

Sir,
Laugier–Hunziker syndrome (LHS) is a rare, acquired, disorder of pigmentation characterized by hyperpigmentation of lips, oral mucosa, and longitudinal melanonychia. The disease is a diagnosis of exclusion and is diagnosed mainly on the basis of clinical and histopathological features. This benign disease mostly manifests in adults with no systemic features or malignant potential. Early identification of LHS helps in ruling out other severe syndromes associated with mucocutaneous hyperpigmentation and malignant potential such as Peutz–Jeghers syndrome. A 19-year-old female attended our department with a history of hyperpigmentation on nails of fingers and toes with longitudinal melanonychia, which had developed gradually over the last 8–9 years [Figure 1a and b]. Patient also complained of pigmentation on the lower lip and tongue for the last 5 years [Figure 2a]. Hyperpigmented lesions were also present over the soft and hard palate. There were no ulcers in the mouth, and orodental hygiene was good. Examination of hands and feet showed hyperpigmented macules over the tips of all fingers and toes. The volar aspect of finger tips and soles also had few pigmented macules [Figure 2b]. She was otherwise healthy with no complaints of any abdominal pain, diarrhoea, vomiting, fatigue, and weight loss; she was not receiving any medication (including antimalarials or minocycline). No family history of any mucocutaneous disorder was present. There was no history of any trauma prior to the pigmentation. General physical and systemic examination was normal. Routine hematological and biochemical investigations were within normal limits. Serum cortisol level was also normal. X-ray of the chest, ultrasound of the abdomen, endoscopy, and echocardiography were normal. The histopathology of skin biopsy taken from the plantar lesion showed hyperkeratosis and acanthosis [Figure 3a]. Basal cell hyperpigmentation was scanty with few melanocytes [Figure 3b]. Patient was diagnosed as a classical case of LHS. LHS is a rare disorder which was first described by Laugier and Hunziker in 1970.[1] It is an acquired, benign pigmentary skin condition characterized by diffuse oral hypermelanosis with pigmentation of the nails and longitudinal melanonychia.[2] Cutaneous manifestations can also be present in other areas of body and the term “idiopathic lenticular mucocutaneous pigmentation” has also been used.[3] This syndrome occurs mainly in middle-aged adults and is progressive. A mean age of 52 years has been reported.[4] However, in our report, the girl presented at an unusual early age of only 10 years. Few cases have been reported at such a young age. Sardana et al.[5] reported one such case in a 12-year-old child. No malignant predisposition has been associated with LHS.[6] The main differential diagnoses are Peutz–Jeghers syndrome, Addison's disease, Mc-Cune–Albright syndrome, LEOPARD syndrome, Gardner's syndrome, drug induced hyperpigmentation, and LAMB/syndrome. Peutz–Jeghers syndrome and Gardner's syndrome are autosomal dominant conditions associated with abdominal pain and intestinal polyps; malignant predisposition may also be present. Addison's disease is associated with findings such as low blood pressure, hyperkalemia, hyponatremia, hypoglycemia, and elevated blood urea nitrogen.[7] McCune–Albright syndrome presents with precocious puberty, polyostotic fibrous dysplasia, and unilateral cafe au lait macules. Therefore, prompt diagnosis of this syndrome helps to rule out the above severe systemic conditions, thus helping the patient from unnecessary investigations and improper treatment.
Figure 1.
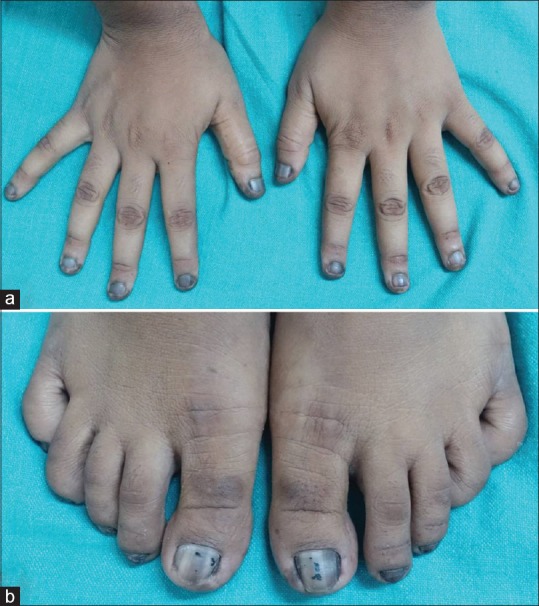
(a) Hyperpigmentation of all nails of hand with longitudinal melanonychia. (b) Hyperpigmentation involving all toe nails
Figure 2.
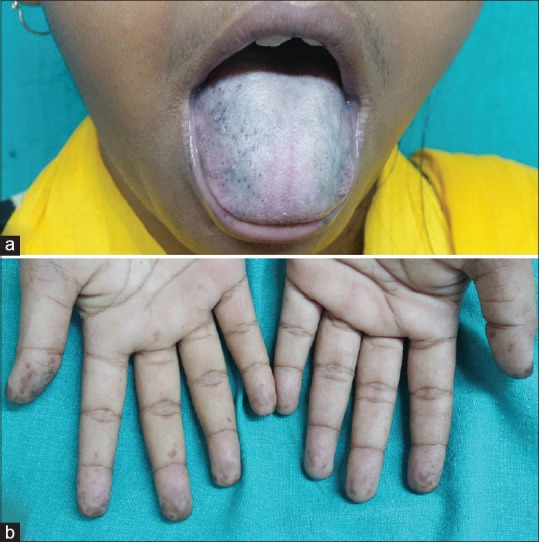
(a) Pigmented macules over tongue and lower lip, upper lip is spared. (b) Pigmented macules over the volar aspect of fingertips
Figure 3.
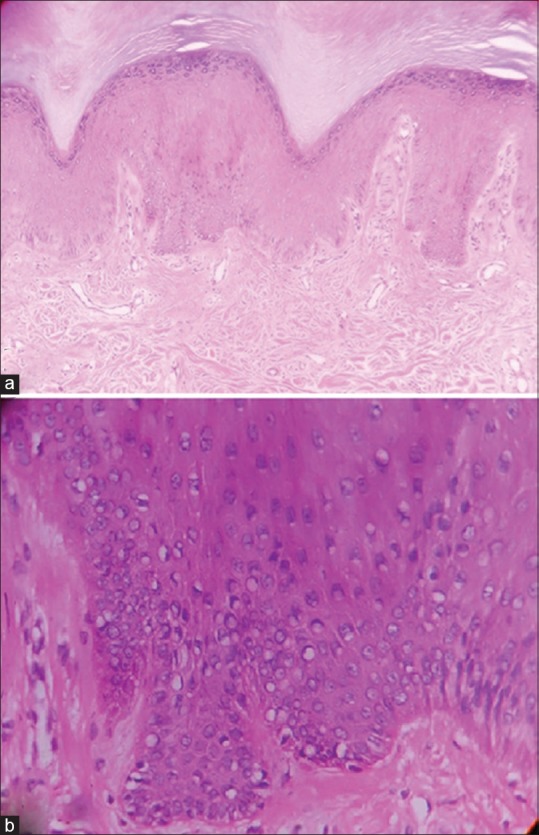
(a) Hyperkeratosis with acanthosis. (Hematoxylin and eosin ×10) (b) Basal cell hyperpigmentation with few melanocytes (Hematoxylin and eosin ×40)
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Montebugnoli L, Grelli I, Cervellati F, Misciali C, Raone B. Laugier-Hunziker syndrome: An uncommon cause of oral pigmentation and a review of the literature. Int J Dent 2010. 2010:525404. doi: 10.1155/2010/525404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nayak RS, Kotrashetti JS, Hosmani JV. Laugier–Hunziker syndrome. J Oral Maxillofac Pathol. 2012;16:245–50. doi: 10.4103/0973-029X.99079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerbig AW, Hunziker T. Idiopathic lenticular mucocutaneous pigmentation or Laugier–Hunziker syndrome with atypical features. Arch Dermatol. 1996;132:844–5. doi: 10.1001/archderm.132.7.844. [DOI] [PubMed] [Google Scholar]
- 4.Sachdeva S, Sachdeva S, Kapoor P. Laugier-Hunziker syndrome: A rare cause of oral and acral pigmentation. J Cutan Aesthet Surg. 2011;4:58–60. doi: 10.4103/0974-2077.79199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sardana K, Mishra D, Garg V. Laugier-Hunziker syndrome. Indian Pediatr. 2006;43:998–1000. [PubMed] [Google Scholar]
- 6.Wang WM, Wang X, Duan N, Jiang HL, Huang XF. Laugier-Hunziker syndrome: A report of three cases and literature review. Int J Oral Sci. 2012;4:226–30. doi: 10.1038/ijos.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nikitakis NG, Koumaki D. Laugier-Hunziker syndrome: Case report and review of literature. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;116:e52–8. doi: 10.1016/j.oooo.2012.12.012. [DOI] [PubMed] [Google Scholar]