

Abstract
The thioredoxin/thioredoxin reductase system (Trx/TrxR) is an attractive drug target because of its involvement in a number of important physiological processes, from DNA synthesis to regulating signal transduction. This study describes the finding of pyrazolone compounds that are active against Staphylococcus aureus. Initially, the project was focused on discovering small molecules that may have antibacterial properties targeting the Mycobacterium tuberculosis thioredoxin reductase. This led to the discovery of a pyrazolone scaffold-containing compound series that showed bactericidal capability against S. aureus strains, including drug-resistant clinical isolates. The findings support continued development of the pyrazolone compounds as potential anti-S. aureus antibiotics.
Keywords: thioredoxin reductase, antibiotic target, molecular docking, enzyme inhibitor, MRSA, pyrazolones
1. Introduction
Thioredoxin reductases (TrxRs) belong to a family of flavoproteins with redox-active cysteine residues that link the pyridine nucleotides with sulfur-containing substrates (thioredoxin proteins) [1]. Each monomer in this homodimeric protein includes an NADPH binding site, a FAD prosthetic group, and an active site containing a redox-active disulfide [2]. TrxRs get their name because they catalyze the reduction of oxidized small protein thioredoxin (Trx), which belongs to a group of small (10–12 kDa) redox-active proteins with a conserved Trp-Cys-Gly-Pro-Cys-Lys catalytic motif [3]. Reduction of the substrate disulfide occurs when electrons are transferred from NADPH via FAD to the sulfur-containing active site of TrxR.
The Trx system—collectively comprised of Trx, TrxR and NADPH—plays an essential role in various physiological processes, including regulating cellular viability and function, redox homeostasis, reducing nucleotides to deoxyribonucleotides, and helping to detoxify the body from oxidants and free radicals [4]. Therefore, it is important as a clinical goal to target the inhibition of TrxR in pathogenic microbes, since it is involved in the above-mentioned physiological/pathological processes.
One particular infectious disease that can be attacked by targeting the Trx system is tuberculosis (TB), untreated infections of which resulted in 9.6 million cases of active TB and 1.5 million deaths in 2014 [5]. Mycobacterium tuberculosis (M. tuberculosis, the causative agent of TB) requires high levels of oxygen, and is primarily a pathogen of the mammalian respiratory lungs [6]. The exact mechanism by which it resists the oxidative killing of immune cells has yet to be fully elucidated; but it is clear that the proteins involved in this defense are attractive drug targets [7].
Another bacterial species that could be targeted by a drug that is specific for the Trx system is Staphylococcus aureus (S. aureus). This Gram-positive bacteria is a significant cause of skin and soft tissue infections as well as life-threatening diseases such as pneumonia, endocarditis, and bloodstream infections [8]. Within the genome of S. aureus, there is a single TrxR gene (trxB), and the Trx system is essential for its growth [9]. Thus, the Trx system is an attractive drug target for multiple infectious diseases.
In recent years, numerous groups have demonstrated the feasibility of targeting the Trx system in bacteria as a unique antibacterial drug discovery approach. Some of the compounds found to inhibit the Trx system have included epigallocatechin-3-gallate [10], ebselen [11,12,13], auranofin [14,15], and gold(I)-alkynyl chromones [16]. The bacterial species that have been targeted by these compounds have included Bacillus subtilis (B. subtilis), Escherichia coli (E. coli), Enterococcus faecalis (E. faecalis), Enterococcus faecium (E. faecium), Helicobacter pylori (H. pylori), M. tuberculosis, and S. aureus. Clearly, the Trx system is a viable target for the discovery of potent antibacterial drugs.
Both high-throughput screening and virtual screening have been successfully used to identify new leads in drug discovery [17]. The program AUTODOCK has been effectively used in various protein systems for virtual screening [18,19]. In the present study, we initially attempted to identify M. tuberculosis TrxR inhibitors by virtual screening of compound libraries, followed by activity assay and in silico structural characterization. After an initial drug lead was identified, derivatives of the lead molecule were found by doing a similar scaffold search from a compound database. Several lead compounds—identified in silico—were screened against six bacterial species that included two mycobacterial species as well as S. aureus and Enterococcus faecalis (E. faecalis). Although none of the lead compounds showed minimum inhibitory concentration (MIC) activity against the two mycobacterial species that were tested, some of them did display antibacterial activity against drug-resistant S. aureus.
2. Results and Discussion
2.1. Initial Virtual Screening and Bacterial Assays
The project started with docking of compounds from the Center for Structure-based Drug Design and Development (CSD3) screening collection against a crystal structure of M. tuberculosis TrxR (PDB accession code 2A87) [7]. Nine compounds with the best-predicted binding energy (Figure 1) were then tested against two Mycobacterium species: Mycobacterium marinum and Mycobacterium smegmatis. Surprisingly, the MIC results did not support the predicted binding energies from the molecular docking (Table 1). We hypothesize that this is probably due to the lack of cell wall penetration, due to the thick mycolic acid lipid layer surrounding the mycobacterial cells.
Figure 1.

Docking energy and binding pose for compound 5376 in its lowest energy and highest populated cluster (Run 36, Cluster 2).
Table 1.
The minimum inhibitory concentration (MIC) results for the compounds tested against a panel of six bacterial species.
| MIC (μg/mL) | ||||||
|---|---|---|---|---|---|---|
| Compound | S. aureus | E. faecalis | E. coli | P. aeruginosa | M. smegmatis | M. marinum |
| 8973 | 64 (64) a | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| 6702 | 64 (64) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| 1108 | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| 2628 | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| 9010 | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | 128 (128) | >128 (>128) |
| 1632 | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| 5376 | 8 (4-8) | 128 (128) | >128 (>128) | >128 (>128) | 128 (128) | >128 (>128) |
| 1882 | 32 (32) | 32 (32) | >128 (>128) | >128 (>128) | 128 (128) | >128 (>128) |
| 3719 | 64 (64) | 128 (128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| Tetracycline | 0.5 (0.25–0.5) | 32 (32) | 2 (1–2) | 16 (16) | ND | ND |
| Isoniazid | ND b | ND | ND | ND | 4 | 8 |
a The MICs represent the modal values (range) from at least three separate runs; b ND = Not determined. E. coli: Escherichia coli; E. faecalis: Enterococcus faecalis; M. marinum: Mycobacterium marinum. M. smegmatis: Mycobacterium smegmatis; P. aeruginosa: Pseudomonas aeruginosa; S. aureus: Staphylococcus aureus.
The same nine compounds were then used to run MIC assays against a panel of four other bacterial species (Table 1). Interestingly, some of the compounds showed activity against both S. aureus and E. faecalis, with compound CSD3 5376 (therein called 5376) having the best MIC of 8 µg/mL against S. aureus bacteria. Compound 1882 had activity against both S. aureus and E. faecalis, although the MIC values demonstrated only a modicum of efficacy against both species. (MIC values of 32 μg/mL for each species). Additional work could be done in the future to assess the efficacy of 1882 against drug-resistant strains of enterococci that include vancomycin-resistant enterococcal species. We chose compound 5376 as a lead compound because the MIC value was less than 10 μg/mL against the ATCC S. aureus strain.
2.2. Ability of Analogues of 5376 to Kill Certain Strains of S. aureus
As a further test, we chose two original compounds, 5376 and 8973, as well as five analogs of the 5376 compound (587084, 5741518, 2082-0182, 2083-1665, and 2083-1773) to perform MIC assays against five well-researched S. aureus strains and four other clinical isolates of S. aureus (Table 2). Oxacillin and vancomycin—two commercially-available anti-staphylococcal drugs—were used as positive controls, and showed good inhibition of bacterial growth with MIC values of 4 and 1 µg/mL, respectively, for all strains. Compound 5376 showed activity against all S. aureus strains that were tested with an MIC50 of 4 μg/mL and an MIC90 of 8 μg/mL. Minimum bactericial concentration (MBC) assays demonstrated that compound 5376 was bactericidal, reducing viable counts by 2 to 2.5 logs. None of the 5376 analogs had activity against any of the S. aureus strains.
Table 2.
MIC results for select compounds tested against nine strains of Staphylococcus aureus.
| MIC (μg/mL) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | ATCC a | MW2 | JE2 | Newman | N315 | MC7606 b | MC7769 | MC7827 | MC7846 |
| 8973 | >128 c (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| 5376 | 8 (4-8) | 4 (4) | 8 (8) | 4 (4) | 4 (2–4) | 2 (2) | 4 (4–8) | 8 (8) | 4 (4) |
| 5741518 | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| 5870804 | 16 (16) | 32 (32) | 32 (32) | 64 (64) | 32 (32) | 4 (4) | 64 (64) | 32 (32–64) | 16 (16) |
| 2082-0182 | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| 2083-1665 | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| 2083-1773 | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| Tetracycline | 0.25 (0.25–0.5) | 0.25 (0.25) | 0.25 (0.25) | 0.25 (0.25) | 0.25 (0.25) | 32 (16–32) | 0.25 (0.25) | 32 (32) | 0.125 (0.125) |
| Oxacillin | 0.25 (0.25) | 8 (8–16) | 8 (8–6) | 0.5 (0.5) | 16 (16) | 4 (4) | 4 (4) | 4 (4) | 4 (4–8) |
| Vancomycin | 0.5 (0.5) | 1 (1) | 1 (1) | 1 (1) | 1 (1) | 1 (1) | 1 (1) | 1 (1) | 4 (4) |
a ATCC is the American Type Culture Collection strain 29213; b Marshfield Clinic S. aureus clinical isolate; c The MICs represent the modal values (range) from at least three separate runs.
Substituted groups in both phenyl X and phenyl Y did not improve the antibacterial activity (Table 3). We found that substituting bromine with a methyl group (compound 5741518) or a hydrogen (compound 5870804) at the same para-position of phenyl X eliminated activity. Other substitutions that eliminated activity were: (1) changing the carboxylate group of phenyl Y to a carboxylate ester (compound 2083-1665) and (2) moving bromine to the meta-position instead of the ortho-position of phenyl X (compound 2083-1773).
Table 3.
Structure–activity relationship of lead compound 5376 and its derivatives targeting drug-resistant Staphylococcus aureus.
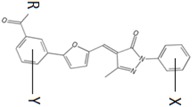 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R | X | Y | MIC (µg/mL) MC7606 | MIC (µg/mL) MC7769 | MIC (µg/mL) MC7827 | MIC (µg/mL) MC7846 | Docking Energy (kcal/mol) |
| 5376 | O | 4-Br | 4-Cl | 2 | 4 | 8 | 4 | −9.3 |
| 5741518 | O | 4-CH3 | 4-Cl | >128 | >128 | >128 | >128 | −8.7 |
| 5870804 | O | 4-Br | H | 4 | 64 | 32 | 16 | −10.9 |
| 2082-0182 | O | H | 4-Cl | >128 | >128 | >128 | >128 | −9.0 |
| 2083-1665 | OCH3 | 4-Br | 4-Cl | >128 | >128 | >128 | >128 | −8.6 |
| 2083-1773 | O | 3-Br | 4-Cl | >128 | >128 | >128 | >128 | −9.8 |
2.3. Molecular Modeling
Docking was performed into two different crystal structures: the structure for S. aureus TrxR (PDB file 4GCM) and the structure for M. tuberculosis TrxR (PDB file 2A87) [7]. Ligand interactions with specific residues of the protein binding site help elucidate the molecular basis of inhibition by our lead compound 5376 with both structures (Figure 2A,B). Based on the molecular model of the compound docked in the M. tuberculosis crystal structure (2A87), the guanidinium group of Arg186 forms an ionic bond with the carboxylate end of the compound (Figure 3). Both Arg127 and Arg292 interact with 5376 via their backbone, while Asn266 is a side chain donor. Interestingly, the 5376 compound docked in a slightly different conformation with the S. aureus crystal structure (4GCM), predicting interactions with different residues (Figure 3B). In this docking pose, Arg175 and Arg180—two positively-charged arginines—participate in ionic interactions.
Figure 2.

Ligand interaction diagram between compound 5376 and two different thioredoxin reductase crystal structures as generated by Molecular Operating System (MOE). (A) Possible active site of M. tuberculosis TrxR (PDB file 2A87):5376 complex; (B) Possible active site of S. aureus TrxR (PDB file 4GCM):5376 complex.
Figure 3.


Possible molecular interactions between compound 5376 and M. tuberculosis thioredoxin reductase (2A87) and S. aureus thioredoxin reductase (4GCM). (A) Molecular model of compound 5376 docked in PDB file 4GCM; (B) molecular model of compound 5376 docked n PDB file 2A87. TrxR is depicted as ribbons, and compound 5376 is shown along the NADPH site with the co-crystallized FAD adjacent to it. The inset shows the interacting residues within 12 Å of the ligand.
2.4. Compound 5376 Inhibits M. tuberculosis TrxR Activity and Binding of M. tuberculosis Thioredoxin C (TrxC) to M. tuberculosis TrxR
A DTNB (5,5′-dithiobis(2-nitrobenzoic acid)) reduction assay was used to measure the inhibitory capability of compound 5376 in vitro. In this assay, thioredoxin (TrxC) is used as a substrate for TrxR. If a compound binds in place of TrxC, it should prevent the production of TNB (1,3,5-trinitrobenzene) in a dose-dependent manner. To test if compound 5376 bound only the NADPH/TrxC binding site, we examined rates of TrxR-catalyzed NADP reduction at various concentrations of TrxC and 5376. Data fit a competitive inhibition model that assumes that both TrxC and 5376 compete for the same binding site on TrxR (Figure 4A). In this model, 5376 is a competitive inhibitor of TrxC, with a Ki (5376) of 1.26 ± 0.11 µM and a Km (TrxC) of 29.7 ± 6.5 µM. Competitive inhibition was also clear when reciprocal data were fit by linear regression on a Lineweaver–Burk plot (Figure 4B).
Figure 4.


Effect of 5376 on M. tuberculosis TrxR-catalyzed DTNB (5,5′-dithiobis(2-nitrobenzoic acid)) reduction by thioredoxin (TrxC). Initial rates measured at indicated TrxC and 5376 concentrations. (A) Data were fitted by non-linear regression; (B) Data on a Lineweaver–Burk plot with lines fitted by linear regression.
Interestingly, other research groups have also shown that the Trx system is a novel drug target. A class of benzoisoselenazol compounds were shown to be potent inhibitors of the thioredoxin reductase of Bacillus anthracis, the causative agent for anthrax [11]. Biochemical studies found that ebselen and its analogs inhibited the activity of TrxR at a subnanomolar concentration. This study also found that these compounds exhibited a high-barrier against resistance development in Bacillus subtilis, S. aureus, Bacillus cereus, and M. tuberculosis. Another study proposed that the mechanism by which ebselen inhibits is by covalent modification of the enzyme’s cysteine residues [12].
In addition, auranofin—an orally bioavailable FDA-approved anti-rheumatic drug—was shown to have potent bactericidal activities against both replicating and non-replicating M. tuberculosis based on a cell-based screen for bactericidal compounds [15]. Auranofin was also active against other Gram-positive bacteria, including B. subtilis, E. faecalis, E. faecium, and S. aureus. Their biochemical study also showed that auranofin inhibited the bacterial TrxR. They hypothesized that auranofin decreases the reducing capacity of target bacteria, thereby sensitizing them to oxidative stress from the host’s immune system.
The efficacy of auranofin and five of its analogs were further evaluated in vitro against H. pylori. Auranofin completely inhibited bacterial growth at 1.2 μM and inhibited the activity purified H. pylori TrxR in a cell-free assay (IC50 ~88 nM). However, the compounds were not as toxic against HEK-293T human embryonic kidney cells. Interestingly, they discovered a synergy between auranofin and amoxicillin, suggesting that H. pylori infections can be treated by targeting both the reductive enzyme TrxR and cell wall synthesis.
We have identified a new class of antibiotics built on a pyrazolone scaffold that inhibits the growth of drug resistant S. aureus strains, potentially by inhibiting the S. aureus TrxR protein. Future work will try to confirm that the mechanism of action of 5376 is indeed the inhibition of the S. aureus TrxR as well as optimize the 5376 lead molecule.
3. Experimental Section
3.1. Bacterial Species
For the routine MIC screening, a panel of four American Type Culture Collection (ATCC) strains was tested. They included S. aureus ATCC 29213, E. faecalis ATCC 29212, E. coli ATCC 25922, and P. aeruginosa ATCC 27853. The following S. aureus strains were then used for additional MIC analysis to assess anti-S. aureus activity: N315 (USA100), MW2 (USA400), and Newman (MSSA) (provided by Jean Lee (Brigham and Young Hospital, Boston, MA, USA)); JE2 (USA300 provided by the Network on Antimicrobial Resistance in Staphylococcus aureus strain repository), and multidrug-resistant MRSA clinical isolates MC7606 (USA300), MC7769 (USA100), MC7827 (USA100), and MC7846 (a vancomycin-intermediate resistance USA100) were supplied by Marshfield Clinic. Strains of M. smegmatis and M. marinum were obtained from the University of Wisconsin-La Crosse culture collection.
3.2. Minimum Inhibitory Concentration (MIC)/Minimum Bactericial Concentration (MBC)
In vitro MIC and MBC determinations were performed on the compounds according to the Clinical and Laboratory Standards Institute (CLSI) guidelines [20] for ATCC strains of S. aureus, E. faecalis, E. coli, and P. aeruginosa that were screened. Tetracycline, oxacillin, and vancomycin (Sigma-Aldrich, St. Louis, MO, USA) were included as control antibiotics and correlated with established MIC values for the ATCC strains. All anti-Mycobacterium MIC evaluations were performed using MIC assays in Middlebrook 7H9 broth with 10% oleic acid albumin dextrose complex (OADC) as previously described [21]. Isoniazid was used as the positive control for the mycobacterial MICs. All MIC numbers are a compilation of modal values plus range from at least three separate runs.
3.3. Thioredoxin Reductase-DTNB Assay
The activity of M. tuberculosis TrxR was examined using a DTNB (5,5′-dithiobis(2-nitrobenzoic acid)) reduction assay [22]. TrxR and TrxC were expressed and purified as previously described [23]. In the assay, TrxR used NADPH to oxidize TrxC, which spontaneously reduced DTNB. The production of TNB (1,3,5-trinitrobenzene) was monitored by measuring its absorbance at 412 nm in a SpectraMax M5 multi-mode plate reader (Molecular Devices, Sunnyvale, CA, USA). The assay was used to examine the inhibition kinetics of 5376. Each 200 µL assay contained 200 mM KPO4 pH 7.2 mM
EDTA, 2 µM bovine serum albumin, 1 mM DTNB, 500 µM NADPH, 10 nM TrxR, and 1% dimethyl sulfoxide. Concentrations of CSD3 5376 tested were 30, 10, 3, and 0 µM, and concentrations of TrxC tested were a seven-point 1:2 dilution series ranging between 16 µM and 0.25 µM, in duplicate Assays were initiated by the addition of TrxR. Assays were done in clear 96-well non-treated microplates (Greiner BioOne 655101, Monroe, LA, USA). Since 5376 has color, compound background absorbance control wells were included that lacked TrxR and TrxC, but included all other assay components. Reaction progress was monitored for 30 min. The compound background absorbance for each concentration was subtracted from experimental wells for each time point before calculating the rate of reaction. The absorbance values were converted to concentration of TNB produced using the extinction coefficient 14,150 M−1·cm−1. The rate of reaction was calculated after 8.25 min to assure a linear rate (see Appendix A Figure A1). The data were fitted to a competitive inhibition model using Graphpad Prism 6.04.
3.4. Docking
The structures of the TrxR proteins in this study were obtained from the protein data bank (www.pdb.org) [24]. The accession code for the M. tuberculosis thioredoxin reductase is 2A87, and the accession code for the S. aureus subsp. aureus Mu50 strain is 4GCM. Compounds were docked into PDB files 2A87 and 4GCM that were stripped of their ligands, water molecules, and counterions. We have decided to dock with FAD included in the structures, since both crystal structures have FAD co-crystallized. Using UCSF Chimera′s Dock Prep module, histidine protonation states were calculated, and incomplete side chains were automatically filled [25]. Three-dimensional conformations of the compounds were generated using Open Babel, and were positioned using a rigid receptor and flexible ligand orienting code in AutoDock 4.2.1 (AD4, Scripps Research Institute, La Jolla, CA, USA) [26]. Both receptor and ligand were seeded with Gasteiger charges. AD4 was used for all dockings in this study. In general, the docking parameters were kept to their default values. However, the number of independent genetic algorithm runs was increased from 10 to 50, and was processed using the built-in clustering analysis with a 2.0 Å cutoff [27].
4. Conclusions
The in silico docking analysis against the M. tuberculosis TrxR identified in vitro enzyme inhibitors, but failed to generate a viable lead compound against M. tuberculosis due to negative MIC results; however, our results have identified and may encourage the development of more potent and specific analogs of a pyrazolone-based lead molecule 5376 as possible S. aureus antibacterial agents. The pyrazolone scaffold of lead compound 5376 showed a positive MIC result that was sufficient to inhibit several strains of S. aureus, including multidrug resistant clinical isolates. Although the 5376 compound did not have the same predicted affinity for the S. aureus TrxR active site as compared to the M. tuberculosis TrxR, it was still a close fit and suggests but does not prove that compound 5376 targets the S. aureus TrxR. Future studies will focus on confirming that the mechanism of action involves inhibiting the S. aureus TrxR enzyme and optimizing the pyrazolone compound to increase efficacy and in vivo properties such as pharmacokinetics.
Acknowledgments
This study was funded in part by National Institutes of Health grants AI101975, GM118304 and HL112639. We wish to thank Terrence Neumann for assistance with the initial M. tuberculosis protein docking; Jean Lee for the MW2, N315, and Newman strains, and NARSA for the JE2 strain.
Abbreviations
The following abbreviations are used in this manuscript:
| DTNB | Ellman′s Reagent (5,5-dithio-bis-(2-nitrobenzoic acid)) |
| DTT | 1,4-Dithiothreitol |
| TrxR | Thioredoxin Reductase |
| Trx | Thioredoxin |
| PDB | Protein Data Bank |
Appendix A
Figure A1.

Reaction progress curves of wells containing varying concentrations of TrxC and 30 μM 5376. The rate of the reaction was calculated after 8.8 minutes of data collection, which is where progress curves become linear.
Author Contributions
L.L. and K.E.E. ran the MIC assays and interpreted the results. W.R.S. designed the MIC experiments, helped interpret the results, and assisted with the editing of the paper. M.E.S. selected the clinical isolates and helped edit the paper. A.M.H. ran the DTNB assays, interpreted the results and helped edit the paper. C.J.B. ran the initial docking of compounds. K.S.K. expressed and purified TrxA and TrxC proteins. N.L.S. ran the succeeding docking of compounds, analyzed the relationships between structure and antibacterial potency and wrote the paper. D.S.S. helped interpret the results and assisted with the editing of the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Finkel T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams C.H., Jr. Mechanism and structure of thioredoxin reductase from Escherichia coli. FASEB J. 1995;9:1267–1276. doi: 10.1096/fasebj.9.13.7557016. [DOI] [PubMed] [Google Scholar]
- 3.Holmgren A. Thioredoxin. Annu. Rev. Biochem. 1985;54:237–271. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- 4.Saccoccia F.A.F., Boumis G., Carotti D., Desiato G., Miele A.E., Bellelli A. Thioredoxin Reductase and Its Inhibitors. Curr. Protein Pept. Sci. 2014;15:621–646. doi: 10.2174/1389203715666140530091910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zumla A., Nahid P., Cole S.T. Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 2013;12:388–404. doi: 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
- 6.Kruh N.A., Troudt J., Izzo A., Prenni J., Dobos K.M. Portrait of a pathogen: The Mycobacterium tuberculosis proteome in vivo. PLoS ONE. 2010;5:e13938. doi: 10.1371/journal.pone.0013938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akif M., Suhre K., Verma C., Mande S.C. Conformational flexibility of Mycobacterium tuberculosis thioredoxin reductase: Crystal structure and normal-mode analysis. Acta Crystallogr. 2005;61:1603–1611. doi: 10.1107/S0907444905030519. [DOI] [PubMed] [Google Scholar]
- 8.Suaya J.A., Mera R.M., Cassidy A., O’Hara P., Amrine-Madsen H., Burstin S., Miller L.G. Incidence and cost of hospitalizations associated with Staphylococcus aureus skin and soft tissue infections in the United States from 2001 through 2009. BMC Infect. Dis. 2014 doi: 10.1186/1471-2334-14-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uziel O., Borovok I., Schreiber R., Cohen G., Aharonowitz Y. Transcriptional regulation of the Staphylococcus aureus thioredoxin and thioredoxin reductase genes in response to oxygen and disulfide stress. J. Bacteriol. 2003;186:326–334. doi: 10.1128/JB.186.2.326-334.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang S.Y., Lin S.Y., Chiang I.C., Shih Y.L. Quantitative inner membrane proteome datasets of the wild-type and the Δmin mutant of Escherichia coli. Data Brief. 2016;8:304–307. doi: 10.1016/j.dib.2016.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gustafsson T.N., Osman H., Werngren J., Hoffner S., Engman L., Holmgren A. Ebselen and analogs as inhibitors of Bacillus anthracis thioredoxin reductase and bactericidal antibacterials targeting Bacillus species, Staphylococcus aureus and Mycobacterium tuberculosis. Biochim. Biophys. Acta. 2016;1860:1265–1271. doi: 10.1016/j.bbagen.2016.03.013. [DOI] [PubMed] [Google Scholar]
- 12.Mukherjee S., Weiner W.S., Schroeder C.E., Simpson D.S., Hanson A.M., Sweeney N.L., Marvin R.K., Ndjomou J., Kolli R., Isailovic D., et al. Ebselen inhibits hepatitis C virus NS3 helicase binding to nucleic acid and prevents viral replication. ACS Chem. Biol. 2014;9:2393–2403. doi: 10.1021/cb500512z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu J., Vlamis-Gardikas A., Kandasamy K., Zhao R., Gustafsson T.N., Engstrand L., Hoffner S., Engman L., Holmgren A. Inhibition of bacterial thioredoxin reductase: An antibiotic mechanism targeting bacteria lacking glutathione. FASEB J. 2013;27:1394–1403. doi: 10.1096/fj.12-223305. [DOI] [PubMed] [Google Scholar]
- 14.Owings J.P., McNair N.N., Mui Y.F., Gustafsson T.N., Holmgren A., Contel M., Goldberg J.B., Mead J.R. Auranofin and N-heterocyclic carbene gold-analogs are potent inhibitors of the bacteria Helicobacter pylori. FEMS Microbiol. Lett. 2016 doi: 10.1093/femsle/fnw148. [DOI] [PubMed] [Google Scholar]
- 15.Harbut M.B., Vilcheze C., Luo X., Hensler M.E., Guo H., Yang B., Chatterjee A.K., Nizet V., Jacobs W.R., Jr., Schultz P.G., et al. Auranofin exerts broad-spectrum bactericidal activities by targeting thiol-redox homeostasis. Proc. Natl. Acad. Sci. USA. 2015;112:4453–4458. doi: 10.1073/pnas.1504022112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hikisz P., Szczupak L., Koceva-Chyla A., Gu Spiel A., Oehninger L., Ott I., Therrien B., Solecka J., Kowalski K. Anticancer and Antibacterial Activity Studies of Gold(I)-Alkynyl Chromones. Molecules. 2015;20:19699–19718. doi: 10.3390/molecules201119647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walters W.P., Stahl M.T., Murcko M.A. Virtual screening—An overview. Drug Discov. Today. 1998;3:160–178. doi: 10.1016/S1359-6446(97)01163-X. [DOI] [Google Scholar]
- 18.Meng X., Zhang H., Mezei M., Cui M. Molecular Docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Discov. 2011;7:146–157. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cosconati S., Forli S., Perryman A.L., Harris R., Goodsell D.S., Olson A.J. Virtual Screening with AutoDock: Theory and Practice. Expert Opin. Drug Discov. 2010;5:597–607. doi: 10.1517/17460441.2010.484460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patel J C.I.F., Bradford P., Eliopoulos G., Hindler J. Performance Standards for Antimicrobial Susceptibility Testing: Seventeenth Informational Supplement. Clinical and Laboratory Standards Institute; Wayne, HJ, USA: 2007. pp. 13–231. [Google Scholar]
- 21.Shahjahan Kabir M., Engelbrecht K., Polanowski R., Krueger S.M., Ignasiak R., Rott M., Schwan W.R., Stemper M.E., Reed K.D., et al. New classes of Gram-positive selective antibacterials: Inhibitors of MRSA and surrogates of the causative agents of anthrax and tuberculosis. Bioorg. Med. Chem. Lett. 2008;18:5745–5749. doi: 10.1016/j.bmcl.2008.09.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akif M., Khare G., Tyagi A.K., Mande S.C., Sardesai A.A. Functional studies of multiple thioredoxins from Mycobacterium tuberculosis. J. Bacteriol. 2008;190:7087–7095. doi: 10.1128/JB.00159-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olson A.L., Neumann T.S., Cai S., Sem D.S. Solution structures of Mycobacterium tuberculosis thioredoxin C and models of intact thioredoxin system suggest new approaches to inhibitor and drug design. Proteins. 2013;81:675–689. doi: 10.1002/prot.24228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berman H., Henrick K., Nakamura H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003 doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- 25.Lang P.T., Brozell S.R., Mukherjee S., Pettersen E.F., Meng E.C., Thomas V., Rizzo R.C., Case D.A., James T.L., Kuntz I.D. DOCK 6: Combining techniques to modelRNA-small molecule complexes. RNA. 2009;15:1219–1230. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morris G.M., Goodsell D.S., Halliday R.S., Huey R., Hart W.E., Belew R.K., Olson A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998;19:1639–1662. doi: 10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B. [DOI] [Google Scholar]