely stressed heart. J. Mol. Cell. Cardiol. 53:469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Jubair S., Levick S. P., and Janicki J. S.. 2016. The autocrine role of tryptase in pressure overload‐induced mast cell activation, chymase release and cardiac fibrosis.IJC Metab. Endocr. 10:16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, C. H. , Akazawa H., Tamagawa M., Ito K., Yasuda N., and Kudo Y., et al. 2010. Cardiac mast cells cause atrial fibrillation through PDGF‐A‐mediated fibrosis in pressure‐overloaded mouse hearts. J. Clin. Invest. 120:242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstedt, K. A. , Wang Y., Shiota N., Saarinen J., Hyytiainen M., Kokkonen J. O., et al. 2001. Activation of paracrine TGF‐beta1 signaling upon stimulation and degranulation of rat serosal mast cells: a novel function for chymase. FASEB J. 15:1377–1388. [DOI] [PubMed] [Google Scholar]
- Liu, W. , Chen J., Xu T., Tian W., Li Y., Zhang Z., et al. 2012. Qiliqiangxin improves cardiac function in spontaneously hypertensive rats through the inhibition of cardiac chymase. Am. J. Hypertens. 25:250–260. [DOI] [PubMed] [Google Scholar]
- Longley, B. J. , Tyrrell L., Ma Y., Williams D. A., Halaban R., Langley K., et al. 1997. Chymase cleavage of stem cell factor yields a bioactive, soluble product. Proc. Natl Acad. Sci. USA 94:9017–9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie, E. K. , Rich S., Levitsky S., and Brundage B. H.. 1992. Doppler echocardiographic demonstration of the differential effects of right ventricular pressure and volume overload on left ventricular geometry and filling. J. Am. Coll. Cardiol. 19:84–90. [DOI] [PubMed] [Google Scholar]
- Lucki, I. , Dalvi A., and Mayorga A. J.. 2001. Sensitivity to the effects of pharmacologically selective antidepressants in different strains of mice. Psychopharmacology 155:315–322. [DOI] [PubMed] [Google Scholar]
- Marone, G. , De Rescenzo G., Patella V., Granata F., Verga L., and Arbustinp E., et al. (2000). Human heart mast cells: immunological characterization in situ and in vitro Pp. 455–477 in Marone G., Lichtenstein L. M., Galli F. J. Mast cells and basophils. Academic Press, London, UK. [Google Scholar]
- Matsumoto, T. , Wada A., Tsutamoto T., Ohnishi M., Isono T., and Kinoshita M.. 2003. Chymase inhibition prevents cardiac fibrosis and improves diastolic dysfunction in the progression of heart failure. Circulation 107:2555–2558. [DOI] [PubMed] [Google Scholar]
- McLarty, J. L. , Melendez G. C., Brower G. L., Janicki J. S., and Levick S. P.. 2011. Tryptase/Protease‐activated receptor 2 interactions induce selective mitogen‐activated protein kinase signaling and collagen synthesis by cardiac fibroblasts. Hypertension 58:264–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez, G. C. , McLarty J. L., Levick S. P., Du Y., Janicki J. S., and Brower G. L.. 2010. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension 56:225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreth, K. , Fischer R., Fuchs H., Gailus‐Durner V., Wurst W., Katus H. A., et al. 2014. High‐throughput phenotypic assessment of cardiac physiology in four commonly used inbred mouse strains. J. Comp. Physiol. B. 184:763–775. [DOI] [PubMed] [Google Scholar]
- Ngkelo, A. , Richart A., Kirk J. A., Bonnin P., Vilar J., Lemitre M., et al. 2016. Mast cells regulate myofilament calcium sensitization and heart function after myocardial infarction. J. Exp. Med. 213:1353–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivetti, G. , Lagrasta C., Ricci R., Sonnenblick E. H., Capasso J. M., and Anversa P.. 1989. Long‐term pressure‐induced cardiac hypertrophy: capillary and mast cell proliferation. Am. J. Physiol. 257:H1766–H1772. [DOI] [PubMed] [Google Scholar]
- Panizo, A. , Mindan F. J., Galindo M. F., Cenarruzabeitia E., Hernandez M., and Diez J.. 1995. Are mast cells involved in hypertensive heart disease? J. Hypertens. 13:1201–1208. [DOI] [PubMed] [Google Scholar]
- Patella, V. , Marino I., Arbustini E., Lamparter‐Schummert B., Verga L., Adt M., et al. 1998. Stem cell factor in mast cells and increased mast cell density in idiopathic and ischemic cardiomyopathy. Circulation 97:971–978. [DOI] [PubMed] [Google Scholar]
- Pejler, G. , Ronnberg E., Waern I., and Wernersson S.. 2010. Mast cell proteases: multifaceted regulators of inflammatory disease. Blood 115:4981–4990. [DOI] [PubMed] [Google Scholar]
- Puxeddu, I. , Piliponsky A. M., Bachelet I., and Levi‐Schaffer F.. 2003. Mast cells in allergy and beyond. Int. J. Biochem. Cell Biol. 35:1601–1607. [DOI] [PubMed] [Google Scholar]
- Rajagopalan, V. , Zhao M., Reddy S., Fajardo G., Wang X., Dewey S., et al. 2013. Altered ubiquitin‐proteasome signaling in right ventricular hypertrophy and failure. Am. J. Physiol. Heart Circ. Physiol. 305:H551–H562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakusan, K. , Sarkar K., Turek Z., and Wicker P.. 1990. Mast cells in the rat heart during normal growth and in cardiac hypertrophy. Circ. Res. 66:511–516. [DOI] [PubMed] [Google Scholar]
- Rao, K. N. , and Brown M. A.. 2008. Mast cells: multifaceted immune cells with diverse roles in health and disease. Ann. N. Y. Acad. Sci. 1143:83–104. [DOI] [PubMed] [Google Scholar]
- Rockman, H. A. , Ono S., Ross R. S., Jones L. R., Karimi M., Bhargava V., et al. 1994. Molecular and physiological alterations in murine ventricular dysfunction. Proc. Natl Acad. Sci. USA 91:2694–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondelet, B. , Dewachter C., Kerbaul F., Kang X., Fesler P., Brimioulle S., et al. 2012. Prolonged overcirculation‐induced pulmonary arterial hypertension as a cause of right ventricular failure. Eur. Heart J. 33:1017–1026. [DOI] [PubMed] [Google Scholar]
- Ruoss, S. J. , Hartmann T., and Caughey G. H.. 1991. Mast cell tryptase is a mitogen for cultured fibroblasts. J. Clin. Invest. 88:493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan, T. , Petrovic O., Dillon J. C., Feigenbaum H., Conley M. J., and Armstrong W. F.. 1985. An echocardiographic index for separation of right ventricular volume and pressure overload. J. Am. Coll. Cardiol. 5:918–927. [DOI] [PubMed] [Google Scholar]
- Shah, A. P. , Siedlecka U., Gandhi A., Navaratnarajah M., Al‐Saud S. A., Yacoub M. H., et al. 2010. Genetic background affects function and intracellular calcium regulation of mouse hearts. Cardiovasc. Res. 87:683–693. [DOI] [PubMed] [Google Scholar]
- Shiota, N. , Jin D., Takai S., Kawamura T., Koyama M., Nakamura N., et al. 1997. Chymase is activated in the hamster heart following ventricular fibrosis during the chronic stage of hypertension. FEBS Lett. 406:301–304. [DOI] [PubMed] [Google Scholar]
- Shiota, N. , Rysa J., Kovanen P. T., Ruskoaho H., Kokkonen J. O., and Lindstedt K. A.. 2003. A role for cardiac mast cells in the pathogenesis of hypertensive heart disease. J. Hypertens. 21:1935–1944. [DOI] [PubMed] [Google Scholar]
- Simon, M. M. , Greenaway S., White J. K., Fuchs H., Gailus‐Durner V., Wells S., et al. 2013. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 14:R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriramula, S. , and Francis J.. 2015. Tumor Necrosis Factor ‐ Alpha Is Essential for Angiotensin II‐Induced Ventricular Remodeling: Role for Oxidative Stress. PLoS ONE 10:e0138372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens, R. L. , and Adachi R.. 2007. Protease‐proteoglycan complexes of mouse and human mast cells and importance of their beta‐tryptase‐heparin complexes in inflammation and innate immunity. Immunol. Rev. 217:155–167. [DOI] [PubMed] [Google Scholar]
- Stewart, J. A. Jr , Wei C. C., Brower G. L., Rynders P. E., Hankes G. H., Dillon A. R., et al. 2003. Cardiac mast cell‐ and chymase‐mediated matrix metalloproteinase activity and left ventricular remodeling in mitral regurgitation in the dog. J. Mol. Cell. Cardiol. 35:311–319. [DOI] [PubMed] [Google Scholar]
- Sun, M. , Chen M., Dawood F., Zurawska U., Li J. Y., Parker T., et al. 2007. Tumor necrosis factor‐alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation 115:1398–1407. [DOI] [PubMed] [Google Scholar]
- Tada, Y. , Laudi S., Harral J., Carr M., Ivester C., Tanabe N., et al. 2008. Murine pulmonary response to chronic hypoxia is strain specific. Exp. Lung Res. 34:313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarnavski, O. 2009. Mouse surgical models in cardiovascular research. Methods Mol. Biol. 573:115–137. [DOI] [PubMed] [Google Scholar]
- Tarnavski, O. , McMullen J. R., Schinke M., Nie Q., Kong S., and Izumo S.. 2004. Mouse cardiac surgery: comprehensive techniques for the generation of mouse models of human diseases and their application for genomic studies. Physiol. Genomics 16:349–360. [DOI] [PubMed] [Google Scholar]
- Urashima, T. , Zhao M., Wagner R., Fajardo G., Farahani S., Quertermous T., et al. 2008. Molecular and physiological characterization of RV remodeling in a murine model of pulmonary stenosis. Am. J. Physiol. Heart Circ. Physiol. 295:H1351–H1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urata, H. , Kinoshita A., Misono K. S., Bumpus F. M., and Husain A.. 1990. Identification of a highly specific chymase as the major angiotensin II‐forming enzyme in the human heart. J. Biol. Chem. 265:22348–22357. [PubMed] [Google Scholar]
- Wei, C. C. , Hase N., Inoue Y., Bradley E. W., Yahiro E., Li M., et al. 2010. Mast cell chymase limits the cardiac efficacy of Ang I‐converting enzyme inhibitor therapy in rodents. J. Clin. Invest. 120:1229–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West, J. D. , Voss B. M., Pavliv L., de Caestecker M., Hemnes A. R., and Carrier E. J.. 2016. Antagonism of the thromboxane‐prostanoid receptor is cardioprotective against right ventricular pressure overload. Pulm Circ 6:211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W. , Chancey A. L., Tzeng H. P., Zhou Z., Lavine K. J., Gao F., et al. 2011. The development of myocardial fibrosis in transgenic mice with targeted overexpression of tumor necrosis factor requires mast cell‐fibroblast interactions. Circulation 124:2106–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, X. Y. , Zhao L. Y., Zheng Q. S., Su J. L., Guan H., Shang F. J., et al. 2008. Chymase induces profibrotic response via transforming growth factor‐beta 1/Smad activation in rat cardiac fibroblasts. Mol. Cell. Biochem. 310:159–166. [DOI] [PubMed] [Google Scholar]