

Abstract
Pediatric Acute liver failure (PALF) is a potentially devastating condition which occurs in previously healthy children of all ages and frequently leads to a rapid clinical deterioration. An identified cause for liver injury is lacking in approximately 30% of cases. Children with undetermined diagnosis have lower spontaneous survival and higher rates of transplantation and death than other diagnostic groups. A single day workshop sponsored by the National Institute of Diabetes and Digestive and Kidney Diseases brought together clinicians and basic scientists to integrate aligned research findings and develop a foundation for new mechanistic studies and future treatment trials. The clinical phenotype of indeterminate PALF shares important similarities to the hyperinflammatory state characteristic of Hemophagocytic lymphohistiocytosis (HLH) and Macrophage Activation Syndrome (MAS). A failure of cytotoxic T cells to limit or contract inflammatory responses may propagate injury and lead to a local and systemic milieu that does not support normal hepatic regeneration. Evidence was presented that bone marrow (BM) derived Sinusoidal endothelial cell PROgenitor Cells (sprocs) play a vital role in hepatic regeneration. Overwhelming systemic inflammatory responses may suppress mobilization of BM sprocs and dampen hepatic recovery. Conclusion: Thus, experience gained through treatment trials of HLH and MAS in childhood may inform study design for therapy of PALF. Successful approaches to limiting neuro-inflammation through reduction of systemic inflammation and standardized neuroprotection protocols that limit glial injury could significantly improve intact survival. Finally, since PALF is a rare disease, investigative efforts must include broad multi-center collaboration and careful stewardship of biorepository specimens.
Acute liver failure in children is a potentially devastating condition occurring in previously healthy children of all ages. Often a viral-like illness preludes the deterioration into jaundice, coagulopathy, encephalopathy and frequently death. Early stages of encephalopathy can be difficult to detect in younger children. Therefore, the broadest, accepted definition of pediatric acute liver failure (PALF) includes both moderate coagulopathy (INR > 1.5) with encephalopathy and severe coagulopathy (INR > 2.0) without evidence of encephalopathy. The precise incidence of PALF is not known, but several cases are seen each year in large pediatric medical centers. According to UNOS/OPTN data from 1/1/11 through 5/31/13, acute liver failure accounted for 11% of pediatric liver transplants.(1) In a large number of PALF cases, the etiology has eluded discovery.
The PALF study group (PALFSG) was formed in 1999 (2), initially as a sub-study within the adult Acute Liver Failure Study Group (ALFSG). It was funded independently through a Cooperative Agreement beginning in 2005. To date, more than 1000 children have enrolled in this study and an etiological diagnosis is lacking in approximately 30% of PALF cases. Among subjects age 1–5 years, this percentage exceeds 60%. Children with liver failure of undetermined cause have lower spontaneous survival and higher rates of transplantation and death than other diagnostic groups. When the diagnosis is undetermined, it may reflect an insufficient evaluation that was inappropriate or interrupted by death or transplant. Within the PALFSG cohort, incomplete evaluations for autoimmune hepatitis, mitochondrial disorders and other metabolic diseases were the most common deficiencies (3). Supplemental testing of PALF repository samples also identified evidence of acute hepatotropic viral infection that was not attributed as the cause of liver injury.(4) To standardize and improve diagnostic testing, recommendations for age appropriate evaluation were adopted by the PALFSG in 2010. Detailed diagnostic recommendations are included elsewhere. (5) It should be noted that children with recurrent episodes of acute liver failure need to be investigated thoroughly for metabolic disorders and for newly described mutations in the Neuroblastoma Amplified Sequence.(6) However, in some cases of undetermined cause, the evaluation is appropriate and complete and the cause is deemed “indeterminate”. Experienced clinicians caring for these patients believe the etiology is truly “indeterminate” in over a third of PALF cases.
A single day PALF workshop sponsored by the National Institute of Diabetes and Digestive and Kidney Diseases took place at the National Institutes of Health in Bethesda on October 16, 2015. The goal of the meeting was to improve diagnostic approaches in PALF and define the phenotype of indeterminate PALF (iPALF). The conference brought together experts from several disciplines to discuss and integrate recent research findings in Hepatology, Immunology, Pathology, Regenerative Medicine, Genetics and Stem Cell Biology. Presentations focused on emerging concepts regarding mechanisms of hepatic injury, new diagnostic tools and options for future treatment trials.
Clinical and Pathophysiological Features of iPALF
The PALFSG has made significant contributions in describing the epidemiology of acute liver failure in childhood and has assembled a large cohort of iPALF cases. iPALF can occur at any age and cases do not segregate by sex or race. Typically, there is a prodromal viral-like illness days to weeks before presentation. The majority of children develop hepatic encephalopathy, but signs can be subtle, especially in infants. Transient bone marrow suppression is common, and some patients progress to aplastic anemia even after successful liver transplantation.
The search for clues to diagnose iPALF cases continues, acknowledging that several distinct causes may be identified. Occult acetaminophen (APAP) injury has gained recognition as one potential cause. APAP-protein adducts are found in serum of 10% of iPALF cases. Adduct positive cases more closely resemble the clinical phenotype (higher transaminases, lower bilirubin and increased spontaneous recovery) of known APAP toxicity cases than that of the remaining iPALF cohort (7). Viral etiologies have also been explored. Wintertime clustering of iPALF supports the theory that community acquired viral infection may play a role, but a novel or common viral etiology has not been identified. Concurrence of aplastic anemia in some cases suggests bone marrow injury that could be secondary to viral infection or auto-inflammatory responses. Yet despite intensified diagnostic efforts over the past five years, many PALF cases cannot be explained by our current understanding of disease mechanisms.
Patients with iPALF have higher rates of death and liver transplantation than those with known causes. Treatments exist for certain diagnoses that may present as PALF. Examples include steroids for autoimmunity and copper chelation for Wilson disease. However, for most PALF cases, general supportive measures and liver transplantation are the mainstays of therapy. The PALFSG conducted a randomized trial of N-acetylcysteine in non-APAP cases that did not demonstrate a treatment benefit (8). Recently, there has been a trend toward utilizing steroid therapy for iPALF cases, especially when they display features of hyperinflammatory immune dysregulation, such as elevated serum soluble interleukin 2 receptor levels. While circumstantial evidence is building that acquired immune dysregulation may contribute to the pathophysiology in PALF, the ability to prove this hypothesis remains challenging (9).
Opportunities to learn from the ALFSG experience are important since they have similarly struggled to understand indeterminate cases. As of January 2014, the ALFSG had enrolled over 2000 patients (10). The etiology of liver failure in adults is distinctly different from children, the majority related to drug induced liver injury including APAP. Yet, the second largest group were indeterminate cases (12% overall). A recent initiative was undertaken to review and adjudicate indeterminate cases to establish if site investigators overlooked a plausible cause. This approach included supplemental testing with newer diagnostic tests, such as APAP adduct testing and more comprehensive testing for viral pathogens. APAP-adducts were positive in 18% suggesting occult APAP toxicity. (11) This led to reassignment of the diagnosis when the clinical characteristics were also consistent with APAP toxicity. Likewise, some cases were reclassified as Autoimmune Acute Liver Failure based on standardized diagnostic criteria (12). These patients had a longer duration from onset of jaundice to encephalopathy, lower alanine aminotransferase, and improved survival when compared to other indeterminate cases. The ALFSG group has also initiated a rigorous viral discovery program, confirming viral cases recognized by enrollment centers, and identifying additional cases of hepatotropic viruses, including Herpes Simplex Virus and Hepatitis C. Ultimately, the adjudication committee ascribed a single diagnosis to 40% of indeterminate cases and multiple plausible diagnoses to 30%. The remaining 30% of indeterminate patients (3% of overall cohort) were classified as having an incomplete work-up or no diagnosis despite a complete evaluation.
Histopathology of Acute Liver Failure
Histopathology may inform potential mechanisms of injury as well as provide prognostic information. Distinct patterns of injury can be recognized, Table 1. Several studies have explored histological features as predictors of prognosis. In a recent examination of transjugular liver biopsy results in 69 adults with acute liver failure, findings altered the diagnosis in 19% of patients and predicted death and transplantation in patients with >75% hepatocyte necrosis (13). Specific histologic features including steatosis and ductular reaction predicted outcomes in suspected drug induced liver injury cases (14).
Table 1.
Common histologic patterns of injury in acute liver failure
| Pattern | Description | Associated Causes |
|---|---|---|
| Coagulative Necrosis | Typically zonal injury including hepatocyte “ghost cells”. Usually accompanied by only minimal inflammatory infiltrate. If the patient recovers, normal architecture is usually restored | Toxins, acetaminophen and ischemic hepatitis |
| Confluent Necrosis | This injury is associated with prominent parenchymal inflammation and an irregular pattern of necrosis. Injury frequently results in parenchymal collapse and is often accompanied by a marked ductular reaction. | Viral hepatitis, drug and indeterminate |
| Acute on Chronic Liver Injury | Hallmarks of this injury include fibrosis, inflammation, and cholestasis | Autoimmune Hepatitis and Wilson disease |
| Microvesicular Steatosis | Evidence of mitochondrial injury with hepatocytes that are generally intact | Primary mitochondrial cytopathy or secondary mitochondrial injury due to aspirin or valproate. |
Analysis of biopsies from 59 adults with acute liver failure (40 of unknown cause) also demonstrated that the degree of necrosis was greater for those who died or received liver transplantation (15). Cases with more severe hepatocyte necrosis had significantly fewer proliferating mature hepatocytes (Ki67 positive) and more hepatic progenitor cells (cytokeratin 7/cytokeratin 19 positive) per high power field. The authors suggested a >50% hepatocyte loss was needed for hepatic progenitor cell activation and that reaching this threshold was associated with significantly decreased proliferative activity of the remaining mature hepatocytes. Patients with >50% necrosis also had lower spontaneous survival, despite robust hepatic progenitor cell activation. Since hepatic progenitor cell response is a characteristic of overwhelming liver injury, it is not unexpected that patients with this response would have a worse prognosis than those capable of repopulating hepatic mass by proliferation of mature hepatocytes. Analysis comparing outcomes by level of hepatic progenitor cell response within the sub-group of patients with extensive (50–75%) hepatic necrosis has not been done.
Inflammatory Milieu in PALF
Recently published studies suggest overzealous systemic inflammatory responses (cellular and humoral) are characteristic of iPALF. Some reports associate PALF with Hemophagocytic Lymphohistiocytosis (HLH) or at least a forme fruste of the disease. HLH is a potentially fatal, hyperinflammatory syndrome caused by excessive activation and expansion of T lymphocytes and macrophages that exhibit hemophagocytic activity (16–18). Familial HLH (FHLH) typically presents in early childhood. This condition is caused by genetic lesions affecting the perforin-mediated cytolytic pathway, typically inherited in an autosomal recessive manner. Normal cytolytic cell function is essential for appropriate termination of immune responses. Defects in the cytolytic pathway seen in FHLH have been associated with over-production of pro-inflammatory cytokines and it has been postulated that failure of cytotoxic T cells to induce apoptosis of target cells delays the contraction phase of the immune response, leading to persistent expansion of activated T cells. This results in escalating production of pro-inflammatory cytokines particularly IFNγ, thus creating a “cytokine storm” (Figure 1). In contrast, non-familial HLH occurs sporadically throughout childhood and may be associated with precipitating conditions, such as malignancies, infections and rheumatologic disease. When HLH presents in the setting of rheumatologic disease it is labeled macrophage activation syndrome (MAS). In MAS, defects in cytolytic pathways are most often not found, even when elevated IFNγ and subsequent organ dysfunction are present. This suggests a different initiating pathophysiology can result in the same end point of HLH like disease.
Figure 1. Proposed Mechanisms of Macrophage Activation in Hemophagocytic Syndromes.
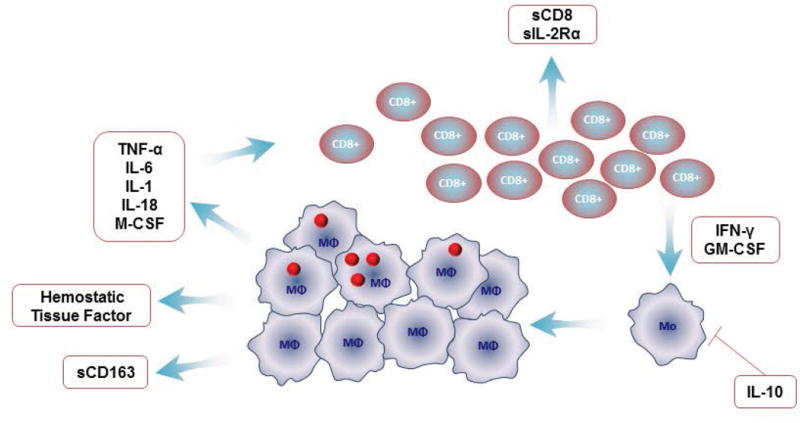
Hemophagocytic syndromes are characterized by an uncontrolled expansion of activated CD8 cells that secrete pro-inflammatory cytokines such as interferon gamma and granulocyte-macrophage colony-stimulating factor. These cytokines cause monocytes to differentiate into macrophages, which then release pro-inflammatory cytokines, thus causing an exaggerated, cyclical cascade. Persistent activation of macrophages leads to massive increase in pro-inflammatory cytokines. The mechanism leading to expansion of activated macrophages is not clear. One possible hypothesis for uncontrolled expansion of T cells is a defect in cytolytic function.
Footnote for Figure 1: Granulocyte-macrophage colony-stimulating factor (GM-CSF), soluble interleukin 2 receptor (sIL-2r), macrophage colony-stimulating factor (M-CSF). Figure courtesy of Dr Alexie Gromm.
Patients with iPALF have a liver injury pattern similar to observations in familial and sporadic HLH and MAS (19–21). However, iPALF patients do not manifest the full spectrum of criteria required for diagnosis of HLH. Like patients with HLH, some patients with iPALF present with elevated ferritin and soluble interleukin 2 receptor levels, low fibrinogen, and numerous infiltrating CD8+ T-cells on liver biopsy. (21) Mouse models of FHLH have been utilized to explore liver injury in this inflammatory state. Data from the Behrens lab demonstrated a role for IFNγ and IL-33 in inflammatory liver injury in FHLH models, leading to cellular infiltration and microvesicular steatosis.(22) Similar IFNγ-driven liver events also occur in the Toll-like Receptor 9 driven model of MAS. While there is no direct genetic or functional evidence to suggest iPALF patients have defects in cytolytic pathways, the liver CD8+ T-cell infiltrate, elevated soluble interleukin 2 receptor and ferritin, and decreased fibrinogen reported in iPALF suggests further consideration should be given to an immunologic cause of iPALF related to HLH physiology.
Mechanisms of Immune Mediated Liver Injury
The cellular and molecular mechanisms that contribute to hepatocyte injury and death during APAP induced liver failure may provide important insights into liver injury of other causes. Intriguingly, the innate immune response plays a major role in hepatocyte injury, recalling some of the processes described in FHLH and MAS associated liver injury.(23) In APAP injury, the initial release of intracellular contents from toxin damaged hepatocytes act as Damage Associated Molecular Patterns (DAMPs) which activate a number of innate immune receptors including Toll-like Receptor 9. The molecules also initiate activation of the inflammasome resulting in production of IL-1β and IL-18, which are cytokines associated with MAS. These pathways result in recruitment of neutrophils and macrophages to the liver. The exact effector functions of these cells, pro-injury versus pro-repair, remain controversial. Observations in these animal models suggest prolonged release of DAMPs, activation of Toll-like Receptor 9, increased cytokine production, and inflammatory infiltrates are prognostic of a poor outcome in acute liver failure, whereas delayed and self-limited immune response correlates with recovery.
Investigators at the University of Pittsburgh examined inflammatory responses in PALF at the systemic level to define mechanisms of injury and predict outcomes.(24) Although inflammatory mediator levels assessed over time did not predict outcomes, dynamic network analysis revealed inflammatory networks were different and more robust in non-survivors versus survivors and transplant recipients. They identified a common inflammatory network regulated via high mobility group box 1 protein and the chemokines interferon-γ-inducible protein 10 and monokine induced by gamma interferon, which may represent a general “liver signature” in inflammatory conditions such as PALF, or which may represent the central contribution of the liver to other inflammatory diseases. PALF non-survivors were more likely to display self-sustaining inflammation networks, whereas survivors and liver transplant recipients displayed lower levels of inflammation and less connectivity within networks.
Hepatic Regeneration
The liver is unique amongst visceral organs in its capacity to regenerate from injury. Research investigating the regulation of liver regeneration has elucidated physiological, cellular, and molecular mechanisms that control this process. For example, such studies have identified humoral signals, including specific growth factors, cytokines, neuroendocrine hormones, and metabolites whose induction in response to liver injury initiates intracellular signaling events that ultimately restore liver mass and function (25). These observations suggest the hepatic regenerative response in acute liver failure could be modifiable and that regenerative biomarkers can be discovered and used to predict acute liver failure outcomes. Consistent with these considerations, a recent report suggested glycemic management can be manipulated to modulate experimental liver regeneration (26). Regenerative recovery of the liver in a partial-hepatectomy model was delayed in dextrose-infused mice (versus vehicle control) implying that hypoglycemia plays an important role in promoting hepatic regeneration. In the same mouse model, comprehensive amino acid analysis revealed both liver and plasma levels of the metabolite, alpha-NH-butyric acid, were elevated early in the course of hepatic regeneration. Levels of alpha-NH-butyric acid were subsequently measured in the serum of 40 PALFSG patients and were significantly greater in patients who survived without transplantation compared to those who died or were transplanted (27). These types of discoveries could allow the development of individualized approaches to enhance hepatic regeneration (28).
During hepatic regeneration, multiple cell compartments must be restored. Regeneration of hepatocytes and biliary epithelial cells are dependent upon proliferation of mature hepatocytes and in more extensive injury, requires hepatic progenitor cell activation. Liver sinusoidal endothelial cells are repaired and replaced by bone marrow Sinusoidal endothelial cell PROgenitor Cells (sprocs) (29, 30). Sprocs are currently defined as CD133+45+31+ cells (31). Bone marrow (BM) sprocs are a major source of hepatocyte growth factor, and therefore, may play a key role in hepatic regeneration following severe acute liver injury. The vascular endothelial growth factor -stromal cell-derived factor 1 pathway is upregulated in the liver following partial-hepatectomy which increases production of BM sprocs, mobilization of sprocs into the circulation and engraftment of sprocs in the liver. The stromal cell-derived factor 1 receptor C-X-C chemokine receptor type 7 (CXCR7) is highly expressed on engrafted BM sprocs and differentiation of engrafted BM sprocs is accompanied by a significant decline in CXCR7 expression. Interventions that suppress recruitment and engraftment of BM sprocs in the liver cause parallel changes in the number of circulating sprocs. In control animals, only 1% of sprocs present in circulation are CXCR7+, whereas after partial-hepatectomy 59% of circulating sprocs are CXCR7+ (31). The implications of these findings are:
Bone marrow injury, as can be seen in systemic inflammatory conditions such as FHLH and MAS, can deplete sprocs or impair their proliferative capacity, and thereby impair hepatic regeneration. Immune therapies that prevent or ameliorate bone marrow injury during systemic inflammation may be able to prevent sproc loss and improve outcome after liver injury.
Quantifying circulating CXCR7+ sprocs could potentially predict hepatic regenerative ability and outcome after liver injury.
Approaches to Discovery of Viral and Genetic Etiologies
Some iPALF cases could be related to unidentified viral infection. Circumstantial evidence, such as prodromal viral-type symptoms, observed in many cases, supports this hypothesis. However, lack of household transmission and known infectious exposures refutes this association. Although screening for common forms of viral hepatitis, including Hepatitis A, B and C, is nearly universal, testing for viruses less frequently considered hepatotropic may not always be complete. A recent analysis of the PALF cohort that included supplemental testing for either causative or associated viral infections identified several potentially causative infections not attributed as the etiology by the managing clinicians. For example, Herpes Simplex Virus infection was noted as the primary diagnosis in only 2 of 13 older subjects who had serologic evidence of recent infection (4). In addition to well characterized, but “overlooked” viral infections, there may be uncharacterized viruses that cause hepatitis in susceptible hosts. Emerging techniques like metagenomic next-generation sequencing, combined with newer bioinformatics methods, such as sequence-based ultrarapid pathogen identification (32) have accelerated the search for these agents. Metagenomic next-generation sequencing is an attractive diagnostic approach because it does not rely on targeted primers or probes; rather, all viruses, bacteria, fungi, and/or parasites are identified in a single assay on the basis of a distinctive sequence signature. These approaches identify pathogens in a variety of tissues and acellular fluids with high sensitivity and within a clinically actionable timeframe. Metagenomic next-generation sequencing has been used to identify potential viral pathogens in a group of 187 ALFSG indeterminate cases. Previously unrecognized viruses were identified in 7 patients including herpes simplex virus -1, parvovirus B19, and human herpesvirus 7. It must be noted the presence of a viral pathogen does not necessarily imply cause. Furthermore, no novel viruses were detected in these patients. This powerful, unbiased approach to finding rare causes of hepatitis has not yet been applied to the PALF population.
Genomic approaches can be leveraged to identify genetic variants that may increase susceptibility to hepatic injury or uncover previously unsuspected Mendelian disorders. Novel statistical methods and analytical tools can reduce the millions of potentially causal genetic variants to a few candidate defects. For example, the Variant Annotation Analysis and Search Tool scores every variant in the genome and compares each variant from a set of case sequences to a set of healthy control sequences (33–35). The output is a list of genes with mutations sorted by probability of phenotype causation, resulting in substantial improvement in statistical power. Power is further increased by incorporating phenotypic information. Whole genome and whole exome sequencing can be powerful approaches to identify novel genetic defects or modifiers that could cause or exacerbate hepatic injury in PALF patients. These analyses also support evaluation of a single proband as is typical in PALF.
Therapeutic Targets for Cytokine Storm, Macrophage Activation Syndrome and Hemophagocytic Lymphohistiocytosis
Review of current therapy for hyperinflammatory states, such as HLH and MAS, may suggest potential targets and novel approaches to treatment of the untethered hepatic inflammatory response in PALF patients. Treatment of FHLH and HLH has previously included non-specific immunosuppression with corticosteroids and more specific suppression of T cell responses with anti-thymocyte globulin, cyclosporine, or Etoposide. The standard approach to therapy of MAS in rheumatologic diseases remains corticosteroids and cyclosporine, but biologic therapy targeting individual cytokines is attractive in this setting as well, Table 3. Therapeutic approaches to block IL-18 are of particular interest. Systemic IL-18 levels are higher in sJIA patients that develop MAS. Also, immunohistochemical staining of bone marrow and liver tissue from these patients reveals the presence of IL-18 in areas of intense macrophage infiltration. Administration of synthetic IL-18 binding protein in a perforin-deficient mouse model of FHLH/MAS did ameliorate the liver damage, but the animals still produced substantial pro-inflammatory cytokines and there was no change in overall survival. However, in a recent case report, treatment with IL-18 binding protein was effective in a patient with a NLR family CARD domain containing 4 protein -associated hyperinflammatory state.(36) These studies suggest further work is needed to better characterize the potential role of anti-cytokine therapy in these disorders.
Table 3.
Therapeutic Strategies in Hyperinflammatory Conditions
| Hemophagocytic lymphohistiocytosis | Macrophage activation syndrome | Systemic juvenile idiopathic arthritis | |
|---|---|---|---|
| Corticosteroids | Standard therapy | Standard therapy | Can be used initially while awaiting response to anti-cytokine therapy. |
| T cell Directed Therapies | |||
| Cyclosporine A | Standard therapy | Standard therapy | Typically only used in patients with SJIA associated MAS. |
| Anti-thymocyte Globulin | Standard therapy | Not indicated | Not indicated |
| Etoposide | Standard therapy | Usually not indicated | Not indicated |
| Anti-cytokine Therapies | |||
| Tumor necrosis factor inhibition | Not Effective | Not Effective | Limited efficacy as compared to other agents. |
| IL-1 inhibition | Not tested | IL-1β blockade with Canakinumab unable to achieve statistically significant effect in treatment or prevention even when SJIA well controlled.(38) IL-1α and β blockade with Anakinra may have dose dependent efficacy.(39, 40) |
IL-1β blockade with Canakinumab effective to induce remission. (41) |
| IL-6 inhibition | Tocilizumab not effective in prevention or treatment.(42) Effective in Chimeric Antigen Receptor T-cell therapy “Cytokine Release Syndrome”.(43) |
Tocilizumab effective in human trials.(44) | |
| IL-18 inhibition | IL-18 binding protein improved liver injury but not survival in familial hemophagocytic lymphohistiocytosis animal model. | Case report showed efficacy of IL-18 binding protein in a single patient with NLRC4-MAS.(36) | Not tested |
Footnote for Table 3: hemophagocytic lymphohistiocytosis (HLH), macrophage activation syndrome (MAS), systemic juvenile idiopathic arthritis (sJIA)
Ongoing Treatment Trials and New Strategies in Acute Liver Failure in Adults and Children
In adult medicine, the majority of trials have been performed within the infrastructure of the ALFSG. A phase II trial of ornithine phenylacetate, an ammonia scavenger, is being completed by this group and detailed results are forthcoming. Preliminary results suggest this treatment is safe and well-tolerated with ammonia levels improving in most patients. Several adult trials of extracorporeal liver support devices are also currently enrolling, the largest of which is a historically-controlled trial of the Extracorporeal Liver Assist System.
Trials currently open to pediatric patients in North America include a single center trial of hepatocyte infusions and a trial of therapy for Amatoxin-induced liver failure. A European trial of ALF-5755, a recombinant version of the secreted human protein, Hepatocarcinoma-Intestine-Pancreas/Pancreatitis Associated Protein, is in progress. This paracrine factor exerts anti-apoptotic, pro-survival, and mitogenic properties on injured hepatocytes. It was also recently shown to display anti-oxidative activity and prolong hepatocyte survival in vitro and in animal models of hepatocyte stress (37).
Treating Neuroinflammation
Results from clinical trials of neuroprotective agents in adults with acute brain injuries, including stroke or traumatic brain injury, provide important lessons for the approach to neuroprotection in PALF. Irreversible central nervous system injury can occur within seconds to minutes, making early recognition and initiation of therapy essential. Therapies targeting a single pathway are unlikely to be effective in this complex system of injury. Multiple studies have determined injury to glial cells including astrocytes, microglia and endothelial cells, play a key role in the neurological complications of acute liver failure. These cells are activated by a combination of insults, including DAMPs secondary to injured hepatocytes, systemic inflammatory response to infection, and by central nervous system-derived cytokines elicited by compromise of the blood brain barrier. These inflammatory processes subsequently lead to loss of tight junction proteins, increase in transforming growth factor beta 1, and activation of matrix metalloproteinase-9. Cytotoxic cerebral edema can occur as expression of aquaporin-4 is altered, pro-inflammatory cytokines are upregulated, and astrocyte swelling occurs. Autoregulation is impaired and synaptic excitability is altered by loss of astrocyte glutamate transporters and changes in GABAergic neurotransmission. Exposure to ammonia either causes or exacerbates many of these pathophysiological processes, Table 3. Drugs and other therapies (e.g. hypothermia) that target these mechanisms are available and in use for clinical indications other than neuroprotection in acute liver failure. Clinical trials in adults with traumatic brain injury have tested anti-inflammatory drugs such as Etanercept, Minocycline, Anakinra, statins, N-acetylcysteine, Erythropoietin and hypothermia. Results suggest such approaches may modulate the inflammatory response, but may not improve survival.
Meeting Summary and Recommendations for Future Research
One of the most pressing challenges within this area of research is the need to better characterize the mechanisms of hepatocyte injury and regeneration in children with PALF. These mechanisms may be significantly impacted by the developing immune system and thus, distinct from what is observed in adults. Knowledge gained would inform the design of treatment trials aimed at limiting ongoing injury and establishing a tissue milieu that supports regeneration. Proposed treatment trials must be feasible and evidence based.
Evidence presented in this conference highlights the similarities between HLH, MAS in rheumatologic diseases and the phenotype of iPALF. These similarities provide support for treatment strategies that manipulate immune responses to ameliorate ongoing liver injury. However, iPALF is fundamentally different from HLH and MAS in that the iPALF phenotype lacks chronicity or recurrence in survivors. Likewise, the iPALF phenotype is different from patients with FHLH in that there is no evidence of genetic defects in perforin and granzyme function and many patients survive spontaneously. Thus, animal models of FHLH may not adequately reflect cellular events in iPALF. Current research efforts would greatly benefit from development of animal models that recapitulate the features that make the “immune dysregulation” phenotype of iPALF distinctive from these other disorders. However, a central goal of investigation is to determine whether iPALF is truly a primary disease process, or an altered response to a variety of liver injuries. This area of investigation will require translational studies in human subjects that assess immune responses throughout the evolution of the injury in patients with known and unknown causes. This work should be embedded within a multi-center treatment trial that includes focused mechanistic aims.
Future studies should consider novel aspects of hepatocytic regeneration, including the relationship between bone marrow derived stem cells and repopulation of hepatic cellular pools, Figure 2. BM sprocs may play a key role in recovery from severe acute liver injury by producing essential growth factors and creating a local milieu that supports hepatic regeneration. Bone marrow suppression, a frequent feature in iPALF, may lead to depletion of sprocs from the bone marrow compartment and compromise hepatic regeneration. Metabolic drivers are likely crucial to the micro-environmental support of hepatic regeneration. Studies to determine whether these influences are uniform across multiple causes of severe liver injury would inform targeted strategies to enhance regeneration. Identification of genetic polymorphisms that suppress regenerative processes would also inform treatment development and improve prognostic models.
Figure 2.
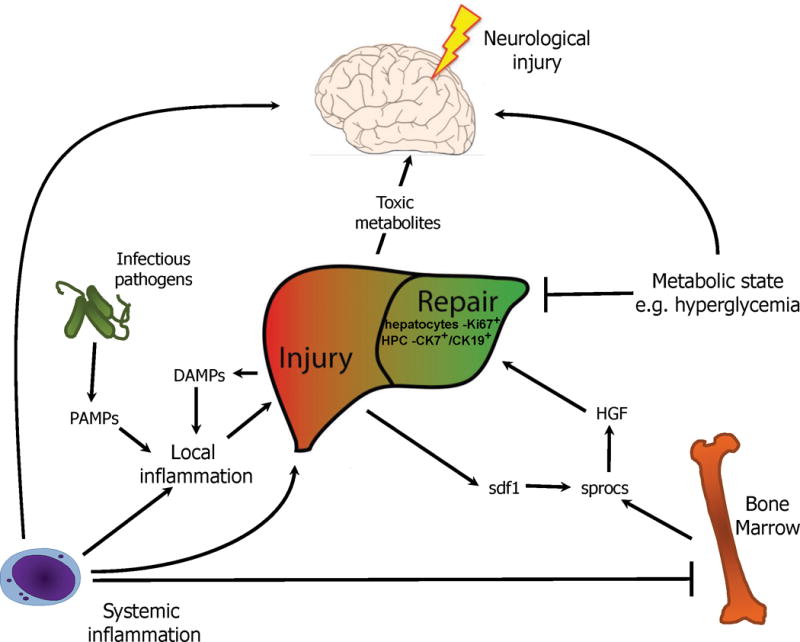
Proposed model of liver injury progression in indeterminate pediatric acute liver failure.
Systemic inflammatory insults may lead to local liver inflammation in conjunction with exogenous local triggers such as viruses that contain PAMPs, and endogenous local triggers in the form of DAMPS generated from liver injury itself. The release of DAMPS from liver injury may promote a feed forward cycle of inflammation. This liver injury ultimately results in the production of sdf1 to promote the recruitment of sprocs and subsequent liver regenerative programs including HGF. As sprocs arise from the bone marrow, systemic inflammation may inhibit this process via suppression of bone marrow stem cells. Altered metabolic state in the form serum glucose may also provide additional signals to promote hepatic regeneration. Brain injury may result from systemic inflammation itself, compounded by toxic metabolite build up from liver injury and altered metabolic state. Together this network of events may lead to the liver, bone marrow, and brain injury often seen in iPALF.
Footnote for Figure 2: pathogen associated molecular patterns (PAMPs), damage associated molecular patterns (DAMPs), sinusoidal endothelial cell PROgenitor cells (sprocs); stromal cell-derived factor 1 (sdf1), hepatocyte growth factor (HGF), indeterminate Pediatric Acute Liver Failure (iPALF), cytokeratin 7 positive (CK7+), cytokeratin 19 positive (CK19+), hepatic progenitor cell (HPC).
Genetic discovery has accelerated during the past ten years making previously expensive and labor intensive approaches more feasible in this population. Bio-repository specimens from the PALFSG should be utilized to complete viral discovery studies and to screen the cohort for previously unrecognized genetic diseases. Despite evidence that diagnostic evaluations for viral pathogens is often incomplete, viruses not commonly associated with significant hepatic injury are unlikely to be singular causes of iPALF. It is more likely such viruses are the first step in a series of events that are modified by genetic variations or defects. Exploring these defects within family pedigrees that include members who were also exposed and infected with these viral pathogens could be an important next step in understanding such purported modifiers.
Finally, discussion following the workshop presentations supported the concern that emerging treatment practices are not evidence based. Specifically, the growing trend toward steroid therapy targeted at suppressing the immune response in patients with iPALF has not been adequately tested. Most importantly, it is yet to be established whether robust or “overzealous” immune responses are primary drivers of the ongoing injury or secondary responses to overwhelming cell death. Treatment trials should be designed to determine the benefit of immune modulation in this rapidly progressive disease and should include mechanistic studies that advance the understanding of disease pathogenesis.
The workshop focused on several approaches to advance these research objectives and explore the role of the immune system in propagating liver injury. These investigations should leverage both basic and translational methodologies. Immune dysregulation is an emerging term describing a phenotype which is observed most commonly in iPALF cases, but which may also be expressed in patients with established etiologies, especially those with evidence of auto-immunity. Development of an immune dysregulation liver injury animal model would accelerate mechanistic studies in this rare disease. A biorepository of human specimens would allow investigators to confirm findings from animal studies and explore unique human responses. Manipulation of individual cell lines responsible for both the innate and adaptive immune response in animal models of overwhelming liver injury could provide important insights into how these responses impact liver regeneration. Examination of circulating T lymphocytes, macrophages and stem cells, particularly BM sprocs from patients with PALF is essential to understanding the relationship between marrow injury and hepatic regeneration. Metagenomics next-generation sequencing of serum and liver tissue should be completed to search for novel hepatotrophic viruses and explore the role of common viral pathogens, such as Herpes Simplex Virus in this disorder. Genomic approaches that maximize the ability to identify genetic modifiers in a single proband should be leveraged. It is also important to examine processes at the tissue level. Liver histopathology is a vital tool to differentiate cell populations. Immunotechniques can further the understanding of cell signaling and identify unexpected patterns of cytokine expression. RNA sequencing can be used to confirm patterns of gene expression leading to immune mediated injury and potentially improve prognostic models.
The overarching goal of these mechanistic studies is to develop targeted therapies. However, emerging experience with empiric immunomodulatory therapy should be tested, even as mechanistic investigations proceed. It is also essential to test supportive care strategies, such as standardized neuroprotection protocols that could enhance survival and functional outcomes. Innovative approaches to study the role of neuroinflammation in the evolution of PALF could inform refinement of these protective strategies and support additional benefits of systemic anti-inflammatory therapy.
Table 2.
Mechanisms of acute liver failure induced central nervous system injury
| MECHANISM | THERAPEUTIC TARGET | |
|---|---|---|
| ASTROCYTE SWELLING | Ammonia accumulation (glutamine metabolism; nitrogen catabolism) | Limit protein intake; scavenge NH3 with lactulose; L-ornithine and phenylacetate |
| EXCITOTOXICITY | Glutamate mediated NMDA-receptor activation; Nitric oxide release | ceftriaxone to restore glutamate transporters |
| CNS SYSTEMIC INFLAMMATION | Peripheral cytokines; Nitric oxide signaling | Hemodialysis/Molecular Adsorbent Recirculation System |
| ACUTE LUNG INJURY/ACUTE RESPIRATORY DISTRESS SYNDROME | Hypercapnia and cerebral vasodilatation | Recruitment; optimize lung compliance Use of Propofol or etomidate during induction Avoid Ketamine (increased intracranial pressure) Do not treat spontaneous hyperventilation |
| CEREBRAL EDEMA | Cytotoxic > vasogenic edema Compromise of AQP4 transporter function Astrocyte and microglial activation Nitric oxide induced cerebral vasodilatation |
Mannitol Minocycline etanercept anakinra Barbiturates Indomethacin Hypothermia/Controlled Normothemia |
| COMPROMISED CEREBRAL METABOLISM | Mitochondrial dysfunction Oxidative stress Induction of mitochondrial permeability transition pore |
Antioxidant therapy (N-acetylcysteine)/Vitamins L-Carnitine Hypothermia/Controlled Normothermia Methylene Blue |
| CEREBRAL ISCHEMIA | Arterial vasodilatation and decreased cerebral perfusion pressure | Vasopressors; treatment of adrenal insufficiency |
| BLOOD BRAIN BARRIER BREAKDOWN | Matrix-metalloproteinase-activation | Neurosteroids/Progesterone TGFB-antagonist (Losartan) |
Footnote for Table 2: N-methyl-D-aspartate (NMDA), aquaporin 4 (AQP4). Table courtesy of Dr Mark Wainwright.
Online Summary.
The cause of liver injury is undetermined in approximately 30% of Pediatric Acute Liver Failure cases. A single day workshop sponsored by the National Institute of Diabetes and Digestive and Kidney Diseases brought together clinicians and basic scientists to integrate aligned research findings and review emerging concepts regarding mechanisms of hepatic injury, new diagnostic tools and options for future treatment trials.
Acknowledgments
Speakers whose lectures contributed to the content of this meeting summary were as follows: Estella M. Alonso, M.D., Ann & Robert H. Lurie Children’s Hospital of Chicago and Northwestern University Feinberg School of Medicine, Chicago, IL; Edward Behrens, M.D., Children’s Hospital of Philadelphia and Perelman School of Medicine at The University of Pennsylvania; Charles Chiu, M.D., Ph.D., University of California San Francisco; Laurie DeLeve, M.D., Ph.D., FAASLD, Keck School of Medicine of University of Southern California; Alexei A. Gromm, M.D., Cincinnati Children’s Hospital Medical Center; Simon Horslen, M.D., Ch.B., FRCPCH, Seattle Children’s Hospital and University of Washington School of Medicine; Harmut Jaeschke, Ph.D., University of Kansas Medical Center; David Kleiner, M.D., Ph.D., National Cancer Institute, Bethesda, MD; David Rudnick, M.D., Ph.D., Washington University School of Medicine; R. Todd Stravitz, M.D., Virginia Commonwealth University, Richmond, VA; Yoram Vodovotz, Ph.D., University of Pittsburgh; Mark Wainwright, M.D., Ph.D., Ann & Robert H. Lurie Children’s Hospital of Chicago and Northwestern University Feinberg School of Medicine, Chicago, IL; Mark Yandell, Ph.D., University of Utah. We would also like to acknowledge Katie Neighbors-Benson MPH1 for support for final review and formatting of the manuscript.
Financial Support
The Pediatric Acute Liver Failure of Undetermined Cause: A Research Workshop was supported by the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health.
List of Abbreviations
- PALF
Pediatric Acute Liver Failure
- PALFSG
Pediatric Acute Liver Failure Study Group
- BM
bone marrow
- ALFSG
Acute Liver Failure Study Group
- iPALF
indeterminate Pediatric Acute Liver Failure
- APAP
acetaminophen
- HLH
hemophagocytic lymphohistiocytosis
- FHLH
familial hemophagocytic lymphohistiocytosis
- MAS
macrophage activation syndrome
- DAMPs
damage associated molecular patterns
- sprocs
sinusoidal endothelial cell PROgenitor cells
- sJIA
systemic juvenile idiopathic arthritis
- CXCR7
C-X-C chemokine receptor type 7
References
- 1.Squires RH, Ng V, Romero R, Ekong U, Hardikar W, Emre S, et al. Evaluation of the pediatric patient for liver transplantation: 2014 practice guideline by the American Association for the Study of Liver Diseases, American Society of Transplantation and the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition. Hepatology. 2014;60:362–398. doi: 10.1002/hep.27191. [DOI] [PubMed] [Google Scholar]
- 2.Squires RH, Jr, Shneider BL, Bucuvalas J, Alonso E, Sokol RJ, Narkewicz MR, et al. Acute liver failure in children: the first 348 patients in the pediatric acute liver failure study group. J Pediatr. 2006;148:652–658. doi: 10.1016/j.jpeds.2005.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Narkewicz MR, Dell Olio D, Karpen SJ, Murray KF, Schwarz K, Yazigi N, et al. Pattern of diagnostic evaluation for the causes of pediatric acute liver failure: an opportunity for quality improvement. J Pediatr. 2009;155:801–806.e801. doi: 10.1016/j.jpeds.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwarz KB, Dell Olio D, Lobritto SJ, Lopez MJ, Rodriguez-Baez N, Yazigi NA, et al. Analysis of viral testing in nonacetaminophen pediatric acute liver failure. J Pediatr Gastroenterol Nutr. 2014;59:616–623. doi: 10.1097/MPG.0000000000000512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Squires R, Alonso E. In: Liver Disease in Children. 4th. Suchy F, Sokol R, Balistreri W, editors. New York: Cambridge University Press; 2014. pp. 32–50. [Google Scholar]
- 6.Haack TB, Staufner C, Kopke MG, Straub BK, Kolker S, Thiel C, et al. Biallelic Mutations in NBAS Cause Recurrent Acute Liver Failure with Onset in Infancy. Am J Hum Genet. 2015;97:163–169. doi: 10.1016/j.ajhg.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alonso E, James L, Zhang S, Squires R, Group PALFS Acetaminophen Adducts Detected in Serum of Pediatric Patients With Acute Liver Failure. J Pediatr Gastroenterol Nutr. 2015;61:102–107. doi: 10.1097/MPG.0000000000000814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Squires RH, Dhawan A, Alonso E, Narkewicz MR, Shneider BL, Rodriguez-Baez N, et al. Intravenous N-acetylcysteine in pediatric patients with nonacetaminophen acute liver failure: a placebo-controlled clinical trial. Hepatology. 2013;57:1542–1549. doi: 10.1002/hep.26001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bucuvalas J, Filipovich L, Yazigi N, Narkewicz MR, Ng V, Belle SH, et al. Immunophenotype predicts outcome in pediatric acute liver failure. J Pediatr Gastroenterol Nutr. 2013;56:311–315. doi: 10.1097/MPG.0b013e31827a78b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ostapowicz G, Fontana RJ, Schiodt FV, Larson A, Davern TJ, Han SH, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002;137:947–954. doi: 10.7326/0003-4819-137-12-200212170-00007. [DOI] [PubMed] [Google Scholar]
- 11.Davern TJ, 2nd, James LP, Hinson JA, Polson J, Larson AM, Fontana RJ, et al. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology. 2006;130:687–694. doi: 10.1053/j.gastro.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 12.Hennes EM, Zeniya M, Czaja AJ, Pares A, Dalekos GN, Krawitt EL, et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology. 2008;48:169–176. doi: 10.1002/hep.22322. [DOI] [PubMed] [Google Scholar]
- 13.Singhal A, Vadlamudi S, Stokes K, Cassidy FP, Corn A, Shrago SS, et al. Liver histology as predictor of outcome in patients with acute liver failure. Transpl Int. 2012;25:658–662. doi: 10.1111/j.1432-2277.2012.01470.x. [DOI] [PubMed] [Google Scholar]
- 14.Kleiner DE, Chalasani NP, Lee WM, Fontana RJ, Bonkovsky HL, Watkins PB, et al. Hepatic histological findings in suspected drug-induced liver injury: systematic evaluation and clinical associations. Hepatology. 2014;59:661–670. doi: 10.1002/hep.26709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katoonizadeh A, Nevens F, Verslype C, Pirenne J, Roskams T. Liver regeneration in acute severe liver impairment: a clinicopathological correlation study. Liver International. 2006;26:1225–1233. doi: 10.1111/j.1478-3231.2006.01377.x. [DOI] [PubMed] [Google Scholar]
- 16.Behrens EM, Cron RQ. Kill or be killed. J Immunol. 2015;194:5041–5043. doi: 10.4049/jimmunol.1500774. [DOI] [PubMed] [Google Scholar]
- 17.Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med. 2012;63:233–246. doi: 10.1146/annurev-med-041610-134208. [DOI] [PubMed] [Google Scholar]
- 18.Ravelli A, Grom AA, Behrens EM, Cron RQ. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immun. 2012;13:289–298. doi: 10.1038/gene.2012.3. [DOI] [PubMed] [Google Scholar]
- 19.Bihl F, Emmenegger U, Reichen J, Neftel KA, Zimmermann A, Cerny A. Macrophage activating syndrome is associated with lobular hepatitis and severe bile duct injury with cholestasis. J Hepatol. 2006;44:1208–1212. doi: 10.1016/j.jhep.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 20.DiPaola F, Grimley M, Bucuvalas J. Pediatric acute liver failure and immune dysregulation. J Pediatr. 2014;164:407–409. doi: 10.1016/j.jpeds.2013.10.044. [DOI] [PubMed] [Google Scholar]
- 21.McKenzie RB, Berquist WE, Nadeau KC, Louie CY, Chen SF, Sibley RK, et al. Novel protocol including liver biopsy to identify and treat CD8+ T-cell predominant acute hepatitis and liver failure. Pediatr Transplant. 2014;18:503–509. doi: 10.1111/petr.12296. [DOI] [PubMed] [Google Scholar]
- 22.Rood J, Rao S, Paessler M, Kreiger P, Chu N, Stelekati E, et al. ST2 contributes to T cell hyperactivation and fatal hemophagocytic lymphohistiocytosis in mice. Blood. 2016;127:426–435. doi: 10.1182/blood-2015-07-659813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaeschke H, Williams CD, Ramachandran A, Bajt ML. Acetaminophen hepatotoxicity and repair: the role of sterile inflammation and innate immunity. Liver International. 2012;32:8–20. doi: 10.1111/j.1478-3231.2011.02501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Azhar N, Ziraldo C, Barclay D, Rudnick DA, Squires RH, Vodovotz Y, et al. Analysis of serum inflammatory mediators identifies unique dynamic networks associated with death and spontaneous survival in pediatric acute liver failure. PLoS ONE [Electronic Resource] 2013;8:e78202. doi: 10.1371/journal.pone.0078202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michalopoulos GK. Principles of liver regeneration and growth homeostasis. Comprehensive Physiology. 2013;3:485–513. doi: 10.1002/cphy.c120014. [DOI] [PubMed] [Google Scholar]
- 26.Huang J, Schriefer A, Cliften P, Dietzen D, Kulkarni S, Sing S, et al. Postponging the hypoglycemic response in partial hepatectomy delays mouse liver regeneration. Am J Pathol. 2016;186:587–599. doi: 10.1016/j.ajpath.2015.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rudnick DA, Dietzen DJ, Turmelle YP, Shepherd R, Zhang S, Belle SH, et al. Serum alpha-NH-butyric acid may predict spontaneous survival in pediatric acute liver failure. Pediatr Transplant. 2009;13:223–230. doi: 10.1111/j.1399-3046.2008.00998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang J, Rudnick DA. Elucidating the metabolic regulation of liver regeneration. Am J Pathol. 2014;184:309–321. doi: 10.1016/j.ajpath.2013.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeLeve LD. Liver sinusoidal endothelial cells and liver regeneration. J Clin Invest. 2013;123:1861–1866. doi: 10.1172/JCI66025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L, Wang X, Xie G, Wang L, Hill CK, DeLeve LD. Liver sinusoidal endothelial cell progenitor cells promote liver regeneration in rats. J Clin Invest. 2012;122:1567–1573. doi: 10.1172/JCI58789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeLeve L, Wang X, Wang L. VEGF-sdf1 recruitment of CXCR7+ bone marrow progenitors of liver sinusoidal endothelial cells promotes rat liver regeneration. Am J Physiol Gastrointest Liver Physiol. 2016;310:G739–746. doi: 10.1152/ajpgi.00056.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naccache SN, Federman S, Veeraraghavan N, Zaharia M, Lee D, Samayoa E, et al. A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res. 2014;24:1180–1192. doi: 10.1101/gr.171934.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu H, Huff CD, Moore B, Flygare S, Reese MG, Yandell M. VAAST 2.0: improved variant classification and disease-gene identification using a conservation-controlled amino acid substitution matrix. Genet Epidemiol. 2013;37:622–634. doi: 10.1002/gepi.21743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu H, Roach JC, Coon H, Guthery SL, Voelkerding KV, Margraf RL, et al. A unified test of linkage analysis and rare-variant association for analysis of pedigree sequence data. Nat Biotechnol. 2014;32:663–669. doi: 10.1038/nbt.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yandell M, Huff C, Hu H, Singleton M, Moore B, Xing J, et al. A probabilistic disease-gene finder for personal genomes. Genome Res. 2011;21:1529–1542. doi: 10.1101/gr.123158.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Canna S, Girard C, Malle L, de Jesus A, Romberg N, Kelsen J, et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with interleukin-18 inhibition. J Allergy Clin Immunol. 2016 doi: 10.1016/j.jaci.2016.10.022. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moniaux N, Song H, Darnaud M, Garbin K, Gigou M, Mitchell C, et al. Human hepatocarcinoma-intestine-pancreas/pancreatitis-associated protein cures fas-induced acute liver failure in mice by attenuating free-radical damage in injured livers. Hepatology. 2011;53:618–627. doi: 10.1002/hep.24087. [DOI] [PubMed] [Google Scholar]
- 38.Grom A, Ilowite N, Brunner H, Ruperto N, Martinin A, Lovell D, et al. Canakinumab in Systemic Juvenille Idiopathic Arthritis: Imact on the rate and clinical presentation of Macrophage Activation Syndrome. Arthritis Rheumatol. 2016;68:218–228. doi: 10.1002/art.39407. [DOI] [PubMed] [Google Scholar]
- 39.Durand M, Troyanov Y, Laflamme P, Gregoire G. Macrophage activation syndrome treated with anakinra. J Rheumatol. 2010;37:879–880. doi: 10.3899/jrheum.091046. [DOI] [PubMed] [Google Scholar]
- 40.Miettunen PM, Narendran A, Jayanthan A, Behrens EM, Cron RQ. Successful treatment of severe paediatric rheumatic disease-associated macrophage activation syndrome with interleukin-1 inhibition following conventional immunosuppressive therapy: case series with 12 patients. Rheumatology. 2011;50:417–419. doi: 10.1093/rheumatology/keq218. [DOI] [PubMed] [Google Scholar]
- 41.Ruperto N, Brunner HI, Quartier P, Constantin T, Wulffraat N, Horneff G, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367:2396–2406. doi: 10.1056/NEJMoa1205099. [DOI] [PubMed] [Google Scholar]
- 42.Yokota S, Itoh Y, Morio T, Sumitomo N, Daimaru K, Minota S. Macrophage Activation Syndrome in Patients with Systemic Juvenile Idiopathic Arthritis under Treatment with Tocilizumab. J Rheumatol. 2015;42:712–722. doi: 10.3899/jrheum.140288. [DOI] [PubMed] [Google Scholar]
- 43.Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127:3321–3330. doi: 10.1182/blood-2016-04-703751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Benedetti F, Brunner HI, Ruperto N, Kenwright A, Wright S, Calvo I, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis.[Erratum appears in N Engl J Med. 2015 Feb 26;372(9):887] N Engl J Med. 2012;367:2385–2395. doi: 10.1056/NEJMoa1112802. [DOI] [PubMed] [Google Scholar]