

Abstract
Background
The SAKK 19/05 trial investigated the safety and efficacy of the combined targeted therapy bevacizumab and erlotinib (BE) in unselected patients with advanced non-squamous non-small cell lung cancer (NSCLC). Although activating EGFR mutations were the strongest predictors of the response to BE, some patients not harboring driver mutations could benefit from the combined therapy. The identification of predictive biomarkers before or short after initiation of therapy is therefore paramount for proper patient selection, especially among EGFR wild-types. The first aim of this study was to investigate the early change in blood gene expression in unselected patients with advanced non-squamous NSCLC treated by BE. The second aim was to assess the predictive value of blood gene expression levels at baseline and 24h after BE therapy.
Methods
Blood samples from 43 advanced non-squamous NSCLC patients taken at baseline and 24h after initiation of therapy were profiled using Affymetrix’ exon arrays. The 24h gene dysregulation was investigated in the light of gene functional annotations using gene set enrichment analysis. The predictive value of blood gene expression levels was assessed and validated using an independent dataset.
Results
Significant gene dysregulations associated with the 24h-effect of BE were detected from blood-based whole-genome profiling. BE had a direct effect on “Pathways in cancer”, by significantly down-regulating genes involved in cytokine–cytokine receptor interaction, MAPK signaling pathway and mTOR signaling pathway. These pathways contribute to phenomena of evasion of apoptosis, proliferation and sustained angiogenesis. Other signaling pathways specifically reflecting the mechanisms of action of erlotinib and the anti-angiogenesis effect of bevacizumab were activated. The magnitude of change of the most dysregulated genes at 24h did not have a predictive value regarding the patients’ response to BE. However, predictive markers were identified from the gene expression levels at 24h regarding time to progression under BE.
Conclusions
The 24h-effect of the combined targeted therapy BE could be accurately monitored in advanced non-squamous NSCLC blood samples using whole-genome exon arrays. Putative predictive markers at 24h could reflect patients’ response to BE after adjusting for their mutational status.
Trial registration ClinicalTrials.gov: NCT00354549
Electronic supplementary material
The online version of this article (doi:10.1186/s12967-017-1174-z) contains supplementary material, which is available to authorized users.
Keywords: Non-small cell lung cancer, Combined targeted therapies, Blood predictive markers, Exon arrays
Background
Combined targeted therapies represent novel therapeutic approaches simultaneously acting on several specific molecular pathways in cancer and having a number of advantages over standard single-targeted agents [1, 2].
Several trials have shown the beneficial effect of epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) in advanced non-small cell lung cancer patients (NSCLC) harboring activating EGFR mutations leading to the adoption of EGFR-TKI as standard treatment in this population [3, 4]. Preclinical studies suggested that the combination of an EGFR-TKI together with an angiogenesis inhibitor (e.g. targeting the vascular endothelial growth factor VEGF) can have a synergistic effect [5, 6]. Recent clinical trials showed superior efficacy of the combined anti-angiogenesis bevacizumab (B) with the TKI erlotinib (E) in EGFR mutated patients compared to E alone [7, 8]. More specifically, these trials showed that first line treatments combining BE improved the progression free survival (PFS)—but not overall survival (OS)—of patients harboring an EGFR driver mutation in comparison with E alone [7, 9].
In unselected patients, BE had better PFS than E alone without improvement of survival in recurrent NSCLC suggesting moderate activity of BE [6]. As first line therapy in unselected patients, the overall response rate of BE was 12%, whereas PFS was 3.5 months, again showing moderate activity [10]. Despite the favorable toxicity profile of BE, these results are inferior to chemotherapy first line or immunotherapy second line. The SAKK 19/05 trial from the Swiss Group for Clinical Cancer Research showed that first-line combined BE treatment followed by chemotherapy regimen is feasible with acceptable toxicity and activity in an unselected advanced non-squamous NSCLC population [11]. On the other hand, the phase II TASK study did not show a benefit in terms of PFS for the combination BE in unselected first line advanced non-squamous NSCLC compared with chemotherapy plus B [10, 12].
Although the presence of EGFR mutations is the strongest predictor of the response to anti-EGFR-TKI, a recent meta-analysis showed that wild-type EGFR patients can benefit from the therapy with an improved OS compared with placebo or standard chemotherapy [hazard ratio 0.780 (95% CI 0.654–0.930)] [13]. Therefore, the identification of very early predictive markers of multiple targeted therapies and the understanding of their mechanisms of action in advanced non-squamous NSCLC is of paramount importance in order to better identify subsets of patients who may still benefit from these treatments.
Blood-based biomarkers in NSCLC are of particular interest as they can be easily and non-invasively accessed [14]. Whole-genome exon arrays provide an ideal platform for the discovery of novel putative biomarkers by investigating expression variations at an exon-level resolution [15]. More specifically, exon arrays allow analyses both at the gene and at the exon level. Exon-level analyses are usually performed to detect alternative splicing events [16].
The aim of the current study was to analyze blood-level exon array profiling data from unselected patients with advanced non-squamous NSCLC before and 24h after initiation of the combined targeted therapy BE. The specific objectives are twofold: (1) uncover which genes from whole blood circulating RNAs are immediately impacted by the effect of the combined therapy BE, and (2) assess the predictive value of these dysregulations.
Methods
Lung cancer dataset
The gene expression data set originated from a translational substudy of the phase II SAKK 19/05 trial from the Swiss Group for Clinical Cancer Research (ClinicalTrials.gov: NCT00354549). In the original study, 103 unselected patients with advanced non-squamous NSCLC were enrolled, among which 101 were evaluable. The experimental design of the trial SAKK 19/05 is summarized in Fig. 1. Patients were treated using the combined targeted therapy BE until disease progression or unacceptable toxicity. At progression, standard platinum-based chemotherapy (CT) was used. The primary endpoint of this trial was disease stabilization 12 weeks after initiation of therapy. Secondary endpoints included tumor shrinkage at 12 weeks (TS12), time to progression under BE (TTPBE), time to progression under CT (TTPCT) and OS. Further detailed information about this trial can be found in previous publications [11, 17, 18]. As part of a translational substudy, blood samples were taken at baseline and 24h after initiation of treatment for gene expression analysis in a subset of 49 patients. The current study was approved by the ethics committee of the canton St. Gallen (EKSG 06/012).
Fig. 1.
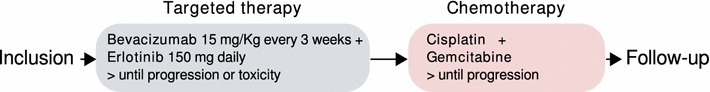
Treatment scheme of the SAKK 19/05 trial. Patients received BE until progression or unacceptable toxicity. Upon disease progression, patients received standard chemotherapy with cisplatin and gemcitabine
Exon array analysis
RNA from whole blood samples was extracted and quality checked. Six pairs of samples had to be excluded from the analysis due to low quality, whereas RNA extracts provided sufficient quality for microarray hybridization in 43 out of 49 pairs of sample. Messenger RNAs were hybridized on Affymetrix Human Exon 1.0 ST arrays (Affymetrix, Santa Clara, CA, USA) following standard recommendations from the manufacturer. This microarray platform measures genome-wide exon-level expression in over 1.4 million probe sets, and allows the investigation of genomic variations both at the gene and at the exon level. For the sake of this analysis, 439,778 exonic probe sets (within 38,900 genes) were kept in the analysis, after filtering out intronic, intergenic and unreliable probe sets (according to the nomenclature defined in the R package annmap [19]). Raw data (Affymetrix CEL files) have been deposited in NCBI’s Gene Expression Omnibus (GEO), and are accessible through GEO Series accession number GSE61676. The exon level probe sets were pre-processed, quality checked and normalized using the RMA procedure (including background correction, quantile normalization and median-polish summarization) as implemented in the R package oligo [20, 21].
Statistical considerations
The current experimental design includes a repeated measurement of genome-wide exon-level expressions in 43 patients at two different time points (baseline and 24h after initiation of therapy). The exon expression data are stored in a pair of fully matched tables. Dually constrained correspondence analysis (DCCA)—an extension of the multivariate technique correspondence analysis—was used to investigate the 24h-change in exon expression levels [16]. DCCA uses observation- and variable-wise linear constraints in order to take into account complex experimental designs including within-patient repeated measurements and variables grouped into hierarchical levels (within-gene exonic structures). The theoretical scheme of DCCA as applied to our experimental design is summarized in Fig. 2. The mathematical underpinnings of DCCA are further described in Additional file 1. In the current work, the following parametrization of DCCA was used: (1) the two fully matched tables of exon-level expression intensities at baseline and 24h were stacked observation-wise; (2) an observation-wise between-time constraint (baseline vs. 24h) was applied after partialling out the within-patient effect; (3) a variable-wise constraint indicating the within-gene exonic structure was applied.
Fig. 2.
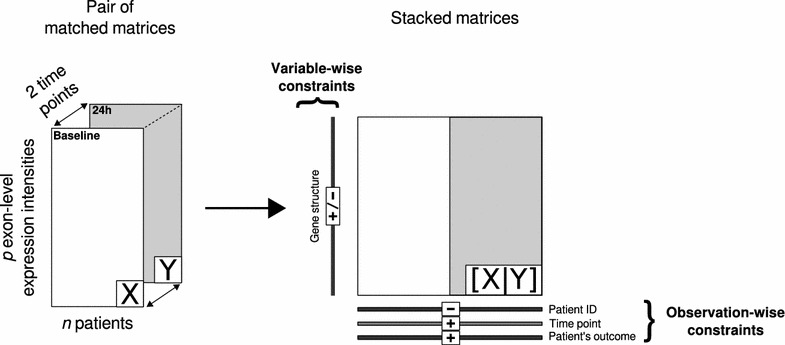
Design of experiment and scheme of dually constrained correspondence analysis. Two matched tables and are analyzed by DCCA. The 2 tables are rearranged into one stacked table. Additional external information on both rows and columns are used as positive and/or negative constraints
Generalized linear (mixed effects) models were used in order to test the predictive value of the identified biomarkers. Binary endpoints were tested using logistic regression, time to event data were modeled using Cox proportional hazards regression, and continuous variables were modeled using (multiple) linear regression. Mixed effects modeling was used when testing associations in the frame of the within-patient repeated measurement design. The adjustment for patients’ EGFR mutational status was done by including the mutational status as covariate in the predictive models. The significance level used for the discovery of the putative predictive markers was set to 0.001. Gene signatures combining the information of the best predictive candidates were built using the metagene approach [22]. In this approach, the linear combination of several genes is calculated as follows: (1) The matrix of normalized gene expression intensities for all patients at a given time point (rows) and for all candidate genes (columns) is analyzed using unscaled principal component analysis (PCA); (2) The row coordinates (scores) on the first PCA axis summarizing the largest amount of variance are extracted (metagene score); (3) Based on the median of the metagene score, a binary score is created to discriminate between low versus high-risk patients.
Computational considerations
All analyses were implemented using the R statistical software [23] including the extension package ade4 [24], as well as dedicated packages from the Bioconductor project [25] such as the package oligo for microarray preprocessing [21] and the annotation package annmap (Homo sapiens database version 86). Functions available in ade4 (including dudi.coa, wca, bca, pcaiv and pcaivortho) can theoretically be used to carry out DCCA. However, for reasons of computational efficiency due to the extensive size of exon array datasets, the DCCA algorithm was substantially optimized and a new R extension package dcca is available (see Additional file 1). Hypothesis testing for the identification of predictive biomarkers was carried out using the following R packages: lme4, coxme and multcomp.
Gene functional annotations and validations
Gene set enrichment analysis was done by interrogating the molecular knowledge databases Kyoto Encyclopedia of Genes and Genomes (KEGG) [26] and WikiPathways [1] using the functional annotation web-service WebGestalt [27]. Enrichment analyses were based on the list of 100 most dysregulated genes (as identified by DCCA), as well as the lists of genes which were significantly predicting patient’s outcome (each of the investigated endpoints). The significance of the enrichment was obtained using hypergeometric tests. Validation was carried out using the lung data set from the Kaplan–Meier Plotter (KMplotter) web tool [28]. KMplotter is a manually curated database including gene expression level information about more than 50,000 Affymetrix probe set IDs together with associated clinical information. The prognostic value of single or multiple genes can be assessed with regard to relapse free and overall survival. Another independent lung cancer dataset was used for external validation. This gene expression microarray dataset includes 85 lung adenocarcinoma tumor samples and is part of the program “Carte d’Identité des Tumeurs” (CIT) from the french national cancer league [29]. Samples were profiled using the Affymetrix Human Genome U133 Plus 2.0 Array and raw data are available in NCBI’s Gene Expression Omnibus through GEO Series accession number GSE30219. Furthermore, the results were discussed in the light of available literature findings.
Results
Patients characteristics
The characteristics of the 43 patients are reported in Table 1. Patients were late stage (91% stage IV/9% stage IIIb) non-squamous NSCLC. Five out of 43 patients had demonstrable EGFR mutations: one on exon 18 (E709A-G719S), three on exon 19 (Del L747-G749, Del E746-A750 and R748-S752) and one on exon 21 (L858R). The median age was 61 years old (IQR 54–66) and the sex ratio was 0.44 (19 males/24 females). Disease stabilization at 12 weeks was reached in 53% of patients. The median tumor shrinkage at 12 weeks was 15.8%. The median overall survival was 11.1 (95% CI 10.1–17.9) months. The median time-to-progression under BE was 4.0 (95% CI 2.8–6.0) months, whereas the median time-to-progression under CT was 2.6 (95% CI 1.7–5.7) months.
Table 1.
Patients characteristics
| Variables | |
|---|---|
| Number of patients (n) | 43 |
| Age (median [range]) | 61 (35–78) |
| Gender (# male [%]) | 19 (44.2%) |
| Stage (n [%]) | IIIb: 4 (9.3%); IV: 39 (90.7%) |
| Demonstrable EGFR mutations (n [%]) | 5 (11.6%) |
| Disease stabilization at 12 weeks (n [%]) | 23 (53.5%) |
| Tumor shrinkage at 12 weeks (median [IQR]), in % of tumor size at baseline | 15.8% (−2.5 to 26.2%) |
| Median overall survival (95% CI), in months | 11.1 (10.1–17.9) |
| Median time-to-progression under BE (95% CI), in months | 4.0 (2.8–6.0) |
| Median time-to-progression under CT (95% CI), in months | 2.6 (1.7–5.7) |
The table summarizes the characteristics of the 43 patients included in the study
24h gene dysregulation
The 100 genes mostly dysregulated (54 up-regulated vs. 46 down-regulated) by the 24h effect of BE are summarized in Table 2. The genes were involved in all aspects of tumor biology. This includes genes involved in mitosis and cell cycle processes such as the cancer susceptibility candidate 5 (CASC5) encoding for a protein influencing the spindle assembly checkpoint during eukaryotic cell cycle; the centromere-associated protein E (CENPE) which accumulates and play a key stabilizing role during mitosis; the protein furry homolog (FRY) which plays a crucial role in the structural integrity of mitotic centrosomes; kinetochore associated 1 (KNTC1) encoding for a protein ensuring proper chromosome segregation during cell division; Phospholipase D1 (PLD1) involved in cancer progression [30] and in the regulation of mitosis in relationship with the “Ras signaling pathway” and “Pathways in cancer”. Other dysregulated genes were involved in energy-dependent metabolisms (ATPase/GTPase). This includes genes from the ATPase Family (ATAD2) known to be related to gastric cancer network, and which may play an important role in cell proliferation and cell cycle progression of breast cancer cells; Guanylate Binding Protein 4 (GBP4) related to GTPase activity and associated with the interferon signaling pathway; dedicator of cytokinesis 10 (DOCK10) acting on GTPase and related to hemostasis and regulation of cell division cycle 42 (CDC42) activity. Mechanisms of cell migration are also controlled by a series of dysregulated genes such as ADAM Metallopeptidase Domain 19 (ADAM19) regulating cell migration, cell adhesion and cell-cell/cell-matrix interactions, supposed to play an important role in pathological processes including cancer; BMX non-receptor tyrosine kinase (BMX) encoding for a protein implicated in several signal transduction pathways regulating tumorigenicity of cancer cells; Epidermal Growth Factor Receptor Pathway Substrate 8 (EPS8) encoding for a protein having functions in part of the EGFR pathway and being related to Tyrosine Kinases/Adaptors and Development FGFR signaling pathways; Fms-Related Tyrosine Kinase 3 (FLT3) encoding for a class III receptor tyrosine kinase regulating hematopoiesis, and whose action is related to apotosis, proliferation and differentiation processes; Interleukin 1 Receptor, Type II (IL1R2) controling many cellular functions including proliferation, differentiation, and cell survival/apoptosis; Matrix Metallopeptidase 9 (MMP9) involved in tissue/matrix remodeling, playing a central role in cell proliferation, migration, differentiation, showing an altered expression in a number of different human cancers with poor prognosis. Apoptosis was regulated through the action of various genes including baculoviral IAP repeat containing 3 (BIRC3) encoding for an inhibitor of apoptosis protein acting on killing tumor cells; death-associated protein kinase 2 (DAPK2) whose overexpression was shown to induce cell apoptosis; Retinoblastoma-Like 1 (RBL1) encoding for a tumor suppressor protein involved in cell cycle regulation; insulin-like growth factor 1 receptor (IGF1R) encoding for a growth factor with tyrosine kinase activity, having an anti-apoptotic effect and being highly overexpressed in most malignant tissues [31].
Table 2.
List of the 100 most dysregulated genes due to the 24h effect of BE
| Ensembl | Gene symbol | Description | Gene dysregulation at 24h |
|---|---|---|---|
| ENSG00000125257 | ABCC4 | ATP-binding cassette, sub-family C (CFTR/MRP), member 4 | Up-regulated |
| ENSG00000114770 | ABCC5 | ATP-binding cassette, sub-family C (CFTR/MRP), member 5 | Down-regulated |
| ENSG00000173208 | ABCD2 | ATP-binding cassette, sub-family D (ALD), member 2 | Up-regulated |
| ENSG00000151726 | ACSL1 | Acyl-CoA synthetase long-chain family member 1 | Down-regulated |
| ENSG00000135074 | ADAM19 | ADAM metallopeptidase domain 19 (meltrin beta) | Down-regulated |
| ENSG00000154027 | AK5 | Adenylate kinase 5 | Up-regulated |
| ENSG00000151150 | ANK3 | Ankyrin 3, node of Ranvier (ankyrin G) | Up-regulated |
| ENSG00000206560 | ANKRD28 | Ankyrin repeat domain 28 | Up-regulated |
| ENSG00000118520 | ARG1 | Arginase, liver | Down-regulated |
| ENSG00000196914 | ARHGEF12 | Rho guanine nucleotide exchange factor (GEF) 12 | Up-regulated |
| ENSG00000156802 | ATAD2 | ATPase family, AAA domain containing 2 | Up-regulated |
| ENSG00000085224 | ATRX | Alpha thalassemia/mental retardation syndrome X-linked (RAD54 homolog, S. cerevisiae) | Up-regulated |
| ENSG00000023445 | BIRC3 | baculoviral IAP repeat-containing 3 | Up-regulated |
| ENSG00000102010 | BMX | BMX non-receptor tyrosine kinase | Down-regulated |
| ENSG00000136492 | BRIP1 | BRCA1 interacting protein C-terminal helicase 1 | Up-regulated |
| ENSG00000197603 | C5orf42 | Chromosome 5 open reading frame 42 | Up-regulated |
| ENSG00000152495 | CAMK4 | Calcium/calmodulin-dependent protein kinase IV | Up-regulated |
| ENSG00000137812 | CASC5 | Cancer susceptibility candidate 5 | Up-regulated |
| ENSG00000138778 | CENPE | Centromere protein E, 312 kDa | Up-regulated |
| ENSG00000198707 | CEP290 | Centrosomal protein 290 kDa | Up-regulated |
| ENSG00000106034 | CPED1 | Cadherin-like and PC-esterase domain containing 1 | Up-regulated |
| ENSG00000103196 | CRISPLD2 | Cysteine-rich secretory protein LCCL domain containing 2 | Down-regulated |
| ENSG00000146122 | DAAM2 | Dishevelled associated activator of morphogenesis 2 | Down-regulated |
| ENSG00000035664 | DAPK2 | Death-associated protein kinase 2 | Down-regulated |
| ENSG00000137628 | DDX60 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 60 | Up-regulated |
| ENSG00000174485 | DENND4A | DENN/MADD domain containing 4A | Up-regulated |
| ENSG00000135905 | DOCK10 | Dedicator of cytokinesis 10 | Up-regulated |
| ENSG00000147459 | DOCK5 | Dedicator of cytokinesis 5 | Down-regulated |
| ENSG00000088387 | DOCK9 | Dedicator of cytokinesis 9 | Up-regulated |
| ENSG00000178904 | DPY19L3 | Dpy-19-like 3 (C. elegans) | Down-regulated |
| ENSG00000134765 | DSC1 | Desmocollin 1 | Up-regulated |
| ENSG00000135636 | DYSF | Dysferlin, limb girdle muscular dystrophy 2B (autosomal recessive) | Down-regulated |
| ENSG00000198919 | DZIP3 | DAZ interacting protein 3, zinc finger | Up-regulated |
| ENSG00000102189 | EEA1 | Early endosome antigen 1 | Up-regulated |
| ENSG00000151491 | EPS8 | Epidermal growth factor receptor pathway substrate 8 | Up-regulated |
| ENSG00000089048 | ESF1 | ESF1, nucleolar pre-rRNA processing protein, homolog (S. cerevisiae) | Up-regulated |
| ENSG00000198734 | F5 | Coagulation factor V (proaccelerin, labile factor) | Down-regulated |
| ENSG00000140525 | FANCI | Fanconi anemia, complementation group I | Up-regulated |
| ENSG00000138829 | FBN2 | Fibrillin 2 (congenital contractural arachnodactyly) | Down-regulated |
| ENSG00000139132 | FGD4 | FYVE, RhoGEF and PH domain containing 4 | Down-regulated |
| ENSG00000122025 | FLT3 | Fms-related tyrosine kinase 3 | Down-regulated |
| ENSG00000161791 | FMNL3 | Formin-like 3 | Up-regulated |
| ENSG00000073910 | FRY | Furry homolog (Drosophila) | Down-regulated |
| ENSG00000162654 | GBP4 | Guanylate binding protein 4 | Up-regulated |
| ENSG00000182885 | GPR97 | G protein-coupled receptor 97 | Down-regulated |
| ENSG00000106070 | GRB10 | Growth factor receptor-bound protein 10 | Down-regulated |
| ENSG00000084110 | HAL | Histidine ammonia-lyase | Down-regulated |
| ENSG00000120694 | HSPH1 | Heat shock 105/110 kDa protein 1 | Up-regulated |
| ENSG00000140443 | IGF1R | Insulin-like growth factor 1 receptor | Down-regulated |
| ENSG00000197081 | IGF2R | Insulin-like growth factor 2 receptor | Down-regulated |
| ENSG00000115604 | IL18R1 | Interleukin 18 receptor 1 | Down-regulated |
| ENSG00000115594 | IL1R1 | Interleukin 1 receptor, type I | Down-regulated |
| ENSG00000115590 | IL1R2 | Interleukin 1 receptor, type II | Down-regulated |
| ENSG00000109452 | INPP4B | Inositol polyphosphate-4-phosphatase, type II, 105 kDa | Up-regulated |
| ENSG00000102445 | KIAA0226L | KIAA0226-like | Down-regulated |
| ENSG00000137261 | KIAA0319 | KIAA0319 | Down-regulated |
| ENSG00000110318 | KIAA1377 | KIAA1377 | Up-regulated |
| ENSG00000054523 | KIF1B | Kinesin family member 1B | Down-regulated |
| ENSG00000138182 | KIF20B | Kinesin family member 20B | Up-regulated |
| ENSG00000139116 | KIF21A | Kinesin family member 21A | Up-regulated |
| ENSG00000068796 | KIF2A | Kinesin heavy chain member 2A | Up-regulated |
| ENSG00000184445 | KNTC1 | Kinetochore associated 1 | Up-regulated |
| ENSG00000126777 | KTN1 | Kinectin 1 (kinesin receptor) | Up-regulated |
| ENSG00000123384 | LRP1 | Low density lipoprotein-related protein 1 (alpha-2-macroglobulin receptor) | Down-regulated |
| ENSG00000186205 | MARC1 | Mitochondrial amidoxime reducing component 1 | Down-regulated |
| ENSG00000257335 | MGAM | Maltase-glucoamylase (alpha-glucosidase) | Down-regulated |
| ENSG00000171843 | MLLT3 | Myeloid/lymphoid or mixed-lineage leukemia (trithorax homolog, Drosophila); translocated to, 3 | Up-regulated |
| ENSG00000196549 | MME | Membrane metallo-endopeptidase | Down-regulated |
| ENSG00000100985 | MMP9 | Matrix metallopeptidase 9 (gelatinase B, 92 kDa gelatinase, 92 kDa type IV collagenase) | Down-regulated |
| ENSG00000051825 | MPHOSPH9 | M-phase phosphoprotein 9 | Up-regulated |
| ENSG00000138119 | MYOF | Myoferlin | Up-regulated |
| ENSG00000049759 | NEDD4L | Neural precursor cell expressed, developmentally down-regulated 4-like | Up-regulated |
| ENSG00000184613 | NELL2 | NEL-like 2 (chicken) | Up-regulated |
| ENSG00000173145 | NOC3L | Nucleolar complex associated 3 homolog (S. cerevisiae) | Up-regulated |
| ENSG00000179299 | NSUN7 | NOL1/NOP2/Sun domain family, member 7 | Down-regulated |
| ENSG00000111581 | NUP107 | Nucleoporin 107 kDa | Up-regulated |
| ENSG00000159339 | PADI4 | Peptidyl arginine deiminase, type IV | Down-regulated |
| ENSG00000123836 | PFKFB2 | 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 2 | Down-regulated |
| ENSG00000075651 | PLD1 | Phospholipase D1, phosphatidylcholine-specific | Down-regulated |
| ENSG00000101868 | POLA1 | Polymerase (DNA directed), alpha 1 | Up-regulated |
| ENSG00000080839 | RBL1 | Retinoblastoma-like 1 (p107) | Up-regulated |
| ENSG00000257743 | RP11-1220K2.2 | Putative inactive maltase-glucoamylase-like protein LOC93432 | Down-regulated |
| ENSG00000226891 | RP11-182I10.3 | Uncharacterized LOC101927084 | Down-regulated |
| ENSG00000258476 | RP11-76E17.3 | NA | Down-regulated |
| ENSG00000140386 | SCAPER | S phase cyclin A-associated protein in the ER | Up-regulated |
| ENSG00000018280 | SLC11A1 | Solute carrier family 11 (proton-coupled divalent metal ion transporters), member 1 | Down-regulated |
| ENSG00000140090 | SLC24A4 | Solute carrier family 24 (sodium/potassium/calcium exchanger), member 4 | Down-regulated |
| ENSG00000112053 | SLC26A8 | Solute carrier family 26, member 8 | Down-regulated |
| ENSG00000157800 | SLC37A3 | Solute carrier family 37 (glycerol-3-phosphate transporter), member 3 | Down-regulated |
| ENSG00000136824 | SMC2 | Structural maintenance of chromosomes 2 | Up-regulated |
| ENSG00000163029 | SMC6 | Structural maintenance of chromosomes 6 | Up-regulated |
| ENSG00000009694 | TENM1 | Teneurin transmembrane protein 1 | Down-regulated |
| ENSG00000169902 | TPST1 | Tyrosylprotein sulfotransferase 1 | Down-regulated |
| ENSG00000198677 | TTC37 | Tetratricopeptide repeat domain 37 | Up-regulated |
| ENSG00000155657 | TTN | Titin | Up-regulated |
| ENSG00000120800 | UTP20 | UTP20, small subunit (SSU) processome component, homolog (yeast) | Up-regulated |
| ENSG00000197969 | VPS13A | Vacuolar protein sorting 13 homolog A (S. cerevisiae) | Up-regulated |
| ENSG00000163625 | WDFY3 | WD repeat and FYVE domain containing 3 | Down-regulated |
| ENSG00000213799 | ZNF845 | Zinc finger protein 845 | Up-regulated |
| ENSG00000167232 | ZNF91 | Zinc finger protein 91 | Up-regulated |
Information includes the Ensembl code, the gene symbol, description and sign of dysregulation. For convenience, the genes are ordered alphabetically
Based on the 100 best candidates identified by DCCA, three main pathways (according to the KEGG functional annotation database) were significantly altered by the 24h effect of the combined therapy BE: Hematopoietic cell lineage (KEGG pathway hsa04640; ); ABC transporters (KEGG pathway hsa02010; ); Pathways in cancer (KEGG pathway hsa05200; ).
Figure 3 displays the gene expression levels of each genes (baseline and 24h) within these dysregulated pathways. Figure 4 shows the detailed KEGG’s “Pathways in Cancer” highlighting the down-regulated genes within the sub-pathways “Cytokine–cytokine receptor information” and “MAPK signaling pathways”. The down-regulation of these sub-pathways contributes to the limitation of cell proliferation.
Fig. 3.
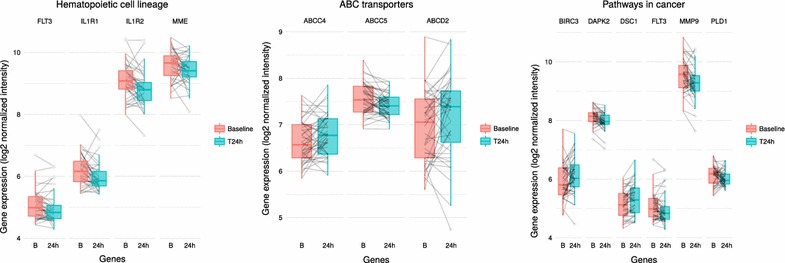
Boxplot representation of the gene expression levels (logarithm base 2 normalized intensity) before (B) and 24h after initiation of bevacizumab/erlotinib in the KEGG pathways “Hematopoietic cell lineage” (hsa04640), “ABC transporters” (hsa02010), and “Pathways in cancer” (hsa05200). The genes depicted in this representation belong the list of the 100 most dysregulated genes
Fig. 4.
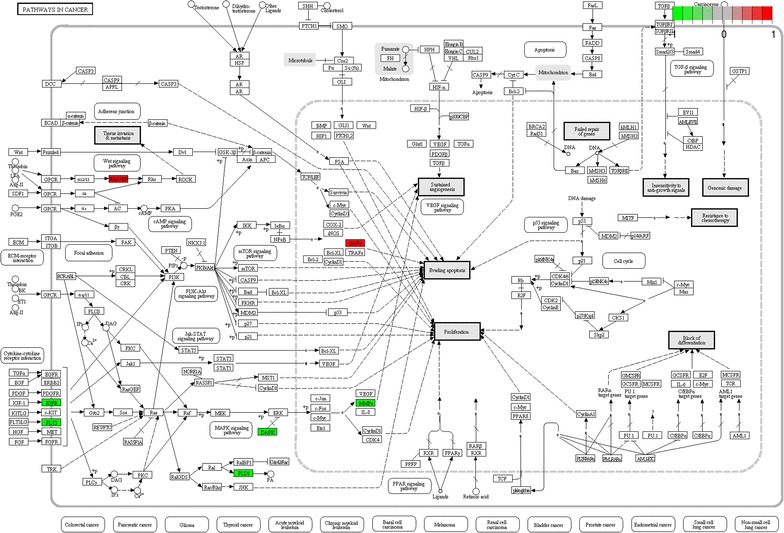
KEGG pathway hsa05200 “Pathways in cancer”. Genes highlighted in red and green were up-regulated and down-regulated due to the 24h action of bevacizumab/erlotinib, respectively
These pathway dysregulations are well in line with the expected effects of both erlotinib (“Pathways in cancer”; “ABC transporters”) and bevacizumab (“Hematopoietic cell lineage”). “Pathways in cancer” is a generic pathway, including genes involved in various aspects of tumorigenesis such as phenomena of proliferation, invasion, resistance and apoptosis. Several diseases are inter-related to this pathway, including non-small cell lung cancer. Most genes from the “Pathways in cancer” (FLT3, IGF1R, DAPK2, PLD1 and MMP9) are inhibited due to the action of BE resulting in an anti-proliferative and pro-apoptotic action of the combined therapy, with the notable exception of the up-regulation of the apoptosis inhibitor BIRC3. The combined action of BE results in the inhibition of all genes that belong to the “Hematopoietic cell lineage” (FLT3, IL1R1, IL1R2, MME). Hematopoietic stem cells play important roles for angiogenesis [32]. The down-regulation of genes within the pathway “Hematopoietic cell lineage” may be related to the specific anti-angiogenic action of bevacizumab [33, 34]. On the other hand, the dysregulation of genes that belong to the pathway “ABC transporters” is probably associated with the energy-related mechanisms of action of erlotinib [35, 36].
Gene set enrichment analysis based on the WikiPathways database provides information on additional cancer- and energy-related activated pathways including “gastric cancer network 2” (WP2363, ), “IL1 megakaryocytes in obesity” (WP2865, ), “apoptosis modulation and signaling” (WP1772, ) and “gastric cancer network 1” (WP2361, ).
Gene expression predictive value
The predictive value of the genes that were mainly dysregulated due to the 24h effect of BE was investigated. The following endpoints were considered: disease stabilization at 12 weeks, tumor shrinkage at 12 weeks, time to progression under BE, time to progression under chemotherapy, and overall survival. The magnitude of the 24h change in expression of the 100 most dysregulated genes was not significantly associated with any of the investigated endpoints, after adjustment for the patients mutational status.
Table 3 summarizes the predictive value of the blood gene expression at baseline and 24h after initiation of BE. Putative markers at baseline that predicted patient’s overall survival included Cancer susceptibility candidate 1 (CASC1). At baseline, 142 genes were identified as putative predictive markers of tumor shrinkage at 12 weeks. Among those genes, there was a significant enrichment of the KEGG signaling pathways “Phagosome” (hsa04145) and “Protein digestion and absorption” (hsa04974).
Table 3.
Putative predictive markers of the patient’s response to bevacizumab/erlotinib
| Time | Endpointa | Gene symbols | KEGG pathway enrichment |
|---|---|---|---|
| Baseline | TS12 | ANO2, VPS13D, EML1, C22orf31, FERMT1, SFRP4, SUPT6H, GYS2, THBS4, ATP6V1B1, TCF21, ESRRB, NEUROD4, SOX15, GATA5, PRRC2B, SLC7A10, LOXL4, DISP2, TRIM7, DRD2, GPHA2, KCTD15, PAQR4, ANKZF1, SHROOM1, FABP7, FBXL21, KCNK5, MBL2, LY6D, VASN, FSTL5, MECP2, ROR2, TPT1-AS1, DCDC1, HS6ST2, HSPB7, NWD2, OR5M3, SLC35E3, UNC119B, OR10AB1P, UMODL1, ERN1, RNF151, CALHM1, TMPRSS12, KLK12, KRT14, LINC01551, NWD1, C15orf52, IL1RAPL2, TRRAP, TPK1, MYO18A, DSCR8, B3GNT6, RNA5SP430, C1orf132, PPAPDC1A, BTBD17, LAYN, CST9LP1, ST8SIA6-AS1, AL592528.1, RP11-337C18.4, GAPDHP44, HAUS7, AC010904.1, CELA3B, RP1-288M22.1, U3, CTA-929C8.7, ARSEP1, UBE2D3P4, RP11-38C18.2, RP11-94M14.2, AC007312.3, LINC00237, RP11-66N5.2, LINC01375, C1DP4, RP11-20P5.2, RP5-888M10.2, AC105443.2, FGFR1OP2P1, RP5-855F14.2, TFAMP1, POLR3KP1, CRYGFP, TBC1D3P7, RP11-336N8.1, DCUN1D2-AS, RP11-528G1.2, ZBTB22, TSPY15P, RP11-98G13.1, AC007679.4, KCNQ1DN, RN7SL549P, SMKR1, PNMA2, PI4KA, RP11-520P18.1, RP11-572M11.3, OR10J7P, RP11-331K21.1, RP11-340A13.1, LINC00977, RP3-368B9.2, SNORA18, HNRNPCP8, MPV17L2, NAV2-AS5, CTD-2517M22.14, ENPP7P5, RP11-150C16.1, HNRNPA3P10, RP11-316E14.2, RP11-566K19.5, RP11-521O16.1, RP11-616M22.7, RP11-523L20.2, AC009120.4, RP11-106M3.3, RP11-553K8.5, RP11-189E14.4, RP11-520P18.5, SAMD11P1, RP11-286N3.2, SNRPCP4, RP11-343K8.3, AC005307.3, AC010524.4, CTB-31C7.3, WI2-80269A6.1, RP11-401N16.1, RP11-416H1.1, RP11-1072C15.7 | Phagosome (hsa04145, ) Protein digestion and absorption (hsa04974, ) |
| DS12 | – | – | |
| OS | CASC1, HIST1H1A, FAM86C2P, Y_RNA, IGHV5-51, RP11-174G6.1, HNRNPA1P63, EEF1B2P1, RP3-403L10.3, RPL7P53, IGKV1-5, MRPS36P2, RP11-415I12.3, RNU6-412P, RNU6-1224P, RP11-61G23.2, CTD-2547H18.1, RP11-295G12.1, ZNF23 | – | |
| TTPBE | BDKRB1, NRN1, LAMC1, ZNF462, LRRC43, SGSM1, FSCN2, C9orf92, RNU6-83P, RPL7AP14, RP11-793K1.1, RP3-433F14.1, RP1-292B18.4, AC098592.7, AC114812.8, TOMM20P1, LL22NC03-88E1.17, C12orf77, RP1-213J1P__B.1, LINC01038, MARK2P12, XRCC6P3, SHC1P1, RP3-463P15.1, LL0XNC01-116E7.2, COL4A2-AS1, CYCSP44, RAC1P8, SLMO2-ATP5E, AF127577.12, STARD13-AS, RN7SL797P, RN7SL598P, RP11-544A12.8, RP11-415I12.3, RP11-61G23.2, RP11-6B19.3, RP11-136F16.2, CASC18, AC002306.1, RP11-552E10.1, RP11-361M10.3, RP11-203B7.2, RP11-475B2.1, RP11-763E3.1, RP13-516M14.2, RP11-820I16.1 | – | |
| TTPCT | KRTAP2-3, AC090957.2, KALP, RP11-157I4.4, TSIX | – | |
| 24h after initiation of BE | TS12 | PDZD4, SH3BP1, CALD1, SPX, MAPKBP1, MMRN1, EGF, PLOD2, ANKRD61, FZD5, CLEC1B, SLC39A13, SMCO4, BCRP2, TSNARE1, TDRP, TOP1MT, TPTE2P3, RNU105C, Y_RNA, RN7SKP257, AC006988.1, ATP5HP3, RNU6-887P, RP11-361F15.2, BANF1P2, AC099344.2, LINC00884, DDX39BP2, RP11-486M23.1, RP11-167N24.3, RP11-693J15.3, RP11-433P17.3 | Pathways in cancer (hsa05200, ) |
| DS12 | – | – | |
| OS | MYO1C, Y_RNA, DEFB134, Y_RNA, IGLV3-21, IGLC7, IGHA2, IGHA1, IGHV3-15, IGHV3-23, IGHV5-51, SOD1P1, AC016768.1, LINC01032, ZSCAN31, RP11-169N13.4, IGKV1-5, IGHV3-65, IGLC4, RP11-280K24.1 | – | |
| TTPBE | TNMD, TSPAN9, PRICKLE3, RAD51, MCM10, TCF3, ATP2A3, OPHN1, FBXL19, PCK2, PLTP, E2F1, PCYT1B, CKM, APBA1, CNTNAP1, FOXM1, KIF20A, CIT, HJURP, E2F8, KLF16, APOC1, ATP8B3, EPHB2, CTIF, TICRR, CTU1, CPXCR1, GRIK4, C16orf78, CKB, VKORC1, TK1, METTL7B, MZB1, ZNF296, RRM2, JUP, PCP2, CADM2, SLC25A22, KLHL28, GJC1, MARCH11, GAS2L1, MITF, HIST1H2BM, SLC25A29, SRC, MYO1C, RNU12-2P, Y_RNA, D86998.1, IGLV6-57, IGLV3-21, IGHG1, IGHJ1, IGHV6-1, IGHV4-28, AP005482.3, RP3-407E4.4, RP11-535M15.1, IPPKP1, FAM195B, RP11-69C17.2, HDGFP1, SCAMP4, RD3L, TUBB4AP1, SDAD1P2, RP11-321L2.1, SPATA31B1P, AC005772.2, RP11-22C8.1, VN1R38P, ITGA9-AS1, NIFK-AS1, B3GNT9, RP11-252M21.6, RN7SL60P, RP11-266N13.2, RP11-517I3.1, RP11-364C11.2, RACGAP1P, RP11-545N8.3, RP11-81A1.3, RP11-2C24.5, AF213884.2, PRKCA-AS1, CTD-2319I12.2 | Melanoma (hsa05218, ) Pancreatic cancer (hsa05212, ) PPAR signaling cancer (hsa03320, ) arginine and proline metabolism (hsa00330, ) Pathways in cancer (hsa05200, ) Pyrimidine metabolism (hsa00240, ) | |
| TTPCT | TTTY14, MTND1P4, RP11-875H7.2, LINC01021, AC008565.1, LINC01486 | – |
Gene significantly predictive of the patient’s response () are reported together with the associated enriched KEGG pathways. Genes that belonged to the KEGG pathway “Pathways in cancer” are highlighted in italic
aTS12: tumor shrinkage at 12 weeks; DS12: disease stabilization at 12 weeks; OS: overall survival; TTPBE: time-to-progression under bevacizumab/erlotinib; TTPCT: time-to-progression under chemotherapy
Putative predictive markers from blood gene expression at 24h after initiation of treatment of the TTPBE included four genes enriched in the pathway “Pathways in cancer”. These genes were the E2F transcription factor 1 (E2F1), RAD51 recombinase (RAD51), the junction plakoglobin (JUP), and the microphthalmia-associated transcription factor (MITF). E2F1 plays a critical role in the control of cell cycle and acts as a tumor suppressor. E2F1 was found to be associated with phenomena of resistance of targeted therapy in breast cancer [37]. There was also a significant enrichment in the signaling pathway “Pathways in cancer” among the predictors of TS12 at 24h: genes frizzled class receptor 5 (FZD5) and epidermal growth factor (EGF). EGF encodes for a protein playing an important role in the cell growth, proliferation and differentiation. It binds with high affinity epidermal growth factor receptor. Its dysregulation has been associated with cancer progression [38]. Other pathways associated with 24h predictive markers of TTPBE included the cancer-related pathways “Melanoma”, “Pancreatic cancer”, “PPAR signaling cancer”, as well as the metabolism-related pathways “arginine and proline metabolism” and “Pyrimidine metabolism”.
All putative predictive markers of TTPBE at 24h were combined into a 91-gene metagene. Patients could be significantly classified into low-risk versus high-risk according to their median metagene score (HR 4.93 [95% CI 2.34 to 10.39], log-rank test: ) (Fig. 5, left panel). The median TTPBE were 2.46 (95% CI 1.54–3.22) months vs. 6.87 (95% CI 4.14–13.31) months in the high-risk and low-risk populations, respectively. This finding was successfully validated using the KMplotter web tool (Fig. 5, central panel) and the external CIT validation dataset (Fig. 5, right panel). The predictive value of the metagene remained significant after adjusting for the patient’s mutational status (Cox proportional hazards regression after adjustment for the mutational status: HR 2.63 [95% CI 1.87–3.70], ). An illustration of selection of responders based on the metagene score is provided in the Results section of the Additioanl file 1.
Fig. 5.
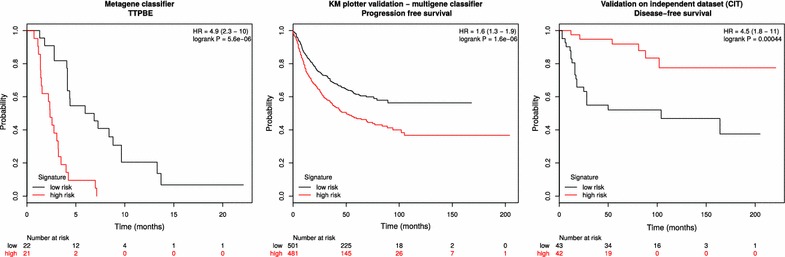
Metagene classifier of time-to-progression under BE. The left panel displays the classification of low- versus high-risk patients based on the 91-gene metagene. The central panel shows the classification obtained by the KMplotter online validation tool using a multigene classifier. The right panel shows the classification obtained by the external CIT validation dataset. Hazard ratios and log rank test p values are reported in the upper right corner of the each panel
Discussion
The analysis of the immediate effect of BE in late stage non-squamous NSCLC reveals a series of important mechanisms dysregulated by the combined action of both therapies. Important activated pathways involved mechanisms such as apoptosis evasion, anti-proliferation and anti-angiogenesis. Interestingly, it was possible to detect these dysregulations directly in the blood, showing that potential biomarkers could be identified at the blood level. The changes measured in the blood over a small time period (24h) were of small magnitude, yet consistent among patients. The use of a within-patient design of experiment including 2 time points before and after treatment helped to characterize these gene variations despite the relatively small sample size.
The choice of the multivariate method DCCA over more common gene-by-gene approaches was driven by the fact that DCCA addresses the problem of the identification of differentially expressed genes (in within-patient repeated measures designs) in a multivariate manner. This is more statisfactory since it allows to take into account potential gene correlations/interactions using a single computationally efficient procedure. DCCA is an exploratory method appropriate for the purpose of the current hypothesis-generating translational study. On the other hand, gene-by-gene approaches are simple and flexible and could be preferred in case of more complex designs or when applied to confirmatory analyses.
Although the magnitude of change of the most dysregulated genes over 24h was not predictive of the patient’s outcome, both the gene expression level at baseline and 24h revealed a series of putative predictive genes. While DS12 was defined as primary clinical endpoint of the original SAKK 19/05 trial, endpoints reflecting the activity of the treatment on the disease were more specifically investigated in the current translational substudy. TTPBE and TS12 are two endpoints which are objectively associated with the direct effect of BE. In both cases, a series of key predictive markers at 24h were enriched within the KEGG pathway “Pathways in cancer”. This pathway appears to play an important role both in the immediate effect of BE as measured in the blood, and in the prediction of the response to BE.
Our findings could be validated using two independent datasets (meta-analysis from the KMplotter web tool and external CIT validation dataset). The combination of the key predictive markers at 24h regarding TTPBE into a metagene was used to generate a gene signature, predicting with high significance patients into high vs. low risk populations. This signature was successfully validated, and could be used independently from the patient’s EGFR mutational status for proper patient selection.
Because our gene signature is independent from the patient’s mutational status, it can be used as predictive marker both in EGFR mutated and wild-type populations. BE has potential to become a standard therapy in NSCLC patients with EGFR mutations, and our signature may help to select patients which may not respond to the therapy despite the presence of the mutation. Inversely, our signature may be useful for proper selection of BE responders among patients not harboring EGFR activating mutation.
Our findings based on exon array data are in essence exploratory and future prospective confirmatory studies are needed to further validate the clinical relevance of our discovery.
Conclusion
The 24h effect of BE could be accurately monitored in peripheral blood using the exon array technology. Genes impacted by the immediate effect of BE belonged to key signaling pathways, according to the expected mechanisms of action of both bevacizumab and erlotinib. Although the magnitude of change over 24h had no predictive value with regard to the investigated endpoints, the blood gene expression level measured 24h after initiation of BE could be used to predict TTPBE independently from the patient’s mutational status. Proper selection of responders to the combined targeted therapy BE could be monitored from blood level gene expression.
Author's contributions
FB implemented the methodology, performed the data analysis and wrote the manuscript. MJ and MF provided oncological expertise. DK coordinated the work and gave methodological input. MB and FZ supervised the work, designed the experiment and gave input in the writing of the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to thank the Swiss Group for Clinical Cancer Research for collecting the samples and financing the microarray experiments, and the Lungenliga St. Gallen for their unconditional support.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Raw data from the exon array analysis have been deposited in NCBI’s Gene Expression Omnibus (GEO), and are accessible through GEO Series accession number GSE61676.
Ethics approval and consent to participate
The clinical trial as well as the current translational substudy were approved by the ethics committee of the canton of St. Gallen (EKSG 06/012). Written informed consent for translational research was obtained from all patients who agreed to have their clinical records used in this study.
Funding
The Swiss Group for Clinical Cancer Research financed the microarray experiments.
Abbreviations
- ABC
ATP binding cassette
- ADAM19
ADAM metallopeptidase domain 19
- ATAD2
ATPase family, AAA domain containing 2
- ATPase
adenosine triphosphatase
- B
bevacizumab
- BE
bevacizumab/erlotinib
- BIRC3
baculoviral IAP repeat containing 3
- BMX
BMX non-receptor tyrosine kinase
- CASC1
cancer susceptibility candidate 1
- CASC5
cancer susceptibility candidate 5
- CDC42
cell division cycle 42
- CENPE
centromere-associated protein E
- CI
confidence interval
- CIT
Carte d’Identité des Tumeurs
- CT
chemotherapy
- DAPK2
death-associated protein kinase 2
- DCCA
dually constrained correspondence analysis
- DOCK10
dedicator of cytokinesis 10
- E
erlotinib
- E2F1
E2F transcription factor 1
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- EKSG
ethics committee of the canton of St. Gallen
- EPS8
epidermal growth factor receptor pathway substrate 8
- FGFR
fibroblast growth factor receptor
- FLT3
Fms-related tyrosine kinase 3
- FRY
protein furry homolog
- FZD5
frizzled class receptor 5
- GBP4
guanylate binding protein 4
- GEO
gene expression omnibus
- GTPase
guanosine triphosphatase
- HR
hazard ratio
- IGF1R
insulin-like growth factor 1 receptor
- IL1R1
interleukin 1 receptor, type I
- IL1R2
interleukin 1 receptor, type II
- IQR
inter-quartile range
- KEGG
Kyoto encyclopedia of genes and genomes
- JUP
junction plakoglobin
- KNTC1
kinetochore associated 1
- MAPK
mitogen-activated protein kinase
- MITF
microphthalmia-associated transcription factor
- MME
membrane metalloendopeptidase
- MMP9
matrix metallopeptidase 9
- mTOR
mechanistic target of rapamycin
- NSCLC
non-small cell lung cancer
- OS
overall survival
- PCA
principal component analysis
- PFS
progression-free survival
- PLD1
phospholipase D1
- RAD51
RAD51 recombinase
- RBL1
retinoblastoma-like 1
- RNA
ribonucleic acid
- SAKK
schweizerische Arbeitsgemeinschaft für klinische Krebsforschung (Swiss Group for Clinical Cancer Research)
- TKI
tyrosine kinase inhibitor
- TS12
tumor shrinkage at 12 weeks
- TTPBE
time to progression under bevacizumab/erlotinib
- TTPCT
time to progression under chemotherapy
- VEGF
vascular endothelial growth factor
Additional file
Additional file 1. R extension package dcca.
Contributor Information
Florent Baty, Email: florent.baty@kssg.ch.
Markus Joerger, Email: markus.joerger@kssg.ch.
Martin Früh, Email: martin.frueh@kssg.ch.
Dirk Klingbiel, Email: dirk.klingbiel@sakk.ch.
Francesco Zappa, Email: francesco.zappa@clinicaluganese.ch.
Martin Brutsche, Email: martin.brutsche@kssg.ch.
References
- 1.Kummar S, Chen HX, Wright J, Holbeck S, Millin MD, Tomaszewski J, Zweibel J, Collins J, Doroshow JH. Utilizing targeted cancer therapeutic agents in combination: novel approaches and urgent requirements. Nat Rev Drug Discov. 2010;9(11):843–856. doi: 10.1038/nrd3216. [DOI] [PubMed] [Google Scholar]
- 2.Qi WX, Wang Q, Jiang YL, Sun YJ, Tang LN, He AN, Min DL, Lin F, Shen Z, Yao Y. Overall survival benefits for combining targeted therapy as second-line treatment for advanced non-small-cell-lung cancer: a meta-analysis of published data. PLoS ONE. 2013;8(2):55637. doi: 10.1371/journal.pone.0055637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, Zhang S, Wang J, Zhou S, Ren S, Lu S, Zhang L, Hu C, Hu C, Luo Y, Chen L, Ye M, Huang J, Zhi X, Zhang Y, Xiu Q, Ma J, Zhang L, You C. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):735–742. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 4.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM, Porta R, Cobo M, Garrido P, Longo F, Moran T, Insa A, De Marinis F, Corre R, Bover I, Illiano A, Dansin E, de Castro J, Milella M, Reguart N, Altavilla G, Jimenez U, Provencio M, Moreno MA, Terrasa J, Munoz-Langa J, Valdivia J, Isla D, Domine M, Molinier O, Mazieres J, Baize N, Garcia-Campelo R, Robinet G, Rodriguez-Abreu D, Lopez-Vivanco G, Gebbia V, Ferrera-Delgado L, Bombaron P, Bernabe R, Bearz A, Artal A, Cortesi E, Rolfo C, Sanchez-Ronco M, Drozdowskyj A, Queralt C, de Aguirre I, Ramirez JL, Sanchez JJ, Molina MA, Taron M, Paz-Ares L. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 5.Pennell NA, Lynch TJ. Combined inhibition of the VEGFR and EGFR signaling pathways in the treatment of NSCLC. Oncologist. 2009;14(4):399–411. doi: 10.1634/theoncologist.2008-0276. [DOI] [PubMed] [Google Scholar]
- 6.Herbst RS, Ansari R, Bustin F, Flynn P, Hart L, Otterson GA, Vlahovic G, Soh CH, O’Connor P, Hainsworth J. Efficacy of bevacizumab plus erlotinib versus erlotinib alone in advanced non-small-cell lung cancer after failure of standard first-line chemotherapy (BeTa): a double-blind, placebo-controlled, phase 3 trial. Lancet. 2011;377(9780):1846–1854. doi: 10.1016/S0140-6736(11)60545-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seto T, Kato T, Nishio M, Goto K, Atagi S, Hosomi Y, Yamamoto N, Hida T, Maemondo M, Nakagawa K, Nagase S, Okamoto I, Yamanaka T, Tajima K, Harada R, Fukuoka M, Yamamoto N. Erlotinib alone or with bevacizumab as first-line therapy in patients with advanced non-squamous non-small-cell lung cancer harbouring EGFR mutations (JO25567): an open-label, randomised, multicentre, phase 2 study. Lancet Oncol. 2014;15(11):1236–1244. doi: 10.1016/S1470-2045(14)70381-X. [DOI] [PubMed] [Google Scholar]
- 8.Stahel R, Dafni U, Gautschi O, Felip E, Curioni-Fontecedro A, Peters S, Massutí B, Cardenal F, Aix S, Früh M, et al. 3ba a phase ii trial of erlotinib (e) and bevacizumab (b) in patients with advanced non-small-cell lung cancer (nsclc) with activating epidermal growth factor receptor (egfr) mutations with and without t790m mutation. the spanish lung cancer group (slcg) and the european thoracic oncology platform (etop) belief trial. Eur J Cancer. 2015;51:711–712. doi: 10.1016/S0959-8049(15)30068-X. [DOI] [Google Scholar]
- 9.Johnson BE, Kabbinavar F, Fehrenbacher L, Hainsworth J, Kasubhai S, Kressel B, Lin CY, Marsland T, Patel T, Polikoff J, Rubin M, White L, Yang JC, Bowden C, Miller V. ATLAS: randomized, double-blind, placebo-controlled, phase IIIB trial comparing bevacizumab therapy with or without erlotinib, after completion of chemotherapy, with bevacizumab for first-line treatment of advanced non-small-cell lung cancer. J Clin Oncol. 2013;31(31):3926–3934. doi: 10.1200/JCO.2012.47.3983. [DOI] [PubMed] [Google Scholar]
- 10.Thomas M, Fischer J, Andreas S, Kortsik C, Grah C, Serke M, von Eiff M, Witt C, Kollmeier J, Muller E, Schenk M, Schroder M, Villalobos M, Reinmuth N, Penzel R, Schnabel P, Acker T, Reuss A, Wolf M. Erlotinib and bevacizumab versus cisplatin, gemcitabine and bevacizumab in unselected nonsquamous nonsmall cell lung cancer. Eur Respir J. 2015;46(1):219–229. doi: 10.1183/09031936.00229014. [DOI] [PubMed] [Google Scholar]
- 11.Zappa F, Droege C, Betticher D, von Moos R, Bubendorf L, Ochsenbein A, Gautschi O, Leibundgut EO, Froesch P, Stahel R, Hess T, Rauch D, Schmid P, Mayer M, Crowe S, Brauchli P, Ribi K, Pless M. Bevacizumab and erlotinib (BE) first-line therapy in advanced non-squamous non-small-cell lung cancer (NSCLC) (stage IIIB/IV) followed by platinum-based chemotherapy (CT) at disease progression: a multicenter phase II trial (SAKK 19/05) Lung Cancer. 2012;78(3):239–244. doi: 10.1016/j.lungcan.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 12.Ciuleanu T, Tsai CM, Tsao CJ, Milanowski J, Amoroso D, Heo DS, Groen HJ, Szczesna A, Chung CY, Chao TY, Middleton G, Zeaiter A, Klingelschmitt G, Klughammer B, Thatcher N. A phase II study of erlotinib in combination with bevacizumab versus chemotherapy plus bevacizumab in the first-line treatment of advanced non-squamous non-small cell lung cancer. Lung Cancer. 2013;82(2):276–281. doi: 10.1016/j.lungcan.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 13.Jazieh AR, Al Sudairy R, Abu-Shraie N, Al Suwairi W, Ferwana M, Murad MH. Erlotinib in wild type epidermal growth factor receptor non-small cell lung cancer: a systematic review. Ann Thorac Med. 2013;8(4):204–208. doi: 10.4103/1817-1737.118503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zander T, Hofmann A, Staratschek-Jox A, Classen S, Debey-Pascher S, Maisel D, Ansen S, Hahn M, Beyer M, Thomas RK, Gathof B, Mauch C, Delank KS, Engel-Riedel W, Wichmann HE, Stoelben E, Schultze JL, Wolf J. Blood-based gene expression signatures in non-small cell lung cancer. Clin Cancer Res. 2011;17(10):3360–3367. doi: 10.1158/1078-0432.CCR-10-0533. [DOI] [PubMed] [Google Scholar]
- 15.Okoniewski MJ, Miller CJ. Comprehensive analysis of affymetrix exon arrays using BioConductor. PLoS Comput Biol. 2008;4(2):6. doi: 10.1371/journal.pcbi.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baty F, Klingbiel D, Zappa F, Brutsche M. High-throughput alternative splicing detection using dually constrained correspondence analysis (DCCA) J Biomed Inform. 2015;58:175–185. doi: 10.1016/j.jbi.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 17.Baty F, Rothschild S, Fruh M, Betticher D, Droge C, Cathomas R, Rauch D, Gautschi O, Bubendorf L, Crowe S, Zappa F, Pless M, Brutsche M. EGFR exon-level biomarkers of the response to bevacizumab/erlotinib in non-small cell lung cancer. PLoS ONE. 2013;8(9):72966. doi: 10.1371/journal.pone.0072966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franzini A, Baty F, Macovei II, Durr O, Droege C, Betticher D, Grigoriu BD, Klingbiel D, Zappa F, Brutsche MH. Gene expression signatures predictive of bevacizumab/erlotinib therapeutic benefit in advanced nonsquamous non-small cell lung cancer patients (SAKK 19/05 trial) Clin Cancer Res. 2015;21(23):5253–5263. doi: 10.1158/1078-0432.CCR-14-3135. [DOI] [PubMed] [Google Scholar]
- 19.Yates T. Annmap: genome annotation and visualisation package pertaining to Affymetrix arrays and NGS analysis. R package version 1.6.0. http://annmap.cruk.manchester.ac.uk (2011).
- 20.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31(4):15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carvalho BS, Irizarry RA. A framework for oligonucleotide microarray preprocessing. Bioinformatics. 2010;26(19):2363–2367. doi: 10.1093/bioinformatics/btq431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bair E, Tibshirani R. Semi-supervised methods to predict patient survival from gene expression data. PLoS Biol. 2004;2(4):108. doi: 10.1371/journal.pbio.0020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.R Core Team. R: a language and environment for statistical computing. R foundation for statistical computing, Vienna, Austria. R foundation for statistical computing. http://www.R-project.org/ (2013).
- 24.Dray S, Dufour A-B. The ade4 package: implementing the duality diagram for ecologists. J Stat Softw. 2007;22(4):1–20. doi: 10.18637/jss.v022.i04. [DOI] [Google Scholar]
- 25.Gentleman RC, Carey VJ, Bates DM. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41(Web Server issue):77–83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gyorffy B, Surowiak P, Budczies J, Lanczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE. 2013;8(12):82241. doi: 10.1371/journal.pone.0082241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rousseaux S, Debernardi A, Jacquiau B, Vitte AL, Vesin A, Nagy-Mignotte H, Moro-Sibilot D, Brichon PY, Lantuejoul S, Hainaut P, Laffaire J, de Reynies A, Beer DG, Timsit JF, Brambilla C, Brambilla E, Khochbin S. Ectopic activation of germline and placental genes identifies aggressive metastasis-prone lung cancers. Sci Transl Med. 2013;5(186):186–266. doi: 10.1126/scitranslmed.3005723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Frohman MA. Cellular and physiological roles for phospholipase D1 in cancer. J Biol Chem. 2014;289(33):22567–22574. doi: 10.1074/jbc.R114.576876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fidler MJ, Shersher DD, Borgia JA, Bonomi P. Targeting the insulin-like growth factor receptor pathway in lung cancer: problems and pitfalls. Ther Adv Med Oncol. 2012;4(2):51–60. doi: 10.1177/1758834011427576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takakura N, Watanabe T, Suenobu S, Yamada Y, Noda T, Ito Y, Satake M, Suda T. A role for hematopoietic stem cells in promoting angiogenesis. Cell. 2000;102(2):199–209. doi: 10.1016/S0092-8674(00)00025-8. [DOI] [PubMed] [Google Scholar]
- 33.Rafii S, Lyden D, Benezra R, Hattori K, Heissig B. Vascular and haematopoietic stem cells: novel targets for anti-angiogenesis therapy? Nat Rev Cancer. 2002;2(11):826–835. doi: 10.1038/nrc925. [DOI] [PubMed] [Google Scholar]
- 34.Ribatti D, Scavelli C, Roccaro AM, Crivellato E, Nico B, Vacca A. Hematopoietic cancer and angiogenesis. Stem Cells Dev. 2004;13(5):484–495. doi: 10.1089/scd.2004.13.484. [DOI] [PubMed] [Google Scholar]
- 35.Shi Z, Peng XX, Kim IW, Shukla S, Si QS, Robey RW, Bates SE, Shen T, Ashby CR, Fu LW, Ambudkar SV, Chen ZS. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res. 2007;67(22):11012–11020. doi: 10.1158/0008-5472.CAN-07-2686. [DOI] [PubMed] [Google Scholar]
- 36.Lainey E, Sebert M, Thepot S, Scoazec M, Bouteloup C, Leroy C, De Botton S, Galluzzi L, Fenaux P, Kroemer G. Erlotinib antagonizes ABC transporters in acute myeloid leukemia. Cell Cycle. 2012;11(21):4079–4092. doi: 10.4161/cc.22382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Louie MC, McClellan A, Siewit C, Kawabata L. Estrogen receptor regulates E2F1 expression to mediate tamoxifen resistance. Mol Cancer Res. 2010;8(3):343–352. doi: 10.1158/1541-7786.MCR-09-0395. [DOI] [PubMed] [Google Scholar]
- 38.Witsch E, Sela M, Yarden Y. Roles for growth factors in cancer progression. Physiology (Bethesda) 2010;25(2):85–101. doi: 10.1152/physiol.00045.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw data from the exon array analysis have been deposited in NCBI’s Gene Expression Omnibus (GEO), and are accessible through GEO Series accession number GSE61676.