

Abstract
Autonomic dysfunction, a relative sedentary lifestyle, a reduced muscle mass and increased adiposity leads to metabolic abnormalities that accelerate the development of coronary artery disease (CAD) in individuals living with spinal cord injury (SCI). An untoward cardiac incident is related to the degree of CAD, suggesting that the occurrence of a significant cardiac event is significantly higher for individuals with SCI. Thus, understanding the fundamental hemodynamic mechanisms mediating the response to myocardial ischemia has the potential to positively impact individuals and families living with SCI. Accordingly, we systematically investigated if thoracic level 5 spinal cord transection (T5X; paraplegia) alters the arterial blood pressure response to coronary artery occlusion and if the different arterial blood pressure responses to coronary artery occlusion between intact and paraplegic mice are mediated by changes in cardiac output and or systemic peripheral resistance and whether differences in cardiac output are caused by changes in heart rate and or stroke volume. To achieve this goal, the tolerance to 3 min of coronary artery occlusion was determined in conscious intact and paraplegic mice. Paraplegic mice had an impaired ability to maintain arterial blood pressure during coronary artery occlusion as arterial pressure fell to near lethal levels by 1.38 ± 0.64 min. The lower arterial pressure was mediated by a lower cardiac output as systemic peripheral resistance was elevated in paraplegic mice. The lower cardiac output was mediated by a reduced heart rate and stroke volume. These results indicate that in paraplegic mice, the arterial pressure response to coronary artery occlusion is hemodynamically mediated primarily by cardiac output which is determined by heart rate and stroke volume.
Keywords: Coronary artery disease, myocardial ischemia, spinal reflex
Introduction
Individuals living with spinal cord injury (SCI) have a significantly greater risk for heart disease and stroke, and cardiovascular disease is the leading cause of death and morbidity (Kessler et al. 1986; Cardus et al. 1992; Whiteneck et al. 1992; DeVivo et al. 1993; DeVivo 1997; Frankel et al. 1998; Groah et al. 2001; West et al. 2012; Cragg et al. 2013). The risk for cardiovascular disease is associated with an unstable autonomic control of the heart and vasculature, a relatively sedentary lifestyle (Inskip et al. 2010) and blood lipid profiles consisting of elevated total and low‐density lipoprotein cholesterol and depressed high‐density lipoprotein cholesterol (Bauman et al. 1992, 1999; Cardus et al. 1992). Relative inactivity associated with SCI also results in a reduced muscle mass and increased adiposity. As such, individuals living with SCI often experience insulin resistance, hyperinsulinemia and an atherogenic profile that contributes to early development of coronary artery disease (CAD). Specifically, autonomic dysfunction, relative inactivity and adverse changes in body composition lead to metabolic changes that promote CAD in individuals living with SCI (Whiteneck et al. 1992; Bauman et al. 1993, 1994; Lee et al. 2006; Bauman and Spungen 2007; Orakzai et al. 2007). An untoward cardiac incident is related to the degree of CAD, suggesting that the occurrence of a significant cardiac event is significantly higher for individuals with SCI (Fleg et al. 1990). Accordingly, understanding the response to myocardial ischemia has the potential to positively impact individuals and families living with SCI and provides an improved approach to clinical care.
In this context, the cardiovascular response to coronary artery occlusion is mediated, in part, by a cardiac spinal reflex as well as the arterial baroreflex. Both reflexes increase heart rate and arterial pressure during reductions in cardiac output caused by the myocardial ischemia‐induced reductions in cardiac performance. Specifically, ischemic metabolites are released during myocardial ischemia that stimulate cardiac spinal afferents (Uchida and Murao 1974; Baker et al. 1980; Malliani et al. 1981; Pan et al. 1999; Fu and Longhurst 2002, 2005; Tjen et al. 2002; Fu et al. 2005) leading to elevations in sympathetic activity, heart rate, and arterial blood pressure (Malliani et al. 1981; Meller and Gebhart 1992; Longhurst et al. 2001). Transection of the dorsal roots disrupts this excitatory cardiac sympathetic reflex and reduces heart rate and arterial pressure during myocardial ischemia (Lujan et al. 2011). In addition, cardiac output and arterial pressure are reduced during myocardial ischemia which initiates a dramatic arterial baroreflex‐mediated increase in sympathetic outflow and decrease in parasympathetic outflow to the heart as well as an increased sympathetic outflow to the vasculature. Importantly, individuals with SCI at thoracic level 5 have a functional loss of the cardiac spinal reflex and the arterial baroreceptor reflex below the level of the injury and thus fail to excite sympathetic preganglionic neurons below T5. Accordingly, the tolerance to coronary artery occlusion is expected to be markedly impaired in T5X mice (Lujan et al. 2011).
Accordingly, we systematically investigated if thoracic level 5 spinal cord transection (T5X; paraplegia) alters the arterial blood pressure response to coronary artery occlusion and if the different arterial blood pressure responses to coronary artery occlusion between intact and paraplegic mice are mediated by changes in cardiac output and or peripheral resistance and whether differences in cardiac output are mediated by variations in heart rate and or stroke volume. To achieve this goal, mice were instrumented to record cardiac output, arterial pressure, and the electrocardiogram (ECG) (Lujan et al. 2012a; Lujan and DiCarlo 2013a, 2014c). In addition, a vascular occluder was placed around the left main coronary artery (Lujan et al. 2012a; Lujan and DiCarlo 2013a, 2014c). Following recovery, mice were subjected to T5X or sham T5X. Two weeks later, the tolerance to 3 min of occlusion of the left main coronary artery was determined by using the coronary vascular occluder.
Materials and Methods
Animals
All procedures involving animals were in accord with The American Physiological Society's Guiding Principles in the Care and Use of Animals. Approval for all procedures and protocols involving animals was provided by the Animal Care and Use Committee of Wayne State University. Twelve adult male C57BL/6 mice 3–4 months of age, (n = 6, midthoracic spinal cord transected [T5X]; and n = 6, sham transected [intact]) were studied 14 days postspinal cord transection or sham transection.
The study was designed to determine the fundamental hemodynamic mechanisms responsible for the impaired capacity to maintain arterial blood pressure during coronary artery occlusion in conscious paraplegic (T5X) mice. Although intact mice tolerate coronary artery occlusion for at least 90 min leading to myocardial infarction (Lujan et al. 2012a); paraplegic mice tolerate coronary artery occlusion for <2 min. Accordingly, a 3‐min period of occlusion was selected for comparison between groups.
Following anesthesia with pentobarbital sodium (60 mg/kg ip), the mice were atropinized (0.05 mg/kg ip), intubated, and prepared for aseptic surgery. Supplemental doses of pentobarbital sodium (10–20 mg/kg ip) were given if the mice responded to tail pinch.
Thoracotomy procedures
Following anesthesia, a thoracotomy was created through the second intercostal space and the sheath of the pericardium that spreads over the ascending aorta was removed. An appropriately sized (1.6‐mm) silicone‐type Doppler ultrasonic flow probe (Iowa Doppler Products, Iowa City, IA) was placed around the ascending aorta as recently described (Lujan and DiCarlo 2013a, 2014c). The flow probe wires were channeled subcutaneously and exited at the dorsal aspect of the neck. Next, a vascular occluder was positioned around the coronary artery as recently described (Lujan et al. 2012a; Lujan and DiCarlo 2013a, 2014c). The ends of the vascular occluder were passed through guide tubing and exited at the dorsal aspect of the neck (Lujan et al. 2012a; Lujan and DiCarlo 2013a, 2014a,c). The local anesthetic, bupivicaine, was injected (sq) at the incision site. Next, ECG electrodes (DataSciences International, Standard Lead Coupler Kit: 276‐0031‐001) were placed in a modified lead II configuration, tunneled subcutaneously and exited at the dorsal aspect of the neck as previously described in mice (Lujan et al. 2012a; Lujan and DiCarlo 2013a, 2014a,c). Finally, a catheter from a telemetry device (Data Sciences International, PA‐C10), for recording arterial pressure, was inserted into the left carotid artery until the tip reached the aortic arch (Kurtz et al. 2014). The transmitter body was placed subcutaneously on the left side.
All animals remained ventilated and on a feedback‐based temperature control system until recovered from the anesthesia. Following recovery from the anesthesia, the mice were housed in a “rodent recovery cage” (Thermocare® Intensive Care Unit, Braintree Scientific, Braintree MA) and returned to the animal facility the following day. Carprofen (5 mg/kg, sq) and cefazolin (10 mg/kg, sq) were continued for 2 days. A minimum ten‐day recovery period was enforced during which the mice were weighed and acclimatized to the laboratory and investigators.
Spinal cord transection (T5X) or Sham transection
Following the minimum ten‐day recovery period, the mice were anesthetized as defined earlier and subjected to complete thoracic level 5 spinal cord transection (T5X) or sham spinal cord transection. Specifically, the spinous process and laminae of the 4th thoracic vertebrae were removed exposing the 5th thoracic spinal cord segment. The 5th thoracic spinal cord segment is positioned below the 4th thoracic vertebral because the spinal cord is shorter than the vertebral column. Sympathetic preganglionic neurons projecting to the heart leave the spinal cord via the 1st through 4th thoracic spinal cord segments (Strack et al. 1988). Accordingly, spinal cord transection between the 5th and 6th thoracic spinal cord segments maintains supraspinal control of preganglionic neurons projecting to the heart but loss of supraspinal control of preganglionic neurons below the thoracic level 5 spinal cord segment. The extensiveness of the spinal cord transection was established by visually inspecting the transection site. Undistinguishable procedures were followed for the sham transected mice, except the spinal cord was not transected. All mice received a supplemented nutritious diets (Bio‐Serv, Frenchtown, NJ).
Experimental procedures
Response to myocardial ischemia
The experimental procedures were conducted in conscious and unrestrained mice. All physiological measurements (cardiac output, arterial blood pressure, and the electrocardiogram) were made as previously described (Lujan et al. 2012a; Lujan and DiCarlo 2013a, 2014a,c; Kurtz et al. 2014) with the mice in their standard home cages during the light cycle. Temperature within the cage was maintained within the thermoneutral zone for mice of approximately 29–31°C (Swoap et al. 2004). Mice adapted to the experimental conditions for approximately 2 h to ensure stable hemodynamic conditions.
Following the adaptation period, the left main coronary artery was temporarily occluded for 3 min using the coronary vascular occluder as previously described (Lujan et al. 2012a; Lujan and DiCarlo 2013a, 2014a,c). Expected responses in the ECG (peaked T wave and S‐T segment elevation), and a rapid fall in cardiac output and arterial pressure documented coronary artery occlusion (Fig. 1) (Lujan et al. 2012a; Lujan and DiCarlo 2013a, 2014a,c). Upon release, intact mice experienced sustained ventricular tachycardia (absence of p wave, wide QRS complex) >900 beats/min as well as a reduced cardiac output and arterial blood pressure (Curtis et al. 2013). Gentle chest compressions or, in some cases, person to mouse ventilation via a small tube over the nose and mouth restored normal sinus rhythm.
Figure 1.
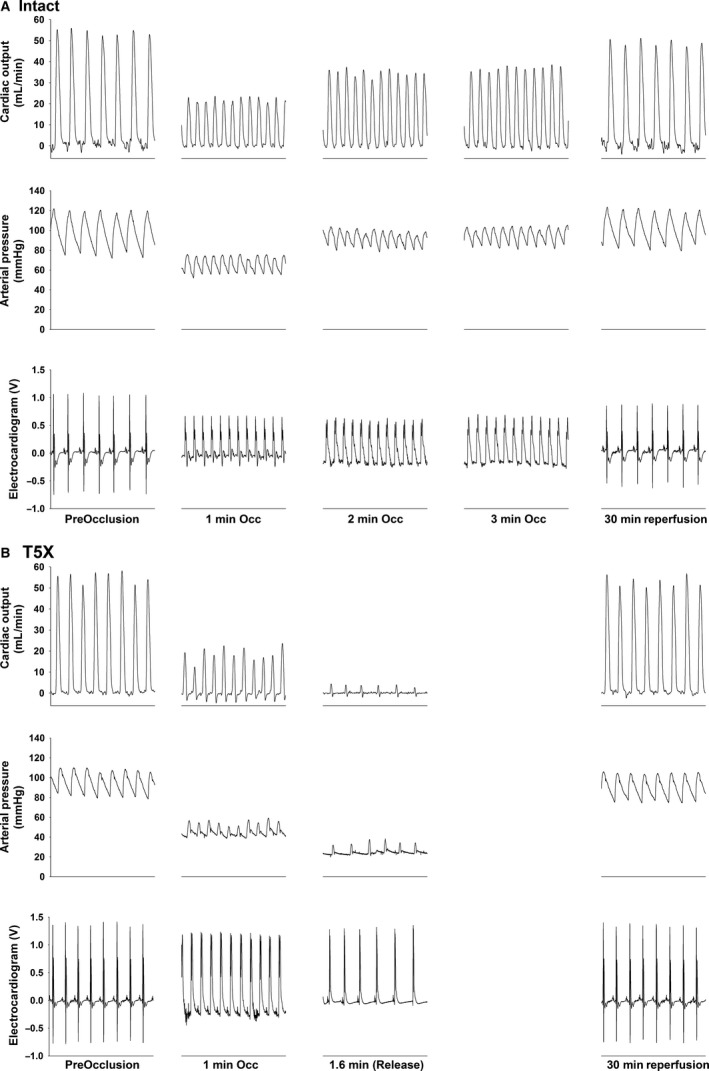
One second original recordings of cardiac output, arterial pressure, and the electrocardiogram for one intact (panel A) and one paraplegic (panel B) mouse are presented. Coronary artery occlusion significantly reduced arterial pressure in paraplegic mice. The reduced arterial pressure was mediated by a reduced cardiac output. None of the T5X mice tolerated 3 min of occlusion. In this example, the duration of the occlusion for the T5X mouse was 1.6 min.
None of the paraplegic mice tolerated the occlusion for more than 2 min. The occlusion was terminated when cardiac output and arterial pressure were virtually zero and the mice required gentle compression of the thorax and person to mouse ventilation using a customized tube over the nose and mouth for survival. One week after the experimental protocol, the hearts were harvested from all mice.
Determination of the ischemic zone (n = 3/group)
One week after completion of the experimental protocols, the mice were deeply anesthetized and the hearts and aortae were removed with the coronary vascular occluder in place. The hearts and aortae were immediately immersed in heparinized saline to remove remaining intracavitary blood. Subsequently, the aorta was cannulated and perfused with 1–2 mL saline until the saline drained clear from the coronary sinus (Bohl et al. 2009). Next, the coronary vascular occluder was tied to block flow through the left main coronary artery. Evans blue dye (250 μL, 0.5%) was then sent via the aorta, so that the dye entered and stained the nonischemic zone of the heart. The ischemic zone remained unstained. The heart was then trimmed leaving only the ventricles. The ventricles were rinsed to remove the excess dye; then sliced from apex to base into approximately four to six sections (∼1 mm thickness). Ventricular sections were compressed between two microscope slides to achieve uniform thickness and images of each ventricular section were obtained with a digital camera mounted on a microscope. The total ventricular section area and the ischemic ventricular zone of each heart section were quantified using the SPOT Imaging software (version 4.7, SPOT Imaging Solutions, Sterling Heights, MI). As noted above, the ischemic ventricular zone was devoid of Evans blue dye. The percentage of the ischemic ventricular zone was calculated for each section by dividing the ischemic ventricular zone by the total ventricular area. The ischemic zones for all ventricular sections were averaged to obtain the percentage of the ventricle that was ischemic. There were no differences in the ischemic ventricular zone between the intact and paraplegic mice (62 ± 5 vs. 60 ± 4%).
Preparation of heart sections (n = 3/group)
One week after completion of the experimental protocols, the mice were deeply anesthetized and the hearts and aortae were removed (Lujan et al. 2012a; Lujan and DiCarlo 2013a, 2014c). The ventricles were quickly rinsed in 10 mmol/L Tris, 0.9% NaCl, 0.05% thimerosal in 10 mmol/L phosphate buffer, pH 7.4 (TPBS), then immersion fixed in formaldehyde/zinc fixative for 60 min, washed in TPBS (3 × 10 min), then cryoprotected overnight in 30% sucrose (prepared in half strength TPBS). The OCT embedded ventricles were sectioned transversely from the apex to the base (short axis) at 10 μ intervals leaving an interval of 300 μ between ventricular sections. Ventricular sections were thaw mounted on Superfrost Plus slides and stained with Masson Trichrome to determine if the vascular occlusion caused cardiac damage.
Data analysis
All physiological recordings were obtained at a sampling frequency of 4 kHz. All data were expressed as means ± SE. A two‐way repeated measures ANOVA was used to determine time and group (intact and T5X) responses in arterial pressure, cardiac output, systemic peripheral resistance, heart rate and stroke volume before coronary artery occlusion, during coronary artery occlusion and 30 min postocclusion.
A second two‐way repeated measures ANOVA was used to compare the change in all hemodynamic variables from preocclusion values during coronary artery occlusion and 30 min postocclusion. Finally, a one‐way repeated measures ANOVA was used to compare all hemodynamic variables from preocclusion values, during coronary artery occlusion and 30 min postocclusion in intact and T5X groups. The Holm‐Sidak post hoc procedure was used for post hoc pair wise comparisons. A value of P < 0.05 was considered statistically significant.
Standard calculations
Cardiac output (Q), in mL/min, was calculated by:
where K = 1.24 × d 2, [d is the cuff diameter of the flow probe, (Haywood et al. 1981)].
Systemic peripheral resistance was calculated by:
Stroke volume (SV), in μL/beat, was calculated by:
Results
Original recordings of cardiac output, arterial blood pressure, and the ECG before coronary artery occlusion (preocclusion) at minutes 1, 2, and 3 of coronary artery occlusion and following 30 min of reperfusion in a conscious intact (Panel A) and paraplegic (Panel B) male mouse are presented in Figure 1. Coronary artery occlusion significantly reduced arterial pressure only in the mouse with paraplegia (Panel B). The ECG showed the expected changes including a peaked T wave and S‐T segment elevation. None of the T5X mice tolerated 3 min of coronary artery occlusion and all mice required resuscitation at the time of release. The duration of occlusion before requiring intervention in this example was 1.6 min for the paraplegic mouse.
Figure 2 presents arterial blood pressure (Panel A), cardiac output (Panel B), systemic peripheral resistance (Panel C), heart rate (Panel D), and stroke volume (Panel E) before coronary artery occlusion (preocclusion), during coronary artery occlusion and following 30 min of reperfusion in conscious intact and paraplegic mice. Preocclusion arterial pressure, cardiac output, and stroke volume were significantly lower while preocclusion heart rate was significantly higher in the T5X mice. There was no difference in preocclusion systemic peripheral resistance.
Figure 2.
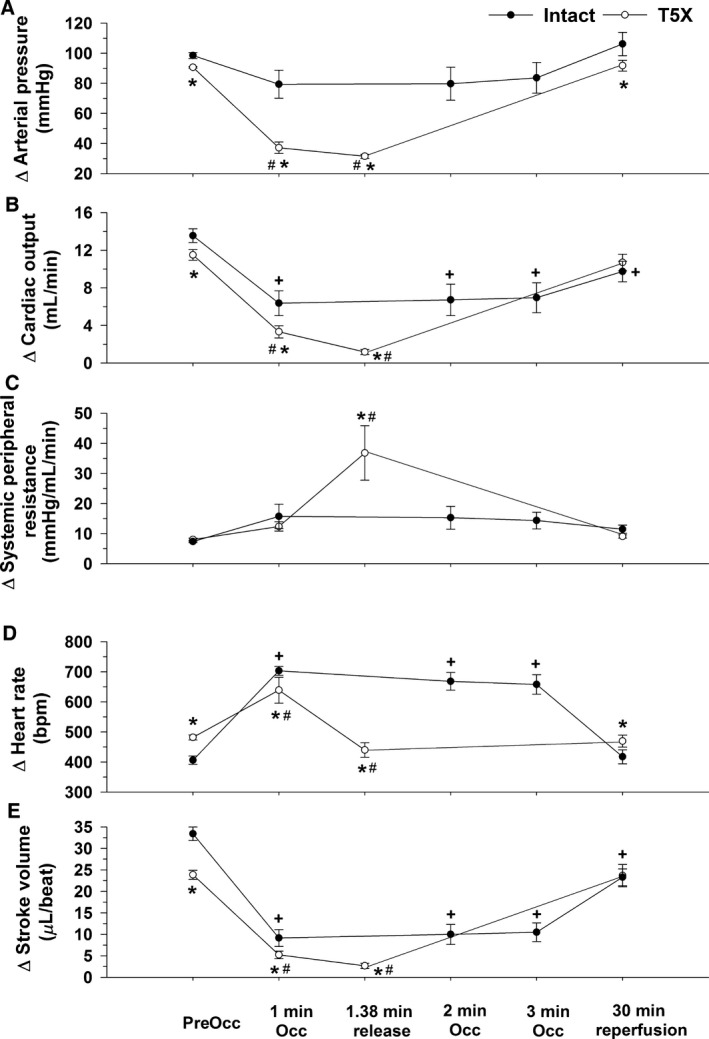
Arterial pressure (Panel A), cardiac output (Panel B), systemic peripheral resistance (Panel C), heart rate (Panel D), and stroke volume (Panel E) in conscious intact and paraplegic mice are presented. Preocclusion arterial pressure, cardiac output, and stroke volume were significantly lower while heart rate was significantly higher in T5X mice. There was no difference in preocclusion systemic peripheral resistance. The lower preocclusion arterial pressure in the mice with paraplegia was the result of a lower cardiac output with no contribution of systemic peripheral resistance. Since preocclusion heart rate was significantly elevated in the mice with paraplegia, the lower preocclusion cardiac output was the result of a lower stroke volume. Moreover, T5X mice had an impaired ability to maintain arterial blood pressure during coronary artery occlusion as arterial pressure fell to near lethal levels by 1.38 ± 0.64 min. The impaired ability to maintain arterial pressure during coronary artery occlusion in paraplegic mice was mediated by a lower cardiac output as peripheral resistance was elevated. The lower cardiac output was mediated by a reduced stroke volume and heart rate. *P ≤ 0.05, T5X versus Intact #P ≤ 0.05, T5X; Preocclusion versus 1 min, 1.38 min release, 30 min reperfusion +P ≤ 0.05, Intact; Preocclusion versus 1 min, 2 min, 3 min, 30 min reperfusion
The lower preocclusion arterial pressure (Panel A) in the mice with paraplegia was the result of a lower cardiac output (Panel B) with no contribution of systemic peripheral resistance (Panel C). Moreover, since preocclusion heart rate was significantly elevated in the mice with paraplegia (Panel D), the lower preocclusion cardiac output was the result of a lower stroke volume (Panel E).
Furthermore, acute coronary artery occlusion significantly reduced arterial blood pressure only in the paraplegic mice (Fig. 2, Panel A). Thus, paraplegic mice had an impaired ability to maintain arterial blood pressure during coronary artery occlusion as arterial pressure fell to near lethal levels by 1.38 ± 0.64 min. In contrast, intact mice tolerated the ischemic insult, without a significant reduction in arterial pressure, for the entire 3 min without incidence (Fig. 2, Panel A).
The impaired ability to maintain arterial blood pressure during coronary artery occlusion in paraplegic mice was mediated by a lower cardiac output. Specifically, the reduction in cardiac output was greater in the paraplegic mice at minute 1 of occlusion and at the time of release compared with the 2‐min time point in the intact mice (Fig. 2, Panel B and Fig. 3, Panel B). Correspondingly, the increase in peripheral resistance was greater in the paraplegic mice at the time of release compared with the 2‐min time point in the intact mice (Fig. 2, Panel C and Fig. 3, Panel C). The lower cardiac output was mediated by a reduced heart rate and stroke volume as the increase in heart rate was smaller in the paraplegic mice (Fig. 2, Panel D and Fig. 3, Panel D) while stroke volume was lower in the paraplegic mice (Fig. 2, Panel E).
Figure 3.
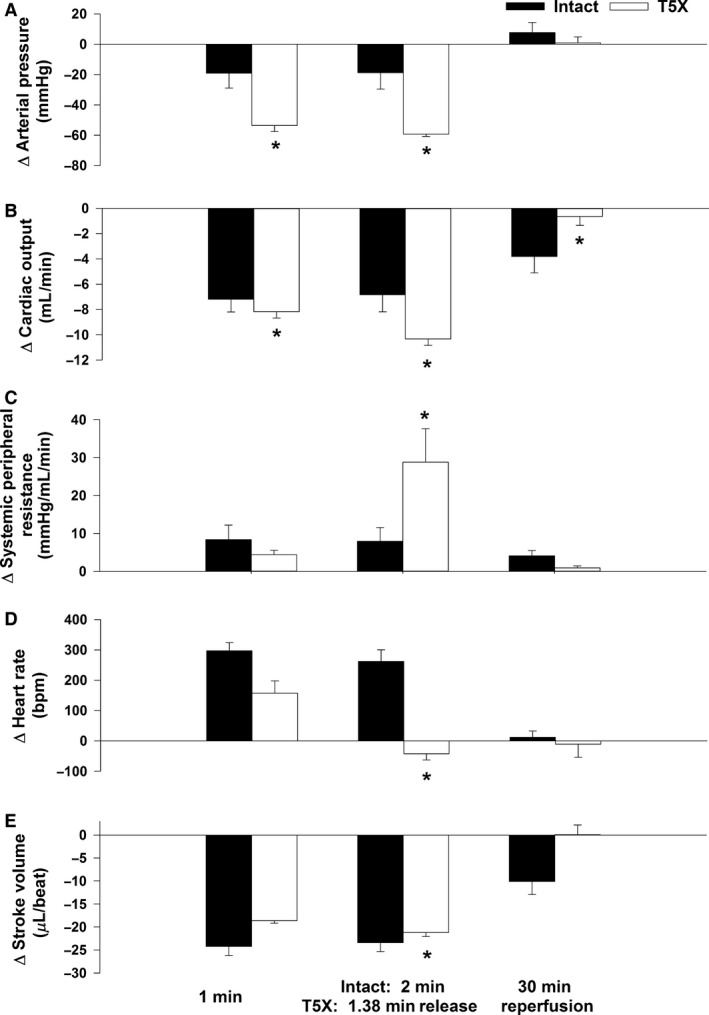
Changes in arterial pressure (Panel A), cardiac output (Panel B), systemic peripheral resistance (Panel C), heart rate (Panel D), and stroke volume (Panel E) from control in conscious intact and paraplegic mice are presented. Paraplegic mice had an impaired ability to maintain arterial blood pressure during coronary artery occlusion as the change in arterial pressure was greater in paraplegic mice (Panel A). The lower arterial pressure was mediated by a larger change in cardiac output (Panel B) as systemic peripheral resistance was elevated in paraplegic mice (Panel C). The lower cardiac output was mediated by a reduced heart rate (Panel D). *P ≤ 0.05, T5X versus Intact
Cardiac output and arterial pressure returned near preocclusion values within 30 min of reperfusion suggesting that little or no tissue damage occurred during the ischemia.
Photomicrographs of ventricular sections from intact and paraplegic mice are presented in Figure 4. The 10 μ sections were obtained from the apex through the base (short axis) at 300 micron intervals and processed with Masson Trichrome stain. The only collagen (i.e., blue stain, documenting tissue injury) shows placement of the coronary artery occluder. Thus, little tissue damage occurred during the ischemia and reperfusion protocol. Furthermore, no differences in the ischemic zone between the intact and paraplegic mice were observed (62 ± 5 vs. 60 ± 4%) documenting that the mice were subjected to the same level of ischemia.
Figure 4.
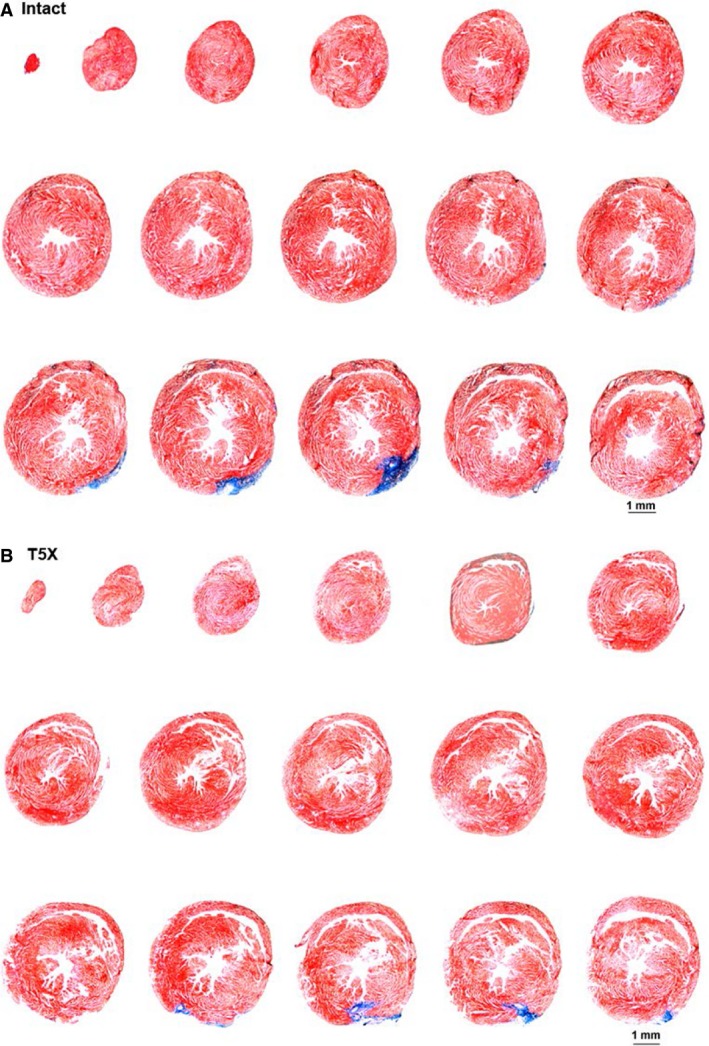
Ventricular sections from one intact and paraplegic mouse are shown. The only collagen (i.e., blue stain, documenting tissue injury) shows placement of the coronary artery occluder. Thus, little tissue damage occurs during the ischemia.
Discussion
Coronary artery occlusion is a major health concern and the principal cause of death in technologically advanced nations. Coronary artery occlusion is also expected to be the main cause of death in the world by the year 2020 (Murray and Lopez 1997). Coronary artery disease is the principal cause of coronary artery occlusion. Importantly, individuals living with SCI have an earlier onset of CAD. In fact, CAD is responsible for nearly 20% of deaths in individuals living with SCI and CAD is expected to become more impactful as the life expectancy of individuals living with SCI increases. Autonomic dysfunction, relative physical inactivity, obesity, hyperlipidemia, insulin resistance, diabetes, and chronic inflammation contribute to CAD in individuals living with SCI (Lee et al. 2005; McKinley et al. 2006).
Importantly, the danger of an untoward cardiac incident is related to the severity of CAD; suggesting that individuals living with SCI may have a greater risk for myocardial infarction or sudden cardiac death. Thus, the management of CAD for individuals living with SCI is a major health concern and therapeutic goal. Primary prevention programs (including smoking cessation, weight control, increasing physical activity, and cholesterol and/or lipid control) are critically important because prevention is the most cost‐effective and harmless strategy. In fact, it has been suggested that the identification of risks for CAD in individuals living with SCI “may be grossly underestimated in those with SCI, requiring a more aggressive approach to determine the presence of atherosclerotic occlusive vascular disease” (Bauman and Spungen 2008).
In this study, we systematically investigated if thoracic level 5 spinal cord transection (T5X; paraplegia) alters the arterial blood pressure response to coronary artery occlusion and if the different arterial pressure responses to coronary artery occlusion between intact and paraplegic mice are mediated by changes in cardiac output, peripheral resistance, or both and whether variations in cardiac output are mediated by variations in heart rate and or stroke volume. Paraplegic mice had an impaired ability to maintain arterial blood pressure during coronary artery occlusion as arterial blood pressure fell to near lethal levels within the first 2 min of occlusion. The lower arterial pressure was mediated by a lower cardiac output as peripheral resistance was elevated in paraplegic mice. The lower cardiac output was mediated by a reduced heart rate and stroke volume. These results indicate that in T5X mice, the arterial blood pressure response to coronary artery occlusion is hemodynamically mediated primarily by cardiac output which is determined by heart rate and stroke volume. Understanding the response to myocardial ischemia in paraplegic mice has the potential to positively impact individuals and families living with spinal cord injury.
The impaired ability to maintain hemodynamic stability may be related to a failure to activate sympathetic outflow below thoracic level 5. This may be due, in part, to the observation that sympathetic preganglionic neurons (SPNs) below the level of the injury undergo morphological changes after SCI. Krassioukov and Weaver documented in rats that SPNs caudal to the lesion lose dendrites and decrease soma size 1 week after the injury; however, the dendritic arbor of SPNs are re‐established 1 month after the injury (Krassioukov and Weaver 1996). There was also a significant decrease in soma size of SPNs below the level of the injury in an individual who survived for 2 weeks following a SCI at T5 (Krassioukov et al. 1999). However, the soma size from SPNs below the level of the injury was not different from SPNs above the level of the injury from a person who survived 23 years after a SCI at T3. Taken together, SPNs in spinal segments caudal to the lesion appear to undergo a significant decrease in size 2 weeks after SCI; however, the atrophy observed at 2 weeks appears transient as there is a re‐establishment of normal SPN dendritic arbor and soma size within 4 weeks. (Krassioukov et al. 1999). The initial atrophy of SPNs is thought to occur as a result of the loss of descending inputs from medullary neurons (Weaver and Polosa 1994). The initial atrophy of SPNs may contribute to the condition of “spinal shock” where there is a flaccid paralysis, with urinary bladder and bowel atonia, and profound reductions of sympathetic activity. Together, the loss of excitatory drive from medullary centers and atrophy of SPNs likely contribute to reduced sympathetic activity below the level of the injury during the first 2 weeks of a SCI (Teasell et al. 2000).
Furthermore, myocardial ischemia produces metabolites that evoke a powerful sympathetic reflex that increases arterial pressure, heart rate, ST‐segment, and double product (Lujan et al. 2011). This cardiac spinal reflex is analogous to the muscle metaboreflex observed in man, rat, and other mammals (Alam and Smirk 1937; Collins and DiCarlo 1993). Transection of the dorsal roots disrupts this excitatory sympathetic reflex and reduces heart rate and arterial pressure during myocardial ischemia (Lujan et al. 2011). In addition, cardiac output and arterial pressure are reduced during myocardial ischemia which initiates a dramatic arterial baroreflex‐mediated increase in sympathetic outflow and decrease in parasympathetic outflow to the heart as well as an increased sympathetic outflow to the vasculature. As noted, individuals with SCI at thoracic level 5 have a functional loss of the cardiac spinal reflex and the arterial baroreceptor reflex below the level of the injury and thus fail to excite sympathetic preganglionic neurons below T5. This failure to excite sympathetic preganglionic neurons below the level of the injury likely contributes to the impaired ability to maintain arterial blood pressure and cardiac output during coronary artery occlusion.
The failure to excite sympathetic preganglionic neurons below the level of the injury and reduced activity of the skeletal muscle pump likely results in a profound reduction in end‐diastolic volume and stroke volume, secondary to a reduction in venous return (Lujan and DiCarlo 2014b). This is suggested because reduced activity of the skeletal muscle pump combined with impaired autonomic control of the circulation below the level of the injury can severely impact venous return and reduce cardiac output. In addition, venous pooling can lead to a hypokinetic circulation (Glaser 1985; Glaser and Davis 1989). Moreover, therapeutic techniques that enhance venous return increase stroke volume and cardiac output for individuals living with SCI (Hopman et al. 1992, 1993; Pitetti et al. 1994; Kerk et al. 1995).
In addition, the loss of supra‐spinal control of sympathetic activity to the vasculature innervated below the level of the spinal cord injury results in hypotension. This hypotension initiates a dramatic arterial baroreflex‐mediated increase in sympathetic outflow and decrease in parasympathetic outflow to the heart (Lujan et al. 2012b). High levels of sympathetic outflow and reduced parasympathetic outflow following SCI induce calcium overload (Sharov and Galakhin 1984), left ventricular dysfunction, cardiac injury, and ST‐segment elevation (Morita et al. 2010). These responses likely contribute to the lower stroke volume in paraplegic mice (Lujan and DiCarlo 2014b).
Loss of supraspinal control of SPNs below the level of the injury would be expected to reduce systemic peripheral resistance and prevent vascular resistance from increasing during coronary artery occlusion (West et al. 2013). However, vascular resistance of the legs is often increased in individuals living with SCI (Hopman et al. 2002; Kooijman et al. 2003; Groothuis et al. 2005; Thijssen et al. 2007). Furthermore, individuals living with thoracic SCI have an increase in leg vascular resistance during head‐up tilt that is comparable with that in able‐bodied individuals (Grimm et al. 1997; Raymond et al. 1999; Groothuis et al. 2005). The mechanism(s) most often defined for mediating the elevated vascular resistance in individuals living with SCI are compensatory alterations in other vasoconstrictor pathways including angiotensin II (Krum et al. 1992; Groothuis et al. 2010) and endothelin I (Thijssen et al. 2007) as well as an enhanced sensitivity to norepinephrine (Mathias et al. 1976; Krum et al. 1992; Arnold et al. 1995).
Myocardial ischemia and reperfusion protocols cannot be tested in man. Accordingly, appropriate complex, conscious animal models are essential. In this context, we recorded the response to myocardial ischemia in conscious, chronically instrumented intact and paraplegic mice. It should be considered that the hemodynamic response to coronary artery occlusion is potentially different between rats and mice (Lujan et al. 2012a; Lujan and DiCarlo 2013a,b, 2014a,c). Specifically, we preformed virtually identical procedures in conscious intact and paraplegic rats and every rat developed sustained ventricular tachycardia within 10 min of coronary artery occlusion (Lujan and DiCarlo 2007; Lujan et al. 2009, 2010). Paraplegic mice however did not develop sustained ventricular arrhythmia as the paraplegic mice tolerated the occlusion for <2 min. It is important to note however that these paraplegic mice were studied 2 weeks post SCI while the paraplegic rats were studied 4 weeks post SCI (Lujan and DiCarlo 2007; Lujan et al. 2009, 2010). Understanding the differences between species and/or duration of injury may advance the concepts and ideas that drive therapeutic intervention to improve the quality of life for individuals and families living with SCI.
Summary and Conclusion
Coronary artery occlusion is a major health concern and a principle cause of death in the world (Murray and Lopez 1997). Autonomic dysfunction, relative inactivity, and adverse changes in muscle mass and adiposity lead to metabolic abnormalities that accelerate the development of CAD; and thus, the potential for myocardial infarction or sudden cardiac death in individuals living with SCI. Accordingly, understanding the response to myocardial ischemia in models of SCI has the potential to be of major importance for advancing the concepts and methods that drive cardiovascular therapies.
Paraplegic mice had an impaired ability to maintain arterial blood pressure during coronary artery occlusion. The lower arterial blood pressure was mediated by a lower cardiac output as peripheral resistance was elevated in paraplegic mice. The lower cardiac output was mediated by a reduced heart rate and stroke volume. These results indicate that in paraplegic mice, the arterial blood pressure response to coronary artery occlusion is hemodynamically mediated primarily by cardiac output which is determined by heart rate and stroke volume.
It should be noted that there are significant differences between mice and man that could profoundly influence the responses. However, studies conducted at many levels, from molecules to man, are critical for finding solutions for individuals with SCI (Lujan et al. 2012a). Accordingly, an extensive array of investigations, rather than a solitary model is required. Accordingly, the conscious mouse provides an additional tool for understanding cardiovascular responses to coronary artery occlusion in models of SCI.
Conflict of Interest
No conflicts of interest, financial or otherwise, are declared by the author(s).
Acknowledgments
We wish to thank Hussein Janbaih for his expert technical assistance.
Lujan H. L., DiCarlo S. E.. Fundamental hemodynamic mechanisms mediating the response to myocardial ischemia in conscious paraplegic mice: cardiac output versus peripheral resistance. Physiol Rep, 5 (6), 2017, e13214, doi; 10.14814/phy2.13214
Funding Information
This work was supported by National Heart, Lung, and Blood Institute Grant RO1HL‐122223
References
- Alam, M. , and Smirk F. H.. 1937. Observations in man upon a blood pressure raising reflex arising from the voluntary muscles. J. Physiol. 89:372–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold, J. M. , Feng Q. P., Delaney G. A., and Teasell R. W.. 1995. Autonomic dysreflexia in tetraplegic patients: evidence for alpha‐adrenoceptor hyper‐responsiveness. Clin. Auton. Res. 5:267–270. [DOI] [PubMed] [Google Scholar]
- Baker, D. G. , Coleridge H. M., Coleridge J. C., and Nerdrum T.. 1980. Search for a cardiac nociceptor: stimulation by bradykinin of sympathetic afferent nerve endings in the heart of the cat. J. Physiol. 306:519–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman, W. , and Spungen A.. 2007. Risk assessment for coronary heart disease in a veteran population with spinal cord injury. Top. Spinal. Cord. Inj. Rehab. 12:35–53. [Google Scholar]
- Bauman, W. A. , and Spungen A. M.. 2008. Coronary heart disease in individuals with spinal cord injury: assessment of risk factors. Spinal Cord 46:466–476. [DOI] [PubMed] [Google Scholar]
- Bauman, W. A. , Spungen A. M., Zhong Y. G., Rothstein J. L., Petry C., and Gordon S. K.. 1992. Depressed serum high density lipoprotein cholesterol levels in veterans with spinal cord injury. Paraplegia 30:697–703. [DOI] [PubMed] [Google Scholar]
- Bauman, W. A. , Raza M., Chayes Z., and Machac J.. 1993. Tomographic thallium‐201 myocardial perfusion imaging after intravenous dipyridamole in asymptomatic subjects with quadriplegia. Arch. Phys. Med. Rehabil. 74:740–744. [DOI] [PubMed] [Google Scholar]
- Bauman, W. A. , Raza M., Spungen A. M., and Machac J.. 1994. Cardiac stress testing with thallium‐201 imaging reveals silent ischemia in individuals with paraplegia. Arch. Phys. Med. Rehabil. 75:946–950. [PubMed] [Google Scholar]
- Bauman, W. A. , Adkins R. H., Spungen A. M., Herbert R., Schechter C., Smith D., et al. 1999. Is immobilization associated with an abnormal lipoprotein profile? Observations from a diverse cohort. Spinal Cord 37:485–493. [DOI] [PubMed] [Google Scholar]
- Bohl, S. , Medway D. J., Schulz‐Menger J., Schneider J. E., Neubauer S., and Lygate C. A.. 2009. Refined approach for quantification of in vivo ischemia‐reperfusion injury in the mouse heart. Am. J. Physiol. Heart Circ. Physiol. 297:H2054–H2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardus, D. , Ribas‐Cardus F., and McTaggart W. G.. 1992. Lipid profiles in spinal cord injury. Paraplegia 30:775–782. [DOI] [PubMed] [Google Scholar]
- Collins, H. L. , and DiCarlo S. E.. 1993. Cardiac afferents attenuate the muscle metaboreflex in the rat. J. Appl. Physiol. 75:114–120. [DOI] [PubMed] [Google Scholar]
- Cragg, J. J. , Noonan V. K., Krassioukov A., and Borisoff J.. 2013. Cardiovascular disease and spinal cord injury: results from a national population health survey. Neurology 81:723–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Hancox J. C., Farkas A., Wainwright C. L., Stables C. L., Saint D. A., et al. 2013. The Lambeth Conventions (II): guidelines for the study of animal and human ventricular and supraventricular arrhythmias. Pharmacol. Ther. 139:213–248. [DOI] [PubMed] [Google Scholar]
- DeVivo, M. J. 1997. Causes and costs of spinal cord injury in the United States. Spinal Cord 35:809–813. [DOI] [PubMed] [Google Scholar]
- DeVivo, M. J. , Black K. J., and Stover S. L.. 1993. Causes of death during the first 12 years after spinal cord injury. Arch. Phys. Med. Rehabil. 74:248–254. [PubMed] [Google Scholar]
- Fleg, J. L. , Gerstenblith G., Zonderman A. B., Becker L. C., Weisfeldt M. L., Costa P. T. Jr, et al. 1990. Prevalence and prognostic significance of exercise‐induced silent myocardial ischemia detected by thallium scintigraphy and electrocardiography in asymptomatic volunteers. Circulation 81:428–436. [DOI] [PubMed] [Google Scholar]
- Frankel, H. L. , Coll J. R., Charlifue S. W., Whiteneck G. G., Gardner B. P., Jamous M. A., et al. 1998. Long‐term survival in spinal cord injury: a fifty year investigation. Spinal Cord 36:266–274. [DOI] [PubMed] [Google Scholar]
- Fu, L. W. , and Longhurst J. C.. 2002. Activated platelets contribute to stimulation of cardiac afferents during ischaemia in cats: role of 5‐HT(3) receptors. J. Physiol. 544:897–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, L. W. , and Longhurst J. C.. 2005. Interactions between histamine and bradykinin in stimulation of ischaemically sensitive cardiac afferents in felines. J. Physiol. 565:1007–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, L. W. , Schunack W., and Longhurst J. C.. 2005. Histamine contributes to ischemia‐related activation of cardiac spinal afferents: role of H1 receptors and PKC. J. Neurophysiol. 93:713–722. [DOI] [PubMed] [Google Scholar]
- Glaser, R. M. 1985. Exercise and locomotion for the spinal cord injured. Exerc. Sport Sci. Rev. 13:263–303. [PubMed] [Google Scholar]
- Glaser, R. , and Davis G. . 1989. Wheelchair‐dependent individuals in Exercise in modern medicine. Williams & Wilkins, Baltimore. [Google Scholar]
- Grimm, D. R. , De Meersman R. E., Almenoff P. L., Spungen A. M., and Bauman W. A.. 1997. Sympathovagal balance of the heart in subjects with spinal cord injury. Am. J. Physiol. 272:H835–H842. [DOI] [PubMed] [Google Scholar]
- Groah, S. L. , Weitzenkamp D., Sett P., Soni B., and Savic G.. 2001. The relationship between neurological level of injury and symptomatic cardiovascular disease risk in the aging spinal injured. Spinal Cord 39:310–317. [DOI] [PubMed] [Google Scholar]
- Groothuis, J. T. , Boot C. R., Houtman S., van Langen H., and Hopman M. T.. 2005. Leg vascular resistance increases during head‐up tilt in paraplegics. Eur. J. Appl. Physiol. 94:408–414. [DOI] [PubMed] [Google Scholar]
- Groothuis, J. T. , Thijssen D. H., Rongen G. A., Deinum J., Danser A. H., Geurts A. C., et al. 2010. Angiotensin II contributes to the increased baseline leg vascular resistance in spinal cord‐injured individuals. J. Hypertens. 28:2094–2101. [DOI] [PubMed] [Google Scholar]
- Haywood, J. R. , Shaffer R. A., Fastenow C., Flink G. D., and Brody M. J.. 1981. Regional blood flow measurements with pulsed Doppler flowmeter in conscious rat. Am. J. Physiol. Heart Circ. Physiol. 241:H273–H278. [DOI] [PubMed] [Google Scholar]
- Hopman, M. T. , Oeseburg B., and Binkhorst R. A.. 1992. The effect of an anti‐G suit on cardiovascular responses to exercise in persons with paraplegia. Med. Sci. Sports Exerc. 24:984–990. [PubMed] [Google Scholar]
- Hopman, M. T. , Kamerbeek I. C., Pistorius M., and Binkhorst R. A.. 1993. The effect of an anti‐G suit on the maximal performance of individuals with paraplegia. Int. J. Sports Med. 14:357–361. [DOI] [PubMed] [Google Scholar]
- Hopman, MT , Groothuis JT, Flendrie M, Gerrits KH, and Houtman S. 2002. Increased vascular resistance in paralyzed legs after spinal cord injury is reversible by training. J. App. Physiol. (Bethesda, Md: 1985) 93: 1966–1972. [DOI] [PubMed] [Google Scholar]
- Inskip, J. , Plunet W., Ramer L., Ramsey J. B., Yung A., Kozlowski P., et al. 2010. Cardiometabolic risk factors in experimental spinal cord injury. J. Neurotrauma 27:275–285. [DOI] [PubMed] [Google Scholar]
- Kerk, J. K. , Clifford P. S., Snyder A. C., Prieto T. E., O'Hagan K. P., Schot P. K., et al. 1995. Effect of an abdominal binder during wheelchair exercise. Med. Sci. Sports Exerc. 27:913–919. [PubMed] [Google Scholar]
- Kessler, K. M. , Pina I., Green B., Burnett B., Laighold M., Bilsker M., et al. 1986. Cardiovascular findings in quadriplegic and paraplegic patients and in normal subjects. Am. J. Cardiol. 58:525–530. [DOI] [PubMed] [Google Scholar]
- Kooijman, M. , Rongen G. A., Smits P., and Hopman M. T.. 2003. Preserved alpha‐adrenergic tone in the leg vascular bed of spinal cord‐injured individuals. Circulation 108:2361–2367. [DOI] [PubMed] [Google Scholar]
- Krassioukov, A. V. , and Weaver L. C.. 1996. Morphological changes in sympathetic preganglionic neurons after spinal cord injury in rats. Neuroscience 70:211–225. [DOI] [PubMed] [Google Scholar]
- Krassioukov, A. V. , Bunge R. P., Pucket W. R., and Bygrave M. A.. 1999. The changes in human spinal sympathetic preganglionic neurons after spinal cord injury. Spinal Cord 37:6–13. [DOI] [PubMed] [Google Scholar]
- Krum, H. , Louis W. J., Brown D. J., and Howes L. G.. 1992. Pressor dose responses and baroreflex sensitivity in quadriplegic spinal cord injury patients. J. Hypertens. 10:245–250. [DOI] [PubMed] [Google Scholar]
- Kurtz, T. W. , Lujan H. L., and DiCarlo S. E.. 2014. The 24 h pattern of arterial pressure in mice is determined mainly by heart rate‐driven variation in cardiac output. Physiol. Rep. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M. Y. , Myers J., Hayes A., Madan S., Froelicher V. F., Perkash I., et al. 2005. C‐reactive protein, metabolic syndrome, and insulin resistance in individuals with spinal cord injury. J. Spinal Cord Med. 28:20–25. [DOI] [PubMed] [Google Scholar]
- Lee, C. S. , Lu Y. H., Lee S. T., Lin C. C., and Ding H. J.. 2006. Evaluating the prevalence of silent coronary artery disease in asymptomatic patients with spinal cord injury. Int. Heart J. 47:325–330. [DOI] [PubMed] [Google Scholar]
- Longhurst, J. C. , Tjen A. L. S., and Fu L. W.. 2001. Cardiac sympathetic afferent activation provoked by myocardial ischemia and reperfusion. Mechanisms and reflexes. Ann. N. Y. Acad. Sci. 940:74–95. [DOI] [PubMed] [Google Scholar]
- Lujan, H. L. , and DiCarlo S. E.. 2007. T5 spinal cord transection increases susceptibility to reperfusion‐induced ventricular tachycardia by enhancing sympathetic activity in conscious rats. Am. J. Physiol. Heart Circ. Physiol. 293:H3333–H3339. [DOI] [PubMed] [Google Scholar]
- Lujan, H. L. , and DiCarlo S. E.. 2013a. Cardiac output, at rest and during exercise, before and during myocardial ischemia, reperfusion, and infarction in conscious mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 304:R286–R295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan, H. L. , and DiCarlo S. E.. 2013b. Mimicking the endogenous current of injury improves post‐infarct cardiac remodeling. Med. Hypotheses 81:521–523. [DOI] [PubMed] [Google Scholar]
- Lujan, H. L. , and DiCarlo S. E.. 2014a. Cardiac electrophysiology and the susceptibility to sustained ventricular tachycardia in intact, conscious mice. Am. J. Physiol. Heart Circ. Physiol. 306:H1213–H1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan, H. L. , and DiCarlo S. E.. 2014b. Increasing venous return as a strategy to prevent or reverse cardiac dysfunction following spinal cord injury. J. Physiol. 592:1727–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan, H. L. , and DiCarlo S. E.. 2014c. Reperfusion‐induced sustained ventricular tachycardia, leading to ventricular fibrillation, in chronically instrumented, intact, conscious mice. Physiol. Rep. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan, H. L. , Chen Y., and DiCarlo S. E.. 2009. Paraplegia Increased Cardiac NGF Content, Sympathetic Tonus and the Susceptibility to Ischemia‐Induced Ventricular Tachycardia in Conscious Rats. Am. J. Physiol. Heart Circ. Physiol. 296:H1364–H1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan, H. L. , Palani G., Zhang L., and DiCarlo S. E.. 2010. Targeted Ablation of Cardiac Sympathetic Neurons Reduces the Susceptibility to Ischemia‐Induced Sustained Ventricular Tachycardia in Conscious Rats. Am. J. Physiol. Heart Circ. Physiol. 298:H1330–H1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan, H. L. , Krishnan S., and DiCarlo S. E.. 2011. Cardiac spinal deafferentation reduces the susceptibility to sustained ventricular tachycardia in conscious rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301:R775–R782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan, H. L. , Janbaih H., Feng H. Z., Jin J. P., and DiCarlo S. E.. 2012a. Myocardial ischemia, reperfusion, and infarction in chronically instrumented, intact, conscious, and unrestrained mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 302:R1384–R1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan, H. L. , Janbaih H., Feng H. Z., Jin J. P., and DiCarlo S. E.. 2012b. Ventricular function during exercise in mice and rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 302:R68–R74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malliani, A. , Lombardi F., and Pagani M.. 1981. Functions of afferents in cardiovascular sympathetic nerves. J. Auton. Nerv. Syst. 3:231–236. [DOI] [PubMed] [Google Scholar]
- Mathias, C. J. , Frankel H. L., Christensen N. J., and Spalding J. N. K.. 1976. Enhanced pressor response to noradrenaline in patients with cervical spinal cord transection. Brain 99:757–770. [DOI] [PubMed] [Google Scholar]
- McKinley, W. , Garstang S, Wieting J, Talavera F, Allen K, and Campagnolo D. 2006. Cardiovascular Concerns in Spinal Cord Injury. eMedicine eMedicine Specialties/Physical Medicine and Rehabilitation/Spinal Cord Injury Available at: http://emedicine.medscape.com/article/321771-overview (accessed 29 December 2016)
- Meller, S. T. , and Gebhart G. F.. 1992. A critical review of the afferent pathways and the potential chemical mediators involved in cardiac pain. Neuroscience 48:501–524. [DOI] [PubMed] [Google Scholar]
- Morita, S. , Inokuchi S., Yamagiwa T., Aoki H., Nakagawa Y., and Yamamoto I.. 2010. Tako‐tsubo‐like left ventricular dysfunction with ST‐segment elevation after central spinal cord injury: a case report. J. Emerg. Med. 39:301–304. [DOI] [PubMed] [Google Scholar]
- Murray, C. J. L. , and Lopez A. D.. 1997. Alternative projection of mortality and disability by cause 1990‐2020: Global burden of disease study. Lancet 349:1498–1504. [DOI] [PubMed] [Google Scholar]
- Orakzai, S. H. , Orakzai R. H., Ahmadi N., Agrawal N., Bauman W. A., Yee F., et al. 2007. Measurement of coronary artery calcification by electron beam computerized tomography in persons with chronic spinal cord injury: evidence for increased atherosclerotic burden. Spinal Cord 45:775–779. [DOI] [PubMed] [Google Scholar]
- Pan, H. L. , Longhurst J. C., Eisenach J. C., and Chen S. R.. 1999. Role of protons in activation of cardiac sympathetic C‐fibre afferents during ischaemia in cats. J. Physiol. 518(Pt 3):857–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitetti, K. H. , Barrett P. J., Campbell K. D., and Malzahn D. E.. 1994. The effect of lower body positive pressure on the exercise capacity of individuals with spinal cord injury. Med. Sci. Sports Exerc. 26:463–468. [PubMed] [Google Scholar]
- Raymond, J. , Davis G. M., Bryant G., and Clarke J.. 1999. Cardiovascular responses to an orthostatic challenge and electrical‐stimulation‐induced leg muscle contractions in individuals with paraplegia. Eur. J. Appl. Physiol. Occup. Physiol. 80:205–212. [DOI] [PubMed] [Google Scholar]
- Sharov, V. G. , and Galakhin K. A.. 1984. Myocardial changes after spinal cord injuries in humans and experimental animals. Arkh. Patol. 46:17–20. [PubMed] [Google Scholar]
- Strack, A. M. , Sawyer W. B., Marubio L. M., and Loewy A. D.. 1988. Spinal origin of sympathetic preganglionic neurons in the rat. Brain Res. 455:187–191. [DOI] [PubMed] [Google Scholar]
- Swoap, S. J. , Overton J. M., and Garber G.. 2004. Effect of ambient temperature on cardiovascular parameters in rats and mice: a comparative approach. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287:R391–R396. [DOI] [PubMed] [Google Scholar]
- Teasell, R. W. , Arnold J. M. O., Krassioukov A., and Delaney G. A.. 2000. Cardiovascular consequences of loss of supraspinal control of the sympathetic nervous system after spinal cord injury. Arch. Phys. Med. Rehabil. 81:506–516. [DOI] [PubMed] [Google Scholar]
- Thijssen, D. H. , Ellenkamp R., Kooijman M., Pickkers P., Rongen G. A., Hopman M. T., et al. 2007. A causal role for endothelin‐1 in the vascular adaptation to skeletal muscle deconditioning in spinal cord injury. Arterioscler. Thromb. Vasc. Biol. 27:325–331. [DOI] [PubMed] [Google Scholar]
- Tjen, A. L. , Fu L. W., and Longhurst J. C.. 2002. Xanthine oxidase, but not neutrophils, contributes to activation of cardiac sympathetic afferents during myocardial ischaemia in cats. J. Physiol. 543:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida, Y. , and Murao S.. 1974. Excitation of afferent cardiac sympathetic nerve fibers during coronary occlusion. Am. J. Physiol. 226:1094–1099. [DOI] [PubMed] [Google Scholar]
- Weaver, L. , and Polosa C.. 1994. Spinal cord circuits providing control of sympathetic preganglionic neurons in Jordan D., ed. The autonomic nervous system control of autonomic functions. Harwood Academic Publishers, London. [Google Scholar]
- West, C. R. , Mills P., and Krassioukov A. V.. 2012. Influence of the neurological level of spinal cord injury on cardiovascular outcomes in humans: a meta‐analysis. Spinal Cord 50:484–492. [DOI] [PubMed] [Google Scholar]
- West, C. R. , Alyahya A., Laher I., and Krassioukov A.. 2013. Peripheral vascular function in spinal cord injury: a systematic review. Spinal Cord 51:10–19. [DOI] [PubMed] [Google Scholar]
- Whiteneck, G. G. , Charlifue S. W., Frankel H. L., Fraser M. H., Gardner B. P., Gerhart K. A., et al. 1992. Mortality, morbidity, and psychosocial outcomes of persons spinal cord injured more than 20 years ago. Paraplegia 30:617–630. [DOI] [PubMed] [Google Scholar]