

Abstract
Photoreceptor degeneration is a cause of irreversible vision loss in incurable blinding retinal diseases including retinitis pigmentosa (RP) and atrophic age-related macular degeneration. We found in two separate mouse models of photoreceptor degeneration that tamoxifen, a selective estrogen receptor modulator and a drug previously linked with retinal toxicity, paradoxically provided potent neuroprotective effects. In a light-induced degeneration model, tamoxifen prevented onset of photoreceptor apoptosis and atrophy and maintained near-normal levels of electroretinographic responses. Rescue effects were correlated with decreased microglial activation and inflammatory cytokine production in the retina in vivo and a reduction of microglia-mediated toxicity to photoreceptors in vitro, indicating a microglia-mediated mechanism of rescue. Tamoxifen also rescued degeneration in a genetic (Pde6brd10) model of RP, significantly improving retinal structure, electrophysiological responses, and visual behavior. These prominent neuroprotective effects warrant the consideration of tamoxifen as a drug suitable for being repurposed to treat photoreceptor degenerative disease.
SIGNIFICANCE STATEMENT Photoreceptor degeneration is a cause of irreversible blindness in a number of retinal diseases such as retinitis pigmentosa (RP) and atrophic age-related macular degeneration. Tamoxifen, a selective estrogen receptor modulator approved for the treatment of breast cancer and previously linked to a low incidence of retinal toxicity, was unexpectedly found to exert marked protective effects against photoreceptor degeneration. Structural and functional protective effects were found for an acute model of light-induced photoreceptor injury and for a genetic model for RP. The mechanism of protection involved the modulation of microglial activation and the production of inflammatory cytokines, highlighting the role of inflammatory mechanisms in photoreceptor degeneration. Tamoxifen may be suitable for clinical study as a potential treatment for diseases involving photoreceptor degeneration.
Keywords: degeneration, microglia, neuroprotection, photoreceptor, retinitis pigmentosa, tamoxifen
Introduction
Degeneration of photoreceptors, the light-sensitive neurons in the retina, is a prominent feature in diseases of the retina, contributing significantly to irreversible blindness worldwide (Congdon et al., 2004; Jonas et al., 2014). Photoreceptor degeneration occurs in a variety of retinal conditions ranging from atrophic age-related macular degeneration (AMD) and diabetic maculopathy, in which death of macular photoreceptors results in central vision loss, to retinitis pigmentosa (RP), in which widespread photoreceptor degeneration across the entire retina leads to total blindness. The cellular mechanisms underlying photoreceptor degeneration in these diseases are incompletely understood and comprehensive treatments that slow down or arrest the progression of degeneration are still unavailable (Holz et al., 2014; Wert et al., 2014). Currently, affected patients such as those with atrophic AMD and RP typically receive no treatment and progress with time to visual deficits ranging from severe vision loss to total blindness.
Studies of human disease and animal models of photoreceptor degeneration have uncovered that photoreceptor loss is often accompanied by chronic neuroinflammatory changes, including increases in retinal levels of proinflammatory cytokines (Yoshida et al., 2013a, 2013b) and retinal microglial infiltration into the photoreceptor layer to come into close proximity with degenerating photoreceptors (Roque et al., 1996; Gupta et al., 2003). In recent work, we have found that these infiltrating retinal microglia contribute actively to photoreceptor demise via the phagocytotic clearance of viable photoreceptors and the secretion of proinflammatory cytokines that potentiate photoreceptor apoptosis (Zhao et al., 2015; Zabel et al., 2016). In rodent models of disease, measures that modulate microglial phagocytic activity (Zabel et al., 2016) or activation status (Peng et al., 2014; Scholz et al., 2015) can effectively ameliorate the rate of photoreceptor loss. Therefore, agents that can diminish microglial contributions to photoreceptor degeneration are promising as potential therapeutic agents, particularly if these are approved drugs with favorable biodistributions and well characterized side-effect profiles and can be readily repurposed for a new indication of photoreceptor neuroprotection (Amantea and Bagetta, 2016).
In this study, we discover that tamoxifen, a well established and approved drug widely used for the treatment of estrogen-receptor-positive metastatic breast cancer, provided potent neuroprotection to photoreceptors in two separate rodent models for photoreceptor degeneration. As a selective estrogen receptor modulator (SERM), tamoxifen is known to exert variable, context-dependent agonist and antagonist effects on estrogen receptor (ER)-expressing cells (Lonard and Smith, 2002); however, its effects on retinal cells are not fully understood. We find here that, contrary to the long-standing association between tamoxifen and retinal toxicity (Nayfield and Gorin, 1996), tamoxifen treatment in the context of retinal degenerative disease models decreased retinal microglia activation and production of inflammatory cytokines and as a consequence reduced microglial-mediated toxicity to photoreceptors. These microglial effects in vivo were correlated with marked rescue of photoreceptor structure and function in an acute model of light-induced photoreceptor injury as well as in a genetic model for RP. Based on these findings, we propose that tamoxifen may be considered as a therapeutic candidate for the neuroprotection of endangered photoreceptors, specifically for inherited retinal degenerations and AMD and also more generally for retinal diseases in which neuroinflammation drives photoreceptor loss (Yu et al., 2015; Russo et al., 2016).
Materials and Methods
Experimental animals.
Young adult wild-type (WT) C57BL/6J mice and mice homozygous for the Pde6brd10 (rd10) mutation were obtained from The Jackson Laboratory. Postnatal rd10 mice (21–50 d of age) and young adult WT mice (2–3 months of age) of both sexes were raised in cyclic light (∼100 lux, 12:12 h) in a National Institutes of Health animal facility. All experiments were conducted according to protocols approved by a local Institutional Animal Care and Use Committee and adhered to the Association for Research in Vision and Ophthalmology statement on animal use in ophthalmic and vision research. For the tamoxifen-treated groups, mice were provided with tamoxifen-supplemented mouse chow (500 mg/kg; Envigo) in place of standard chow for the duration of experiment.
Mouse model of retinal light injury (LI).
Experimental animals were dark adapted in a dark room for 7 d and then subjected to pupillary dilation with topical tropicamide (1%; Alcon) and phenylephrine (10%; Alcon). After full dilation, animals were exposed to 2 × 104 lux of diffuse white fluorescent light (Sunlite Manufacturing) for 2 h. After light exposure, mice were maintained in typical conditions of ambient cyclic light (∼100 lux, 12:12 h) under which the animals were housed. For tamoxifen treatment groups, animals were fed a tamoxifen-supplemented chow beginning 7 d before LI and maintained on the same diet thereafter. Control groups not administered tamoxifen were fed standard mouse chow in separate cages at all time points.
In vivo optical coherence tomographic (OCT) imaging and fundus autofluorescence imaging.
Mice were anesthetized with intraperitoneal ketamine (90 mg/kg) and xylazine (8 mg/kg) and their pupils were dilated. Retinal structure was assessed using an OCT imaging system (Bioptigen; Diver Software). To document retinal changes in the LI model, animals were imaged before dark adaptation and subsequently at different times after light exposure. Volume scans of 1.4 mm by 1.4 mm centered on the optic nerve (1000 A-scans/horizontal B-scan, 33 horizontal B-scans, average of three frames per B-scan, each spaced 0.0424 mm apart) were captured. Retinal thickness measurements in each quadrant of a circular grid of diameter 1.2 mm were computed using the manufacturer's software. Total retinal thickness, measured from the nerve fiber layer to the retinal pigment epithelium (RPE) layer, and outer retinal thickness, measured from the outer plexiform layer to the inner surface of the RPE layer, were calculated from OCT images after automated retinal segmentation (Bioptigen; Diver Software). To document retinal changes in rd10 mice, additional horizontal and vertical linear scans (1.4 mm width, 1000 A-scans/B-scan, average of 20 frames per B-scan) were obtained with the scan centered on the optic nerve head. In these vertical and horizontal B-scans, the area of outer nuclear layer in the scan was circumscribed and measured manually. Average outer retinal thickness was computed by dividing the outer retinal area with the length of measured retina. Fundus autofluorescence imaging was performed using a confocal scanning laser ophthalmoscopy (cSLO; Heidelberg Engineering) with 488 nm wavelength excitation. Fundus images were obtained over the central 30° angle with the field centered on the optic nerve head.
Electroretinographic (ERG) analysis.
ERGs were recorded using an Espion E2 system (Diagnosys). Mice were anesthetized as described above after dark adaptation overnight. Pupils were dilated and a drop of proparacaine hydrochloride (0.5%; Alcon) was applied on cornea for topical anesthesia. Flash ERG recordings were obtained simultaneously from both eyes with gold wire loop electrodes, with the reference electrode placed in the mouth and the ground subdermal electrode at the tail. ERG responses were obtained at increasing light intensities over the ranges of 1 × 10−4 to 10 cd/s/m2 under dark-adapted conditions and 0.3 to 100 cd/s/m2 under a rod-saturating background light. The stimulus interval between flashes varied from 5 s at the lowest stimulus strengths to 60 s at the highest ones. Two to 10 responses were averaged depending on flash intensity. ERG signals were recorded with 0.3 Hz low-frequency and 300 Hz high-frequency cutoffs sampled at 1 kHz. Analysis of a-wave and b-wave amplitudes was performed using customized Espion ERG Data Analyzer software (version 2.2) that digitally filters out high-frequency oscillatory potential wavelets. The a-wave amplitude was measured from the baseline to the negative peak and the b-wave was measured from the a-wave trough to the maximum positive peak. For the LI model, ERGs were recorded at 1 week after light exposure. For rd10 mice, ERGs were recorded at postnatal day 29 (P29) and P50. Statistical significance between nontreated control and tamoxifen-treated mice was analyzed using a two-way ANOVA.
To test whether tamoxifen treatment affected rod photoreceptor dark-adaptation function, animals administered the tamoxifen diet for 7 d and age-matched control animals (not fed tamoxifen) were dark adapted overnight and then subjected to moderate visual pigment bleaching using the background light of a Ganzfeld chamber (1000 cd/m2) for 30 s. Immediately after the bleach, single-flash ERGs at a flash intensity of 10 cd/s/m2 were recorded every 2 min for 40 min to chart the dynamics of the recovery of the a-wave amplitude. To analyze the data, the recovery of a-wave amplitude after bleaching is plotted against the post-bleach time. Statistical significance between nontreated control and tamoxifen-treated mice was analyzed using a two-way ANOVA.
Measurement of optokinetic response (OKR).
OKRs of awake, behaving mice to visual stimuli in the form of a moving grating were measured using a custom-designed apparatus (Kretschmer et al., 2015). OKRs were measured in rd10 mice with tamoxifen treatment starting at P21 and nontreated control at P49 (n = 7 animals in control group, n = 9 animals in tamoxifen-treated group) as described previously (Wang et al., 2016). Briefly, mice were positioned on a platform and presented with sinusoidal gratings at maximum contrast at spatial frequencies of 0.025, 0.05, 0.1, 015, 0.2, 0.25, 0.3, 0.35, 0.4, 0.425, and 0.45 cycles/degree at a stimulus speed of 12deg/s on 4 LCD screens that surrounded the test animal. Stimuli presentation was controlled using an open source software program to produce a virtual cylinder that maintains a constant distance between the grating and horizontal visual field of the tested animal located at the center of the apparatus.
To measure elicited optokinetic motor reflexes (OMR), the head movements of the unrestrained tested animal were video-recorded by a camera placed above the animal and analyzed by an algorithm that tracks the position of the mouse's head. These data are used to automatically readjust the presentation of the stimuli to changing head positions to keep the size of the gratings constant (the analysis software used, OmrArena, was modified from a version previously published; Kretschmer et al., 2013). OMR measurements were obtained objectively using an automated approach during offline analysis and do not involve subjective grading or input from a human observer. OMR tracking behavior was quantified as the ratio of the total amount of time the animal's head moved in the stimulus direction and the amount of time that the head moved against the stimulus direction (Tcorrect/Tincorrect). Each animal was measured under each condition 5 times for 1 min. OMR was recorded under photopic (9 × 1010 Q/s/cm2) light conditions. Estimations of visual threshold, defined as the spatial frequency corresponding to 25% of the maximum optomotor response, were also calculated. Statistical significance between nontreated control and tamoxifen-treated mice was analyzed using unpaired t test.
Immunohistochemistry and TUNEL of retinal tissue.
Mice were killed by CO2 inhalation and their eyes removed. Enucleated eyes were dissected to form posterior segment eye cups, which were then fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) for 2–4 h at 4°C. Eye cups were either processed for vibratome sectioning (100-μm-thick sections, VT1000; Leica) or dissected to form retinal flat mounts. Flat-mounted retinas or retinal sections were blocked overnight in blocking buffer containing 6% normal donkey serum and 0.5% Triton X-100 in PBS at room temperature. Primary antibodies (Iba1, 1:500, catalog #019-19741, Wako; CD68, 1:500, catalog #MCA1957, RRID:AB_322219, Bio-Rad, glial fibrillary acidic protein (GFAP, 1:800, catalog #13-0300, RRID:AB_86543Invitrogen) were diluted in blocking buffer and applied overnight for sections and flat mounts at 4°C on a shaker. After washing in 1× PBS with 0.5% Triton X-100, sections were incubated overnight with secondary antibodies (Alexa Fluor-488 (or Alexa Fluor 594)-conjugated donkey anti-rabbit or rat IgG, respectively (Jackson ImmunoResearch Laboratories catalog #711-546-152, RRID:AB_2340619, catalog #711-586-152 RRID:AB_2340622, catalog #712-546-153 RRID:AB_2340686), and DAPI (1:500, catalog #D3571, RRID:AB_2307445, Thermo Fisher Scientific) to label cell nuclei. Apoptosis of retinal cells was assayed using a terminal deoxynucleotidyl transferase dUTP Nick End Labeling (TUNEL) assay (in situ cell death detection kit, TMR red; Roche) according to the manufacturer's specifications. Stained retinal sections were imaged with confocal microscopy (FluoView 1000; Olympus). Multiplane z-series were collected using a 20× objective; each z-series spanned 20 μm in depth, with each section spaced 1 μm apart. Confocal image stacks were viewed and analyzed with FV100 Viewer Software (Olympus) and ImageJ. Mean thickness measurements of the outer nuclear layer (ONL) and cell densities (TUNEL+, Iba1+, and CD68+ cells) were computed over 20× imaging fields obtained in the mid-peripheral retina in the superotemporal quadrants.
Cell culture and measurement of cell viability.
Retinal microglia were isolated from 1- to 2-month-old C57BL/6J wild-type mice as described previously (Ma et al., 2013). Briefly, retinal cells were dissociated by digestion in 2% papain, followed by trituration and centrifugation. Resuspended cells were transferred into 75 cm2 flasks containing Dulbecco's Modified Eagle Medium (DMEM): NutrientMixture F-12 medium with 10% fetal bovine serum (FBS) (Invitrogen) and nonessential amino acids solution (Sigma-Aldrich). After overnight culture, the medium and any floating cells were discarded and replaced with fresh medium. When the cells grew to confluence, culture flasks were shaken gently to detach microglial cells, which were subcultured in six-well plates. Subcultured microglia (2.5 × 105 cells/well in a 6-well plate) were exposed to tamoxifen (0, 1, 5, and 10 μg/ml; Sigma-Aldrich) for 2 h, followed by 1 μg/ml lipopolysaccharide (LPS; Sigma-Aldrich) for 16 h and then assessed for protein expression of inflammatory cytokines.
Murine microglial cell line (BV-2) cells and photoreceptor-like cell line (661W, gift from Dr. Muyyad Al-Ubaidi, University of Oklahoma Health Sciences Center) were used in this study. BV-2 cells were plated in 6-well plates at the density of 4 × 105/well and cultured for 24 h in DMEM (Life Technologies) containing 5% heat inactivated FBS (Life Technologies) at 37°C in a humidified atmosphere of 5% CO2. BV-2 cells were preincubated for 24 h with tamoxifen (1, 5, or 10 μg/ml) or 0.5% ethanol as a vehicle control. After preincubation with the proteasome inhibitor ALLN (100 μg/ml; Santa Cruz Biotechnology) for 30 min, BV-2 cells were stimulated with 50 ng/ml LPS for 6 h before conditioned media were collected. 661W cells, which were maintained in DMEM (Invitrogen) containing 15% FBS, 165 nm hydrocortisone21-hemisuccinate (Sigma-Aldrich), 255 nm progesterone (Sigma-Aldrich), 0.4 mm putrescine (Sigma-Aldrich), 0.008% β-mercaptoethanol, and 2% penicillin-streptomycin, were transferred into 96-well plates at 4 × 104 cells/well and allowed to reach 80–90% confluence before further use. In experiments to investigate the ability of tamoxifen to decrease microglia-mediated neurotoxicity, 661W cells were incubated for 48 h with culture supernatants from BV2 cells cultured under the following conditions: control, 5 μg/ml tamoxifen, 50 ng/ml LPS, 1 μg/ml tamoxifen + 50 ng/ml LPS, 5 μg/ml tamoxifen + 50 ng/ml LPS, or 10 μg/ml tamoxifen + 50 ng/ml LPS. Cell viability of 661W photoreceptors were assessed using a MTT cell proliferation assay kits (ATCC) following the manufacturer's specifications. In pharmacological experiments to phenocopy or inhibit the neuroprotective effect of tamoxifen, the following reagents were used: β-estradiol, 100 nm and 10 μm (Sigma-Aldrich); diethylstilbestrol, 1 μm (Sigma-Aldrich); 4,4′,4′′-(4-propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol (PPT), 1 μm; diarylpropionitrile (DPN), 10 nm; 7α,17β-n[9-[(4,4,5,5,5-pentafluoropentyl)sulfinyl]nonyl]estra-1,3,5(10)-triene-3,17-diol (ICI 182,780), 1 μm; 1,3-bis(4-hydroxyphenyl)-4-methyl-5-[4-(2-piperidinylethoxy)phenol]-1H-pyrazole dihydrochloride (MPP), 1 μm; and 4-[2-phenyl-5,7-bis(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-3-yl]phenol (PHTPP), 1 μm (all from Tocris Bioscience).
Measurement of cytokine levels.
Cultured cells or retinas were lysed by trituration in protein lysate buffer (Complete Ultra; Roche) with proteinase inhibitor mixture (Calbiochem) at 4°C. After sonication and centrifugation, protein concentration was measured (BCA protein assay kit; Pierce). Cytokine levels were determined using a Milliplex VR assay kit (Milliplex MAP mouse cytokine/chemokine magnetic bead panel, #MCYTOMAG-70K; Millipore) using the Luminex MAPIX system with data analysis using xPONENT 4.2 software (Luminex).
Measurement of tamoxifen levels.
Adult 8-week-old C57BL6 mice (n = 8) were maintained on tamoxifen-supplemented mouse chow (500 mg/kg) for 1–2 weeks and their retinas harvested, weighed, and stored at −80°C. Retinas from animals fed standard chow without tamoxifen served as negative controls (n = 3). Retinas were homogenized and centrifuged and 50 μl of the supernatant was dried down and reconstituted in 50 μl of 70:30 5 mm ammonium formate:acetonitrile. Samples were subjected with high-pressure liquid chromatography (LC) analysis (Model 2795; Waters) using a 2.1×50 mm Kinetex C18 2.6 μm 100A column (Phenomenex). Mass spectroscopy (Quattro Premier; Waters) with electrospray ionization in positive ion mode was performed. Tamoxifen MRM transition data were collected at 372.3→72.4 m/z and tamoxifen IS was monitored at 375.2→75.4 m/z both with cone voltage of 30, collision energy 25, and dwell time 0.2 s. Waters MassLynx version 4.1 software as used for Data acquisition was performed with MassLynx software (version 4.1; Waters) using the Quanlynx application manager (Waters) for quantitation.
Statistical analysis.
Statistical analyses were performed using statistical software (GraphPad). For comparisons involving two data columns, t tests (paired or unpaired) or nonparametric tests (Mann–Whitney) were used, depending on whether the data followed a Gaussian distribution as determined by normality tests. A normality test (D'Agostino and Pearson) was used to analyze the distribution of all datasets. For comparisons involving three or more data columns, a one-way ANOVA (with Dunnett's multiple-comparisons test) was used if the data followed a Gaussian distribution and a nonparametric Kruskal–Wallis test (with Dunn's multiple-comparisons test) was used if it did not. Datasets from tamoxifen-treated versus control diet-treated animals were compared using a two-way ANOVA. The χ2 statistic was used to compare the prevalence of retinal detachments in tamoxifen-treated versus control diet-treated animals. p < 0.05 was set as the basis for rejecting the null hypothesis. Error bars in graphs indicate SE.
Results
Model of light-induced injury results in photoreceptor apoptosis and loss of photoreceptor function
Wild-type adult (2–3 months old) C57BL/6J mice were subjected to LI and the effects of LI were analyzed using in vivo OCT imaging and histology. OCT imaging in the central retina revealed widespread overall retinal thinning 7 d after LI that was present most prominently in the superotemporal quadrant of the retina (Fig. 1A). OCT B-scans obtained in the superotemporal quadrant demonstrated a progressive thinning of the ONL over the first 7 d after LI that stabilized thereafter (Fig. 1B). At 7 d, shallow detachments of the retina from the RPE were also observed in areas of ONL thinning. Histological analysis in retinal sections obtained in equivalent retinal positions showed the prominent emergence of apoptotic TUNEL-positive photoreceptors at 1 and 3 d after LI, which decreased after 7 d (Fig. 1C,D). Quantification of ONL thicknesses in histological analyses of retinal sections corroborated the temporal pattern of retinal thinning observed on in vivo OCT imaging (Fig. 1E). LI also induced significant decrements in dark- and light-adapted ERG a- and b-wave amplitudes 7 d after LI (Fig. 1F,G), which indicated concurrent losses in rod and cone photoreceptor function. This current model of light-induced injury in adult C57BL/6J mice serves as an effective model of acute, time-limited, and photoreceptor-specific injury that is evident structurally as apoptotic photoreceptor loss and functionally as decreased responses to light stimuli.
Figure 1.
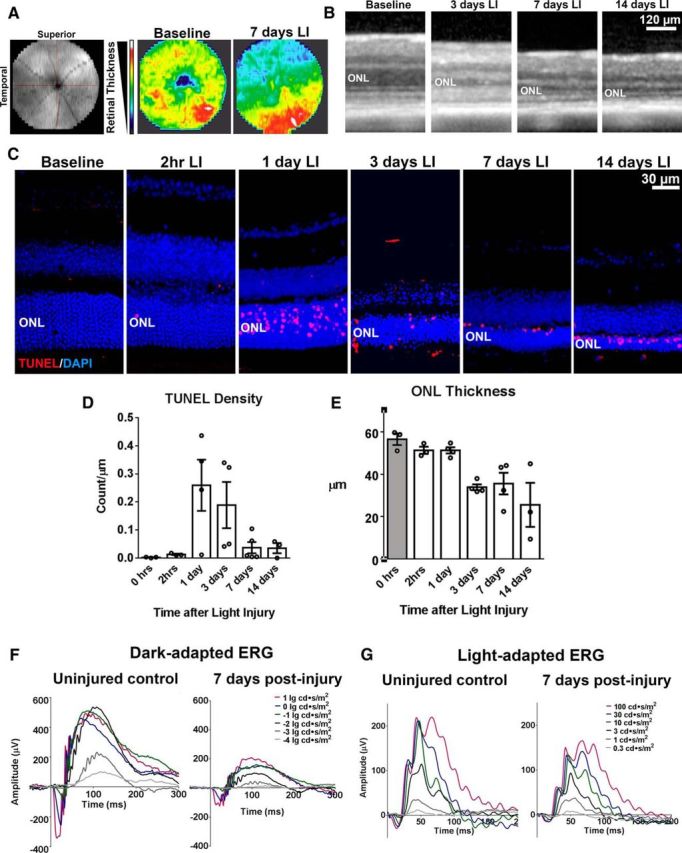
In vivo mouse model of acute light exposure-induced photoreceptor injury involves photoreceptor apoptosis, retinal atrophy, and loss of photoreceptor cell function. Young adult (2–3 months old) C57BL6J mice were dark reared for 1 week before being subjected to pupillary dilation and exposure to ambient white light at 20 × 103 lux for 2 h. The effects on retinal structure and function were evaluated at time points from 2 h to 14 d after LI. A, B, Retinal thickness and lamination was evaluated in vivo using OCT; 1.4 mm wide scan fields centered on the optic nerve were obtained. Heat maps (A) representing total retinal thickness of OCT images taken at baseline (before LI) and 7 d after LI demonstrated retinal thinning that was most marked in the superior temporal retina. Individual OCT B-scans from the superior temporal retina (B) show progressive thinning of the ONL from 3 d after LI. C–E, Histological analysis of retina in the superotemporal quadrant (1.25 mm from the optic nerve) revealed prominent emergence of apoptotic photoreceptors (as marked by TUNEL, red) in the ONL starting at 1 d after LI. Significant thinning of the ONL was observed starting at 3 d after LI. Plots in D and E show the time course of changes in the density of TUNEL-positive photoreceptors and ONL thickness after LI (column heights and error bars represent mean and SEM; n = 3–4 animals per time point). F, G, Representative ERG recordings demonstrating functional decreases at 7 d after LI relative to uninjured control mice for a- and b-wave amplitudes in dark- and light-adapted responses.
Tamoxifen administration provides complete structural and functional photoreceptor rescue in the model of LI
Experimental animals were administered tamoxifen as a supplement in standard mouse chow (estimated 80 mg/kg/d intake of tamoxifen) beginning 1 week before LI and maintained after LI. Age-matched control animals were provided with standard chow (without added tamoxifen) and housed in separate cages alongside tamoxifen-treated animals in similar lighting environments. Tamoxifen levels in the retina measured using high-pressure LC and mass spectroscopy after 1–2 weeks of tamoxifen administration in uninjured animals (n = 8) was found to be 13.6 ± 4.4 μg/g (mean ± SD) of retina weight. At 7 d after LI, the typical thinning of the retina centered in the superotemporal quadrant that was observed in control animals on OCT imaging was absent in all tamoxifen-treated animals (Fig. 2A). Similarly, the thinning of the ONL layer and the presence of shallow retinal detachments observed on individual OCT B-scans in control animals were also generally absent in tamoxifen-treated animals (Fig. 2B). Quantification of total retinal thicknesses and outer retinal thicknesses from OCT images after automated segmentation in 8 separate retinal positions (100–300 μm and 300–600 μm in each of the superior, inferior, temporal, and nasal quadrants around the optic nerve) demonstrated significantly greater retinal thicknesses in all retinal positions at 7 d in tamoxifen-treated animals relative to controls (Fig. 2C). In tamoxifen-treated animals, the laminated appearance of the retina and retinal thicknesses at 7 d after LI were nearly identical to those obtained at baseline.
Figure 2.
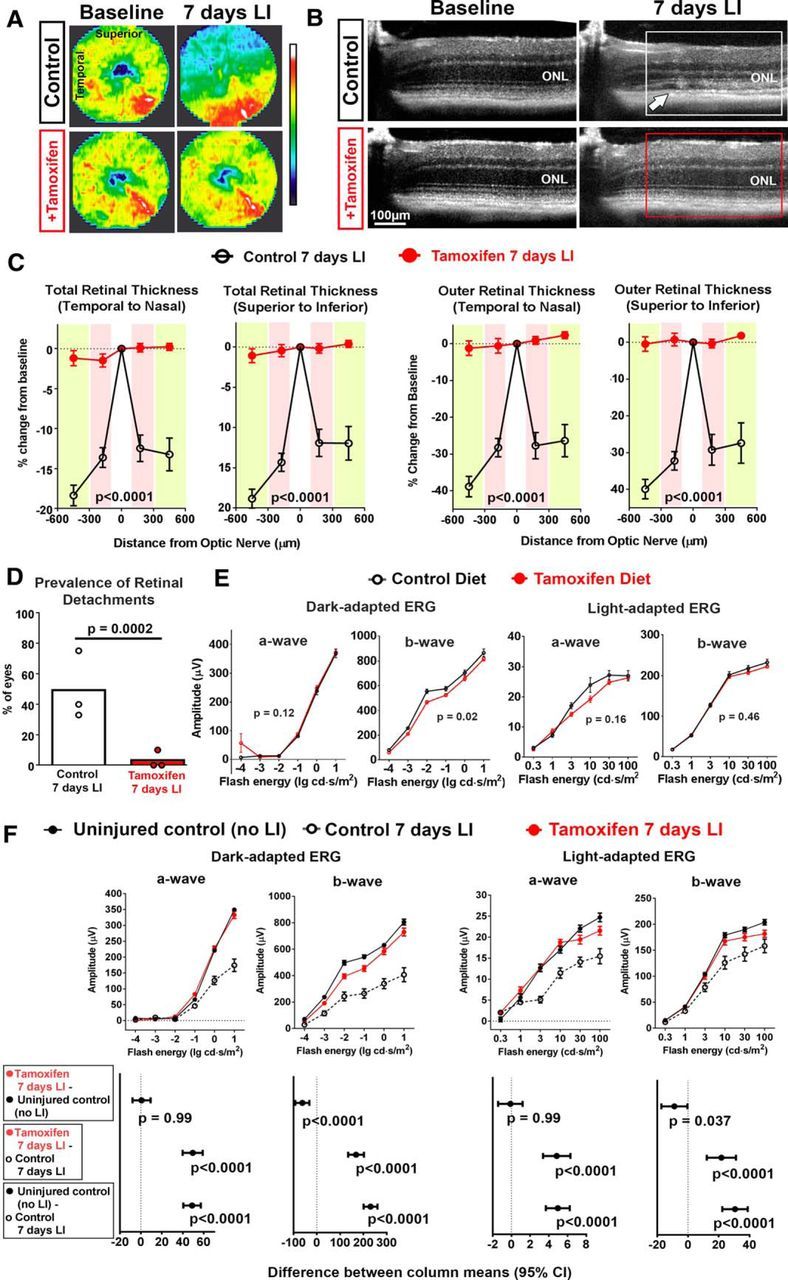
Tamoxifen administration before acute LI results in near complete rescue of structural and functional retinal damage. Adult mice were administered daily oral tamoxifen (added to standard animal chow, 500 mg/kg chow, estimated intake of 80 mg/kg body weight/d) 1 week before LI and thereafter; control animals were fed standard chow without tamoxifen at all time points. A, Total retinal thickness in the central retina (radius 600 μm around optic nerve) as measured by OCT are depicted in heat maps; control animals demonstrate extensive retinal thinning (blue areas) at 7 d after LI that was absent in tamoxifen-treated animals. B, Individual OCT B-scans in the superior retina demonstrated the following: (1) loss of outer segments, (2) thinning of the ONL layer (white box), and (3) emergence of areas of retinal detachment (white arrow); these features were absent in tamoxifen-treated animals (red box). C, Quantification of mean OCT-derived total (top row) and outer retina thickness (bottom row) in inner (pink quadrants) and outer (yellow quadrants) in the horizontal (left) and vertical (right) axes demonstrate marked thinning 7 d after LI in control animals (black lines) but no significant changes from baseline in tamoxifen-treated animals (red lines) (n = 27 eyes from 14 animals of mixed gender for each group, 2-way ANOVA). D, Prevalence of localized retinal detachments, scored at 7 d after LI, was significantly lower in tamoxifen-treated versus control groups (n = 27 eyes in each group in 3 independent experiments, χ2 statistic). E, ERG measurements of uninjured animals administered standard chow (control diet) versus oral tamoxifen for 14 d demonstrated that tamoxifen did not exert a large change on the magnitude of response amplitudes (2-way ANOVA, n = 6 animals in each group, 3 male and 3 female, 2.5–3 months old). F, ERG measurements in the LI model demonstrated that a- and b-wave amplitudes for dark- and light-adapted functional responses obtained 7 d after LI in tamoxifen-treated animals (red lines) were significantly greater than those in untreated controls (black dashed lines). a-wave amplitudes for both light- and dark-adapted responses in tamoxifen-treated animals were statistically similar to uninjured controls (p > 0.99 for both comparisons), indicating full protection. b-wave amplitudes approached those in uninjured controls, but did not reach full protection (p < 0.05 for both comparisons). Top, Data points and error bars represent mean and SEM; n = 12, 7, and 7 animals for uninjured control, untreated control 7 d after LI, and tamoxifen-treated animals 7 d after LI, respectively. Bottom, Difference in column means between the three groups, all comparisons made with two-way ANOVA, error bars indicate 95% confidence intervals.
We also assessed the prevalence of shallow separation of the neural retina from the RPE (i.e., retinal detachments) in the experimental groups using OCT. Retinal detachment is a feature of outer retina degeneration that is also observed in mouse models of inherited photoreceptor degeneration (Pennesi et al., 2012). The prevalence of retinal detachments in three separate experimental repeats were all significantly lower in tamoxifen-treated versus control animals (Fig. 2D), with the overall prevalence being 1/27 versus 13/27 in tamoxifen-treated versus control animals, respectively (χ2 statistic = 13.9, p = 0.0002).
ERG evaluations were performed in wild-type animals fed standard chow (control diet) versus tamoxifen-supplemented diet for 14 d in the absence of LI; these established that tamoxifen feeding alone did not exert a large positive or negative effect on ERG amplitudes (Fig. 2E). After LI, tamoxifen-treated animals demonstrated significantly greater a- and b-wave amplitudes for both dark- and light-adapted ERGs relative to control animals not provided tamoxifen (Fig. 2F). The dark- and light-adapted a-wave amplitudes in tamoxifen-treated animals were in fact statistically similar those in uninjured controls, indicating a complete protection from LI. The dark- and light-adapted b-wave amplitudes in tamoxifen-treated animals, although improved relative to controls after LI, approached but did not reach full recovery compared with uninjured controls.
In analysis of data collected separately from female (n = 17 control, 17 tamoxifen-treated) and male (n = 10 control, 10 tamoxifen-treated) animals, we noted similar protective effects on retinal structure in animals of both sexes (Fig. 3). Tamoxifen administration provided near-complete structural rescue in the model of light-induced photoreceptor injury in a manner that was independent of the sex of the animal.
Figure 3.

Tamoxifen-mediated rescue of photoreceptor degeneration in LI model is not dependent on the sex of experimental animals. Young adult (2–3 months old) female and male mice were subjected to LI with and without pretreatment with tamoxifen. OCT evaluation of total retinal thickness (top) and outer retinal thickness (bottom) demonstrated that significant protection was conferred in both male and female animals treated with tamoxifen relative to sex-matched untreated controls (p-values from 2-way ANOVA, n = 17 treated and 17 untreated control female animals, 10 treated and untreated 10 control male animals).
Tamoxifen treatment reduces microglial infiltration and activation induced by LI
In vivo fundus autofluorescence imaging was performed in tamoxifen-treated and control animals before and at different times following LI. Before LI, minimal autofluorescence was observed on fundus imaging. In animals fed a control diet, imaging performed 3 d after LI demonstrated the appearance of isolated autofluorescent spots near the optic nerve. These spots subsequently increased in number and intensity, particularly in the superotemporal quadrant at 7 d after LI, before decreasing in intensity and number at 14 d after LI (Fig. 4A). In tamoxifen-treated animals, minimal to no fundus autofluorescent signals were observed at all time points monitored. To investigate the basis for these autofluorescent spots, flat-mounted retina tissue was prepared from control animals at 14 d after LI. On confocal imaging, autofluorescent spots matching the size and distribution of those observed on in vivo fundus imaging were observed at the level of the outer retina (Fig. 4B) and colocalized well with Iba1 immunopositivity, indicating that they had arisen from autofluorescent retinal microglia that had infiltrated into the outer retina after LI, as observed in other models of subretinal microglia accumulation (Ma et al., 2013).
Figure 4.
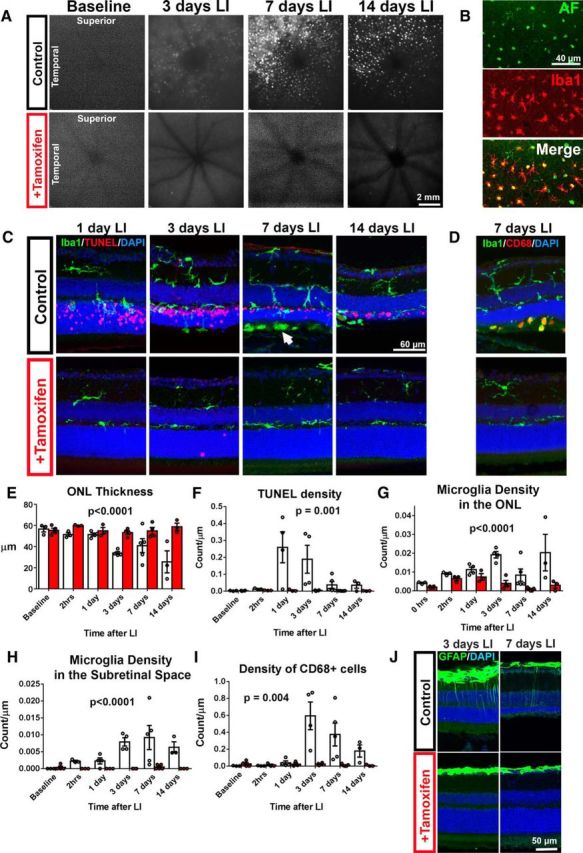
Tamoxifen administration reduced microglial activation and infiltration induced by acute LI. A, In vivo fundus autofluorescence (FAF) imaging after LI revealed the emergence of a punctate hyperautofluorescent pattern centered in the superotemporal quandrant at the level of the subretinal space that peaked in intensity at 7 d after LI; this pattern of fundus autofluorescence was not observed in tamoxifen-treated animals across the same time points after LI. B, Immunohistochemical analysis of RPE flat mounts in control animals 14 d after LI demonstrated that the subretinal hyperautofluorescent spots (green) observed in FAF imaging in vivo and on histological analysis corresponded to Iba1-immunopositive microglia (red), indicating that microglial infiltration into the outer retina was induced by LI. C, Histological analysis of retinal sections from experimental animals after LI showed early (1 d after LI) infiltration of the ONL by Iba1-positive microglia (green) by microglia from the inner retina; this occurred concurrently with the emergence of TUNEL staining (red) in the ONL. Microglia infiltration increased at subsequent time points with additional accumulation of Iba1+ cells in the subretinal space (arrow). In comparison, microglia infiltration into the outer retina was markedly decreased by tamoxifen treatment. D, Infiltrating microglia in the outer retina showed expression of the activation marker, CD68 (red) in untreated control animals, which were largely absent in tamoxifen-treated animals. E, F, Quantitative histological analyses showing the time course of photoreceptor atrophy (as revealed by ONL thinning) (E) and photoreceptor apoptosis (as revealed by TUNEL staining) (F) in control animals, which were both markedly reduced in tamoxifen-treated animals. Morphological rescue of photoreceptors in tamoxifen-treated animals was correlated with decreases in the numbers of infiltrating microglia in the ONL (G) and subretinal space (H) and activated CD68-immunpositive microglia (I). n = 3–5 animals per time point in control and treated groups, p-values correspond to comparisons between control and treated groups, 2-way ANOVA. J, Histological analysis of retinal sections from animals 3 and 7 d after LI in control versus tamoxifen-treated groups revealed a low-level induction of GFAP immunopositivity in Müller cell processes in control animals that was absent in tamoxifen-treated animals.
To characterize the distribution and activation of retinal microglia after LI and the effect of tamoxifen administration on these features, retinal microglia were examined in retinal sections at different times after LI using Iba1 immunohistochemistry. In control animals, beginning as early as 1 d after LI, microglia in the inner retina were observed to extend their processes into the ONL and begin infiltrating the photoreceptor layer concurrent with the emergence of TUNEL-positivity in the ONL (Fig. 4C). Microglia infiltration in control animals continued to persist in the ONL (Fig. 4G) and the subretinal space (Fig. 4H) for up to 14 d after LI. The number of activated CD68-immunopositive microglia also increased in the subretinal space beginning at 3 d after LI (Fig. 4D,I). These observations indicate that the infiltration of activated microglia into the photoreceptor layer occurred early in LI-induced degeneration and was present concurrently during the period of photoreceptor apoptosis and degeneration. In the tamoxifen-treated group, minimal decreases in ONL thickness were observed after LI, corroborating observations on OCT (Fig. 4E). Minimal TUNEL was also observed (Fig. 4F). Morphology, distribution, and activation status (as revealed by CD68 labeling) of microglia were also minimally changed from uninjured controls at all time points after LI (Fig. 4G–I). These data demonstrate that, whereas microglial activation and infiltration were early and prominent features in the LI model, these microglial changes were largely inhibited with tamoxifen treatment. Analysis of GFAP, a marker of Müller cell gliosis, by immunohistochemistry revealed a low-level, patchy labeling in radial Müller cell processes at 3 and 7 d after LI in the control group that was absent in the tamoxifen-treated group (Fig. 4J), indicating an amelioration of a slight macroglial gliotic response with tamoxifen treatment.
Tamoxifen administration suppresses microglial activation and inflammatory cytokine production and reduces microglial toxicity to photoreceptors
To assess directly the effect of tamoxifen on microglia, we exposed cultured retinal microglia to tamoxifen and evaluated the effect of tamoxifen pretreatment on LPS-induced microglial upregulation of key inflammatory cytokines. We found that tamoxifen administration alone, in the absence of LPS stimulation, had a minimal effect on cytokine expression (Fig. 5A). However, tamoxifen pretreatment of microglia significantly inhibited the upregulation of cytokine expression in response to LPS. These tamoxifen effects were dose dependent, with higher doses inducing greater inhibition to LPS stimulation in the 1–10 μg range, indicating that tamoxifen can act directly on microglial cells to reduce their proinflammatory responses to injury-related stimuli. We also assessed whether tamoxifen treatment before LI also decreased proinflammatory cytokine production in the retina in vivo. Protein assessment of cytokine levels in whole retinas from tamoxifen-treated animals relative to control diet-treated animals, although not decreased to a statistically significant extent, demonstrated a general trend of decrease toward baseline levels in the noninjured retina (Fig. 5B), indicating that tamoxifen pretreatment may be able to inhibit microglial upregulation of cytokines in responses to LI in vivo.
Figure 5.

Tamoxifen administration suppresses microglial activation and inflammatory cytokine production and reduces microglial toxicity to photoreceptors. A, Effect of tamoxifen (TMX) on retinal microglia activation and inflammatory cytokine expression was assessed in vitro. Microglia cultured from the retinas of young adult mice (1–2 months of age, 2.5 × 105 cells/well in a 6-well plate) were exposed to tamoxifen (0, 1, 5, and 10 μg/ml) for 2 h, followed by 1 μg/ml LPS for 16 h, and then assessed for protein expression of inflammatory cytokines. In the absence of LPS, protein levels of inflammatory cytokines in microglia cell lysates were at low basal levels and were unchanged with exposure to tamoxifen (5 μg) alone. LPS-induced expression of cytokines in microglia was significantly reduced by tamoxifen pretreatment in a dose-dependent manner (n = 3 repeats per condition, *p < 0.05 for comparisons relative to the LPS only group, 1-way ANOVA). B, Ability of tamoxifen treatment to influence inflammatory cytokine production in the retina in vivo was evaluated in the acute LI model. Inflammatory cytokines levels were assessed in retinal tissue from 2- to 3-month-old control animals (not subject to LI), light-injured controls not treated with tamoxifen (3 d after LI), and tamoxifen-treated animals (3 d after LI). Although comparisons did not reach statistical significance, multiple cytokines demonstrated increases after LI in the absence of tamoxifen that were reduced to close to baseline levels with tamoxifen treatment (n = 3–6 animals in 2 separate trials). C, Ability of tamoxifen to decrease microglia-mediated neurotoxicity was evaluated in a microglia-photoreceptor interaction model. 661W photoreceptors cultured in 96-well plate (4 × 104 cells/well) were exposed to conditioned medium from LPS-stimulated (50 ng/ml) BV2 microglia (cultured in 6-well plate with a cell density of 4 × 105 cells/well) for 48 h. Before LPS stimulation, BV2 microglia were pretreated with varying doses of tamoxifen (1, 5, or 10 μg/ml). Cell viability of 661W photoreceptors were assessed using a MTT assay. Pretreatment with tamoxifen (5 and 10 μg/ml) significantly reduced neurotoxicity of microglia-conditioned medium. *p < 0.0001, 1-way ANOVA, comparison with LPS-only control, n = 16 independent replicates. Exposure of 661W photoreceptors to vehicle (0.5% ethanol) or tamoxifen (5 μg/ml) alone had no effect on viability.
To relate the inhibition of microglial activation to photoreceptor degeneration, we performed an in vitro microglia–photoreceptor interaction assay in which 661W cells from a photoreceptor cell line were exposed to conditioned medium from LPS-stimulated BV2 microglia. Although control medium from LPS-stimulated microglia could induce decreased photoreceptor viability, this negative effect was reduced by the pretreatment of BV2 cells with tamoxifen (Fig. 5C), with larger doses (5–10 μg) providing a greater rescue of 661W viability. Tamoxifen (5 μg) on its own did not exert any effect on the viabilility of 661W photoreceptors in the range of concentrations tested. These results indicate that tamoxifen treatment can limit the activation of microglia to injury signals and thus reduce the consequent proinflammatory neurotoxic effects on photoreceptors.
Tamoxifen administration provides structural and functional rescue of photoreceptor degeneration in the rd10 mouse model of RP
To evaluate whether tamoxifen treatment can provide photoreceptor protection in other etiologies of photoreceptor degeneration, such as RP, we investigated the effect of tamoxifen supplementation in rd10 mice, a mouse model of RP that is induced by a mutation in the photoreceptor-specific Pde6b gene (Chang et al., 2007), a causative gene in human RP (McLaughlin et al., 1993). In our previous work, we found that activated microglia infiltrating the photoreceptor layer in the rd10 retina contribute non-cell autonomously to the rate of photoreceptor demise via phagocytic and proinflammatory mechanisms (Zhao et al., 2015; Zabel et al., 2016). Therefore, we hypothesized that the inhibition of microglial activation by tamoxifen may be helpful in delaying photoreceptor degeneration in RP. We provided postweaning P21 rd10 mice with the tamoxifen-supplemented chow and used littermates that were fed standard chow as controls. OCT measurements at P42 and P49 demonstrated that outer retina thicknesses in tamoxifen-treated animals were significantly greater than those in littermate controls (Fig. 6A,B). The prevalence of shallow retinal detachments at P49 were also lower in tamoxifen-treated versus control animals (2/18 vs 14/14, χ2 statistic = 24.9, p = 0.00001). ERG evaluations at P29 and P50 demonstrated significantly larger b-wave amplitudes in dark- and light-adapted ERGs in tamoxifen-treated animals relative to littermate controls (Fig. 6C). To determine whether these rescue effects relate to an improved visual capability of treated animals, we assessed optomotor responses in experimental animals to presentations of visual stimuli. We found that tamoxifen-treated animals at P49 demonstrated improved photopic optomotor responses over a broad range of spatial frequencies compared with littermate controls (Fig. 6D). The mean visual threshold, which approximates the maximum spatial frequency in the visual stimuli grating sufficient to elicit an optomotor response, was also significantly greater in tamoxifen-treated animals. Although tamoxifen treatment did not completely prevent progressive structural and functional deterioration in rd10 mice, it enabled statistically significant improvements in photoreceptor thickness and ERG responses relative to controls, which also translated to improved visual capability on a functional visual task.
Figure 6.
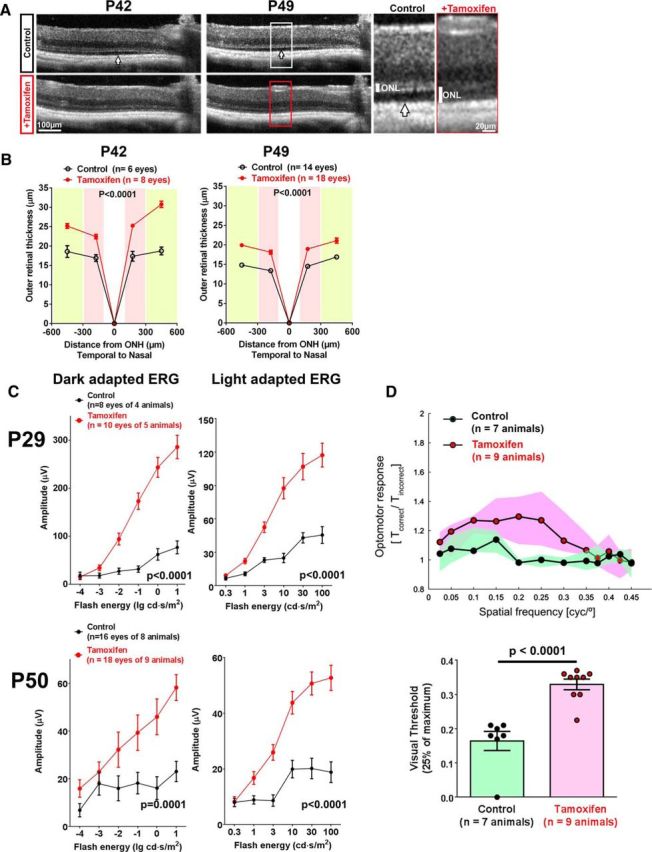
Tamoxifen administration provides structural and functional rescue of photoreceptor degeneration in rd10 mouse model for RP. rd10 mice were provided either standard chow (control group) or tamoxifen-supplemented chow from P21 and evaluated at different time points during photoreceptor degeneration. A, In vivo OCT imaging at P42 and P49 demonstrated in control rd10 animals a marked thinning of the ONL and the emergence of local retinal detachments (white arrow). Comparison of equivalent retinal areas in tamoxifen-treated rd10 animals revealed a greater preservation of ONL thickness (insets show magnified views of the ONL and retinal detachment). B, Quantification of OCT-derived outer retinal thicknesses showed significantly greater preservation of the photoreceptor layer in tamoxifen-treated animals (red lines) versus control animals (black lines) (p-values from 2-way ANOVA). C, ERG evaluation demonstrated significantly greater b-wave amplitudes in light- and dark-adapted responses at both P29 and P50 in tamoxifen-treated animals (red lines) versus control animals (black lines). p-values from 2-way ANOVA. D, Visual acuity capabilities of P49 rd10 animals were evaluated under photopic conditions by automated assessment of optomotor responses. Sinusoidal gratings, rotating in a virtual cylinder at 12°/s, were presented at different spatial frequencies to each awake and unrestrained animal tested and resulting optomotor responses were quantitated from the tracking of head movements as the ratio of the time during which head movement occurred in the same direction with stimulus movement to the time during which it occurred in the opposite direction (Tcorrect/Tincorrect). Data points (top) indicate median ratios at each grating spatial frequency, with the colored areas indicating the upper and lower quartiles of the dataset. Comparison of responses indicated that tamoxifen-treated animals had greater optomotor responses to moving stimuli relative to control animals over a range of spatial frequencies presented. Estimations of visual threshold (bottom), defined as the spatial frequency corresponding to 25% of the maximum optomotor response, significantly higher for tamoxifen-treated animals (p-value from unpaired t test with Welch correction), indicating that tamoxifen treatment resulted in a greater preservation of visual acuity function relative to controls.
Evaluation of mechanism underlying tamoxifen-related photoreceptor neuroprotection
Because tamoxifen administration decreased markers of microglial activation in vitro and in vivo, we investigated the possibility that these effects may be mediated via microglia-expressed ERs or estrogen-related receptors (ERRs) with which tamoxifen may interact (Coward et al., 2001; Tremblay et al., 2001a; McDonnell et al., 2002; Greschik et al., 2004). However, we found that agonists to ERα and ERβ receptors (estradiol, 100 nm and 10 μm; PPT 1 μm; DPN, 10 nm) did not phenocopy the ability of tamoxifen to rescue 661W photoreceptors from microglia-mediated neurotoxicity and antagonists to these receptors (ICI, 1 μm; MPP, 1 μm; PHTPP, 1 μm), when coapplied with tamoxifen, also failed to decrease the extent of tamoxifen-mediated rescue (Fig. 7). Consistent with this, the application of diethylstilbestrol, an inverse agonist of ERRs (Tremblay et al., 2001b), did not influence the results of neurotoxicity assays either when applied alone or together with tamoxifen. These findings indicate that tamoxifen may exert its effects on microglia activation independently of ER and ERRs in this context, as was reported previously (Suuronen et al., 2005).
Figure 7.

Effects of agonism and antagonism of ERs and ERR on BV2 microglia-mediated neurotoxicity to 661W photoreceptors. 661W photoreceptors cultured in 96-well plate (4 × 104 cells/well) were exposed to conditioned medium from LPS-stimulated (50 ng/ml) BV2 microglia (cultured in 6-well plate with a cell density of 4 × 105 cells/well) for 24 h. Before LPS stimulation, BV2 microglia were pretreated with tamoxifen (TMX, 5 μg/ml). Cell viability of 661W photoreceptors was assessed using an MTT assay. Pretreatment with tamoxifen significantly reduced neurotoxicity of microglia-conditioned medium. Similar pretreatment with the ER agonist estradiol (E2, 100 nm and 10 μm), the ERα agonist PPT (1 μm), and the ERβ agonist DPN (10 nm) did not significant alter the neurotoxicity of microglia-conditioned media. Addition of ER agonist (E2, 10 μm) or antagonist (ERα/β antagonist, ICI, 1 μm; ERα antagonist, MPP, 1 μm; ERβ antagonist PHTPP, 1 μm), together with TMX (5 μg/ml), did not result in 661W survival levels that were significantly different from when TMX was applied alone. Pretreatment of diethylstilbestrol (DES, 1 μm), an inhibitor of ERRs, did not alter the neurotoxicity of activated microglia, nor did it significantly change the effect of TMX when applied in combination. n.s. indicates p > 0.05, 1-way ANOVA, Sidak's multiple-comparison test, n = 6 independent replicates.
Because ERs and ERRs are additionally expressed by nonmicroglia CNS cell types, the neuroprotective effects of tamoxifen may also entail mechanisms involving neurons and macroglia cells via ER-dependent (Elzer et al., 2010) and ER-independent (Zhang et al., 2007) pathways. In the rodent and human retina, ERs are expressed in multiple neuronal cell types and the RPE (Ogueta et al., 1999; Cascio et al., 2007) and ERRβ in rod photoreceptors (Blackshaw et al., 2001; Onishi et al., 2010). Therefore, it is plausible that tamoxifen may signal directly to photoreceptors to exert its protective effects. It has been previously reported that modulation of the dynamics of the visual cycle can confer significant in vivo protection in a LI model (Li et al., 2015); however, we found that administration of tamoxifen at the protective dose did not exert significant changes on the rate of photoreceptor dark adaptation (Fig. 8A), indicating that tamoxifen is unlikely to exert neuroprotection through this particular mechanism. In addition, we found that, although subjecting 661W photoreceptors to LI decreased photoreceptor survival in a dose-dependent manner in vitro, direct application of tamoxifen did not increase 661W photoreceptor survival in the dose range tested (Fig. 8B). This indicates that tamoxifen does not exert a prosurvival effect directly on 661W photoreceptors. We also addressed the possibility that tamoxifen may act directly on 661W photoreceptors to increase their ability to resist the deleterious effects of BV2-conditioned medium. We exposed 661W photoreceptors initially to 0, 1, or 5 μg/ml tamoxifen for 24 h, followed by exposure to BV2-conditioned medium. We found that tamoxifen pretreatment did not boost 661W resistance to BV2-conditioned medium, but rather decreased it when the tamoxifen concentration was increased from 1 to 5 μg/ml (Fig. 8C). This indicated that tamoxifen can actually decrease 661W photoreceptor survival directly when combined with an additional inflammatory insult or when increased to higher concentrations (>15 μg/ml; Fig. 8D). Whereas the involvement of direct mechanisms involving photoreceptor neurons in tamoxifen-mediated photoreceptor rescue cannot be completely ruled out (Cascio et al., 2015), we did not find support for this mechanism in our experiments.
Figure 8.

Evaluation of the effects of tamoxifen on photoreceptor survival and function. A, Measurement of ERG recovery after a moderate bleaching flash in animals pretreated for oral tamoxifen for 7 d compared with age-matched control fed a standard control diet. The amplitudes of the a-wave of ERGs after bleach were recorded every 2 min at a flash intensity of 10 cd/s/m2 for the two groups of animals and mean a-wave recovery with time was plotted. Mean a-wave amplitudes recovered to baseline conditions at similar times and the dynamics of recovery were not significantly different between the 2 groups (2-way ANOVA, p = 0.493), indicating that tamoxifen pretreatment did not affect photoreceptor dark adaptation function. B, Direct effect of tamoxifen on photoreceptor survival was evaluated in vitro using a light-induced injury model. 661W photoreceptors cultured in a 96-well plate (4 × 104 cells/well) were subjected to two regimens of photic injury; the conditions were as follows: (1) control conditions (no LI), (2) under 35 × 103 lux for 90 min (severe injury), and (3) under 60 × 103 lux for 30 min (moderate injury). Cultured photoreceptors were either cultured under standard conditions or pretreated with tamoxifen (5 μg/ml for 24 h) before LI. Cell survival was measured 12 h after injury using an MTT assay. Tamoxifen pretreatment did not result in any significant improvements in photoreceptor survival relative to controls under any condition. n = indicates independent replicates, n.s. indicates p > 0.05, 1-way ANOVA, Sidak's multiple-comparisons test. C, Effect of tamoxifen pretreatment on the ability of 661W photoreceptors to resist the deleterious effects of BV2 microglia-conditioned medium was evaluated. 661W photoreceptors were cultured in the absence or presence of tamoxifen (1 and 5 μg/ml) for 24 h and then transferred into conditioned medium from LPS-treated BV2 microglia for another 48 h. Cell viability was assessed using an MTT assay and is expressed as a percentage of the control. Tamoxifen pretreatment of 661W photoreceptors did not confer protection from BV2 conditioned medium at 1 μg/ml and conversely decreased survival at 5 μg/ml. *p < 0.05, Kruskal–Wallis 1-way ANOVA with Dunn's multiple-comparisons test, n = 8 replicates in each condition. D, Direct effect of tamoxifen on the viability of 661W photoreceptors in vitro. 661W photoreceptors were cultured in the absence and presence of tamoxifen (1–15 μg/ml) for 24 h and viability of 661W photoreceptors assessed using an MTT assay. The experiment was repeated in an independent replicate and the results were pooled. n = 20 wells in 2 independent replicates, *p < 0.05, Kruskal–Wallis test with Dunn's multiple-comparisons test. Tamoxifen decreased the survival of 661W photoreceptors at a concentration of 15 μg/ml.
Discussion
Tamoxifen, a drug approved for the treatment of metastatic breast cancer, exerts mixed agonist and antagonist actions at ERs, depending on the target tissue and cell type (Riggs and Hartmann, 2003), and influences signaling through ERRs (Tremblay et al., 2001a) and the G-protein coupled receptor GPR30 (Vivacqua et al., 2006a). Within the CNS, tamoxifen, as well as other SERMs, have been documented to exert complex effects that culminate in neuroprotective effects in injury models including spinal cord injury (Ismailoğlu et al., 2010; de la Torre Valdovinos et al., 2016), penetrating brain injury (Arevalo et al., 2012; Franco Rodríguez et al., 2013; Barreto et al., 2014), and radiation injury (Liu et al., 2010). Although therapeutic application of SERMs for brain and spinal cord neurodegeneration have been proposed (Chakrabarti et al., 2014; Villa et al., 2016), their potential use for retinal degeneration is unknown. On the contrary, reported effects of tamoxifen in the retina have centered on tamoxifen-related retinal toxicity, which appears as retinal crystalline deposits, macular edema, or cystic cavitations (Nayfield and Gorin, 1996; Gualino et al., 2005; Bourla et al., 2007). Functionally, variable visual symptoms and physiological changes, such as decreased multifocal ERG amplitudes (Gorin et al., 1998; Salomão et al., 2007; Watanabe et al., 2010), have been reported. The prevalence of toxicity is thought to increase with higher doses of >120 mg/d (Kaiser-Kupfer and Lippman, 1978; Bourla et al., 2007), but remains detectable (6–12%) at lower doses of 20–40 mg/d (Pavlidis et al., 1992; Noureddin et al., 1999). The mechanism for clinical toxicity is unclear; proposed mechanisms include the induction of lysosomal dysfunction (Imperia et al., 1989; Kim et al., 2014), excessive autophagy (Cho et al., 2012), and oxidative stress (Toler, 2004). Tamoxifen, by acting as an inverse agonist of rod-expressed ERRβ, may also induce rod death by dysregulating ATP utilization (Onishi et al., 2010). As a result of these clinical observations and reports of tamoxifen inducing retinal cell damage in vitro (Cho et al., 2012; Kim et al., 2014), neuroprotective effects of tamoxifen that prevent or retard photoreceptor degeneration have been unanticipated and have not to our knowledge been investigated previously.
Contrary to clinical reports, we did not find evidence in mouse models for ocular toxicity of tamoxifen. Tamoxifen administered daily for 7 d at a dose of 80 mg/kg (equivalent in adult mice to a 6- to 7-fold increase over the standard 40 mg dose for an adult human) to either uninjured (Fig. 2E) or light-injured animals did not exert marked changes to retinal structure or ERG function relative to untreated controls. In previous studies involving even greater tamoxifen exposure (500 mg/kg oral dose every 5 d for 30 d), retinal pathology or marked ERG changes were also undetected (Wang et al., 2016). Direct ocular administration of tamoxifen as an eye drop (5 mg/ml delivered as a single 10 μl drop thrice daily for 5 d) at doses high enough to induce Cre recombination via the Cre-ERT system (Hayashi and McMahon, 2002) similarly did not induce detectable retinal structural or functional changes (Boneva et al., 2016). The reason for this disparity between mice and human responses is unclear, but may relate to an increased vulnerability of the human macula or to an interspecies differences in the metabolism and/or biodistribution of tamoxifen in the retina. Further development of tamoxifen as neuroprotective therapy for photoreceptors in retinal disease will likely involve dose-ranging studies, alternative methods of targeted delivery, and the evaluation of the neuroprotective properties of alternative SERMs.
The cellular mechanism underlying tamoxifen-mediated photoreceptor neuroprotection likely involves the modulation of retinal microglial activation. Studies from our group and others have demonstrated that the specific modulation of microglial phagocytosis and activation results in structural and functional amelioration of photoreceptor degeneration in mouse models of RP (Peng et al., 2014; Zhao et al., 2015; Zabel et al., 2016). Inhibition of microglia activation using pharmacological means has also been reported to improve photoreceptor survival in models of light-induced degeneration (Zhang et al., 2004; Ni et al., 2008; Collier et al., 2011). We found here that tamoxifen directly suppressed proinflammatory cytokine production in activated retinal microglia and decreased microglial toxicity to 661W photoreceptor cells. These studies corroborate findings in which tamoxifen suppresses activation in microglia cultured from the brain (Suuronen et al., 2005; Tapia-Gonzalez et al., 2008) and reduces microglial neurotoxicity in vitro (Ishihara et al., 2015) and in vivo models of brain injury (Khan et al., 2015). We have found previously that inhibition of IL1β signaling in mouse models of RP decreases photoreceptor apoptosis and rescues retinal function (Zhao et al., 2015); tamoxifen, by reducing microglial production of inflammatory cytokines including IL1β, may have contributed to photoreceptor rescue through this mechanism.
However, the molecular mechanisms underlying how tamoxifen influences microglia activation are not fully elucidated. Because tamoxifen can modulate ER signaling, primarily by acting as an agonist as in the CNS (Zhang et al., 2009), we investigated whether tamoxifen's effect may be phenocopied by agonists to ERα or ERβ receptors. In addition, because tamoxifen has been reported to be capable of binding and deactivating ERRs (Coward et al., 2001; Tremblay et al., 2001a; Greschik et al., 2004), we investigated whether tamoxifen-induced effects on microglia may be induced by ERRs. Our pharmacological studies, however, did not implicate either ER or ERRs as potential mechanisms of action. Alternative mechanisms for tamoxifen's effect remain possible; SERMs have been described to confer neuroprotection independently of ER binding or classical ER activity, activating instead associated kinase signaling cascades via pathways involving the G-protein coupled receptor GPR30 (Abdelhamid et al., 2011). Macrophages and microglia have been reported to express GPR30 (Blasko et al., 2009; Schaufelberger et al., 2016), which can bind tamoxifen (Méndez-Luna et al., 2015), enabling tamoxifen actions on microglia that are independent of ERs (Vivacqua et al., 2006a; Vivacqua et al., 2006b; Fitts et al., 2011; Tsai et al., 2013). Our future work will explore how these alternative mechanisms may be operational in our experimental systems.
We also investigated the possibility that tamoxifen may act directly on photoreceptors to exert its protective effects. However, we did not find data to support a direct effect of tamoxifen on photoreceptors; instead we found that: (1) tamoxifen administration did not influence electroretinographic or dark adaptation parameters in uninjured animals, (2) direct tamoxifen application to 661W photoreceptors subjected to LI in vitro does not result in photoreceptor protection, and (3) tamoxifen administration to 661W photoreceptors before exposure to BV2 microglia-conditioned medium does not boost the ability of photoreceptors to resist microglia-mediated neurotoxicity. Although molecules expressed by photoreceptors such as ER and ERRs may be molecular candidates for tamoxifen-mediated protection, we have so far not uncovered evidence showing that these protective effects are mediated via direct signaling of tamoxifen to photoreceptors. In the case of the rod-enriched ERR-beta receptor, tamoxifen, in acting to deactivate ERRs, would be expected to augment, not ameliorate, photoreceptor degeneration, as demonstrated in experiments that induce genetic or pharmacological disruptions of ERRβ function (Onishi et al., 2010). Therefore, tamoxifen's neuroprotective effects may not occur directly on photoreceptors, but instead via its effects of other retinal cells such as microglia.
Beyond the therapeutic implications of its neuroprotective effects, tamoxifen is also commonly used experimentally to manipulate gene expression in transgenic mice expressing tamoxifen-dependent Cre recombinases [CreER(T)s] that enable DNA recombination upon tamoxifen administration (Feil et al., 1997). Transgenic mice with microglia-specific promoters driving CreER(T) expression and genetic recombination in microglia in the brain (Goldmann et al., 2013; Parkhurst et al., 2013; Yona et al., 2013) and the retina (Zhao et al., 2015; Wang et al., 2016) have been developed and studied. Our findings that tamoxifen administration markedly suppresses of microglia activation and exerts neuroprotective effects in injury models suggest potential confounding effects in the interpretation of results obtained in CreER(T) systems and the need to institute experimental safeguards and controls. Because tamoxifen applied topically to the eye appears capable of inducing CreER(T) activation without providing protection against induced photoreceptor apoptosis (Boneva et al., 2016), investigation of the appropriate dose ranges and delivery routes for tamoxifen may be necessary to find experimental parameters at which CreER(T) activation can be induced without incurring other CreER(T)-independent biological effects (Denk et al., 2015).
In conclusion, we discovered here that tamoxifen, a drug previously associated with retina toxicity, confers significant structural and functional protection to photoreceptors in both acute and genetic models of photoreceptor degeneration. The action of tamoxifen in suppressing retinal microglial activation and proinflammatory cytokine expression likely contributes to this protection, as supported by recent studies documenting the involvement of microglia in the acceleration of photoreceptor demise (Scholz et al., 2015; Zhao et al., 2015; Zabel et al., 2016). Proof-of-concept clinical trials involving microglial modulation for retinal diseases constitute an active area of investigation (Cukras et al., 2012) and studies targeting microglial activation in RP are currently under way (www.clinicaltrials.gov, NCT02140164). Tamoxifen may constitute a candidate therapeutic agent for clinical investigation for the treatment of photoreceptor-degenerative diseases such as RP.
Footnotes
This work was supported by funds from the National Eye Institute Intramural Research Program–National Institutes of Health.
A provisional patent for the use of tamoxifen for the treatment of retinal degenerations is currently being filed by the National Eye Institute, National Institutes of Health.
References
- Abdelhamid R, Luo J, Vandevrede L, Kundu I, Michalsen B, Litosh VA, Schiefer IT, Gherezghiher T, Yao P, Qin Z, Thatcher GR (2011) Benzothiophene selective estrogen receptor modulators provide neuroprotection by a novel GPR30-dependent mechanism. ACS Chem Neurosci 2:256–268. 10.1021/cn100106a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amantea D, Bagetta G (2016) Drug repurposing for immune modulation in acute ischemic stroke. Curr Opin Pharmacol 26:124–130. 10.1016/j.coph.2015.11.006 [DOI] [PubMed] [Google Scholar]
- Arevalo MA, Diz-Chaves Y, Santos-Galindo M, Bellini MJ, Garcia-Segura LM (2012) Selective oestrogen receptor modulators decrease the inflammatory response of glial cells. J Neuroendocrinol 24:183–190. 10.1111/j.1365-2826.2011.02156.x [DOI] [PubMed] [Google Scholar]
- Barreto GE, Santos-Galindo M, Garcia-Segura LM (2014) Selective estrogen receptor modulators regulate reactive microglia after penetrating brain injury. Front Aging Neurosci 6:132. 10.3389/fnagi.2014.00132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackshaw S, Fraioli RE, Furukawa T, Cepko CL (2001) Comprehensive analysis of photoreceptor gene expression and the identification of candidate retinal disease genes. Cell 107:579–589. 10.1016/S0092-8674(01)00574-8 [DOI] [PubMed] [Google Scholar]
- Blasko E, Haskell CA, Leung S, Gualtieri G, Halks-Miller M, Mahmoudi M, Dennis MK, Prossnitz ER, Karpus WJ, Horuk R (2009) Beneficial role of the GPR30 agonist G-1 in an animal model of multiple sclerosis. J Neuroimmunol 214:67–77. 10.1016/j.jneuroim.2009.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boneva SK, Groß TR, Schlecht A, Schmitt SI, Sippl C, Jägle H, Volz C, Neueder A, Tamm ER, Braunger BM (2016) Cre recombinase expression or topical tamoxifen treatment do not affect retinal structure and function, neuronal vulnerability or glial reactivity in the mouse eye. Neuroscience 325:188–201. 10.1016/j.neuroscience.2016.03.050 [DOI] [PubMed] [Google Scholar]
- Bourla DH, Sarraf D, Schwartz SD (2007) Peripheral retinopathy and maculopathy in high-dose tamoxifen therapy. Am J Ophthalmol 144:126–128. 10.1016/j.ajo.2007.03.023 [DOI] [PubMed] [Google Scholar]
- Cascio C, Russo D, Drago G, Galizzi G, Passantino R, Guarneri R, Guarneri P (2007) 17beta-estradiol synthesis in the adult male rat retina. Exp Eye Res 85:166–172. 10.1016/j.exer.2007.02.008 [DOI] [PubMed] [Google Scholar]
- Cascio C, Deidda I, Russo D, Guarneri P (2015) The estrogenic retina: The potential contribution to healthy aging and age-related neurodegenerative diseases of the retina. Steroids 103:31–41. 10.1016/j.steroids.2015.08.002 [DOI] [PubMed] [Google Scholar]
- Chakrabarti M, Haque A, Banik NL, Nagarkatti P, Nagarkatti M, Ray SK (2014) Estrogen receptor agonists for attenuation of neuroinflammation and neurodegeneration. Brain Res Bull 109:22–31. 10.1016/j.brainresbull.2014.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B, Hawes NL, Pardue MT, German AM, Hurd RE, Davisson MT, Nusinowitz S, Rengarajan K, Boyd AP, Sidney SS, Phillips MJ, Stewart RE, Chaudhury R, Nickerson JM, Heckenlively JR, Boatright JH (2007) Two mouse retinal degenerations caused by missense mutations in the beta-subunit of rod cGMP phosphodiesterase gene. Vision Res 47:624–633. 10.1016/j.visres.2006.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KS, Yoon YH, Choi JA, Lee SJ, Koh JY (2012) Induction of autophagy and cell death by tamoxifen in cultured retinal pigment epithelial and photoreceptor cells. Invest Ophthalmol Vis Sci 53:5344–5353. 10.1167/iovs.12-9827 [DOI] [PubMed] [Google Scholar]
- Collier RJ, Wang Y, Smith SS, Martin E, Ornberg R, Rhoades K, Romano C (2011) Complement deposition and microglial activation in the outer retina in light-induced retinopathy: inhibition by a 5-HT1A agonist. Invest Ophthalmol Vis Sci 52:8108–8116. 10.1167/iovs.10-6418 [DOI] [PubMed] [Google Scholar]
- Congdon N, O'Colmain B, Klaver CC, Klein R, Muñoz B, Friedman DS, Kempen J, Taylor HR, Mitchell P; Eye Diseases Prevalence Research Group (2004) Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol 122:477–485. 10.1001/archopht.122.4.477 [DOI] [PubMed] [Google Scholar]
- Coward P, Lee D, Hull MV, Lehmann JM (2001) 4-Hydroxytamoxifen binds to and deactivates the estrogen-related receptor gamma. Proc Natl Acad Sci U S A 98:8880–8884. 10.1073/pnas.151244398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukras CA, Petrou P, Chew EY, Meyerle CB, Wong WT (2012) Oral minocycline for the treatment of diabetic macular edema (DME): results of a phase I/II clinical study. Invest Ophthalmol Vis Sci 53:3865–3874. 10.1167/iovs.11-9413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre Valdovinos B, Duenas Jimenez JM, Estrada IJ, Banuelos Pineda J, Franco Rodríguez NE, Lopez Ruiz JR, Osuna Carrasco LP, Candanedo Arellano A, Duenas Jimenez SH (2016) Tamoxifen promotes axonal preservation and gait locomotion recovery after spinal cord injury in cats. J Vet Med 2016:9561968. 10.1155/2016/9561968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk F, Ramer LM, Erskine EL, Nassar MA, Bogdanov Y, Signore M, Wood JN, McMahon SB, Ramer MS (2015) Tamoxifen induces cellular stress in the nervous system by inhibiting cholesterol synthesis. Acta Neuropathol Commun 3:74. 10.1186/s40478-015-0255-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elzer JG, Muhammad S, Wintermantel TM, Regnier-Vigouroux A, Ludwig J, Schütz G, Schwaninger M (2010) Neuronal estrogen receptor-alpha mediates neuroprotection by 17beta-estradiol. J Cereb Blood Flow Metab 30:935–942. 10.1038/jcbfm.2009.258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil R, Wagner J, Metzger D, Chambon P (1997) Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Commun 237:752–757. 10.1006/bbrc.1997.7124 [DOI] [PubMed] [Google Scholar]
- Fitts JM, Klein RM, Powers CA (2011) Tamoxifen regulation of bone growth and endocrine function in the ovariectomized rat: discrimination of responses involving estrogen receptor alpha/estrogen receptor beta, G protein-coupled estrogen receptor, or estrogen-related receptor gamma using fulvestrant (ICI 182780). J Pharmacol Exp Ther 338:246–254. 10.1124/jpet.110.173955 [DOI] [PubMed] [Google Scholar]
- Franco Rodríguez NE, Dueñas Jiménez JM, De la Torre Valdovinos B, López Ruiz JR, Hernández Hernández L, Dueñas Jiménez SH (2013) Tamoxifen favoured the rat sensorial cortex regeneration after a penetrating brain injury. Brain Res Bull 98:64–75. 10.1016/j.brainresbull.2013.07.007 [DOI] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Müller PF, Wolf Y, Varol D, Yona S, Brendecke SM, Kierdorf K, Staszewski O, Datta M, Luedde T, Heikenwalder M, Jung S, Prinz M (2013) A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci 16:1618–1626. 10.1038/nn.3531 [DOI] [PubMed] [Google Scholar]
- Gorin MB, Day R, Costantino JP, Fisher B, Redmond CK, Wickerham L, Gomolin JE, Margolese RG, Mathen MK, Bowman DM, Kaufman DI, Dimitrov NV, Singerman LJ, Bornstein R, Wolmark N, Kaufmann D (1998) Long-term tamoxifen citrate use and potential ocular toxicity. Am J Ophthalmol 125:493–501. 10.1016/S0002-9394(99)80190-1 [DOI] [PubMed] [Google Scholar]
- Greschik H, Flaig R, Renaud JP, Moras D (2004) Structural basis for the deactivation of the estrogen-related receptor gamma by diethylstilbestrol or 4-hydroxytamoxifen and determinants of selectivity. J Biol Chem 279:33639–33646. 10.1074/jbc.M402195200 [DOI] [PubMed] [Google Scholar]
- Gualino V, Cohen SY, Delyfer MN, Sahel JA, Gaudric A (2005) Optical coherence tomography findings in tamoxifen retinopathy. Am J Ophthalmol 140:757–758. 10.1016/j.ajo.2005.04.042 [DOI] [PubMed] [Google Scholar]
- Gupta N, Brown KE, Milam AH (2003) Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp Eye Res 76:463–471. 10.1016/S0014-4835(02)00332-9 [DOI] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP (2002) Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 244:305–318. 10.1006/dbio.2002.0597 [DOI] [PubMed] [Google Scholar]
- Holz FG, Strauss EC, Schmitz-Valckenberg S, van Lookeren Campagne M (2014) Geographic atrophy: clinical features and potential therapeutic approaches. Ophthalmology 121:1079–1091. 10.1016/j.ophtha.2013.11.023 [DOI] [PubMed] [Google Scholar]
- Imperia PS, Lazarus HM, Lass JH (1989) Ocular complications of systemic cancer chemotherapy. Survey Ophthalmol 34:209–230. 10.1016/0039-6257(89)90105-7 [DOI] [PubMed] [Google Scholar]
- Ishihara Y, Itoh K, Ishida A, Yamazaki T (2015) Selective estrogen-receptor modulators suppress microglial activation and neuronal cell death via an estrogen receptor-dependent pathway. J Steroid Biochem Mol Biol 145:85–93. 10.1016/j.jsbmb.2014.10.002 [DOI] [PubMed] [Google Scholar]
- Ismailoğlu O, Oral B, Görgülü A, Sütçü R, Demir N (2010) Neuroprotective effects of tamoxifen on experimental spinal cord injury in rats. J Clin Neurosci 17:1306–1310. 10.1016/j.jocn.2010.01.049 [DOI] [PubMed] [Google Scholar]
- Jonas JB, Bourne RR, White RA, Flaxman SR, Keeffe J, Leasher J, Naidoo K, Pesudovs K, Price H, Wong TY, Resnikoff S, Taylor HR; Vision Loss Expert Group of the Global Burden of Disease S (2014) Visual impairment and blindness due to macular diseases globally: a systematic review and meta-analysis. Am J Ophthalmol 158:808–815. 10.1016/j.ajo.2014.06.012 [DOI] [PubMed] [Google Scholar]
- Kaiser-Kupfer MI, Lippman ME (1978) Tamoxifen retinopathy. Cancer Treat Rep 62:315–320. [PubMed] [Google Scholar]
- Khan MM, Wakade C, de Sevilla L, Brann DW (2015) Selective estrogen receptor modulators (SERMs) enhance neurogenesis and spine density following focal cerebral ischemia. J Steroid Biochem Mol Biol 146:38–47. 10.1016/j.jsbmb.2014.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim LA, Amarnani D, Gnanaguru G, Tseng WA, Vavvas DG, D'Amore PA (2014) Tamoxifen toxicity in cultured retinal pigment epithelial cells is mediated by concurrent regulated cell death mechanisms. Invest Ophthalmol Vis Sci 55:4747–4758. 10.1167/iovs.13-13662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmer F, Kretschmer V, Kunze VP, Kretzberg J (2013) OMR-arena: automated measurement and stimulation system to determine mouse visual thresholds based on optomotor responses. PLoS One 8:e78058. 10.1371/journal.pone.0078058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmer F, Sajgo S, Kretschmer V, Badea TC (2015) A system to measure the optokinetic and optomotor response in mice. J Neurosci Methods 256:91–105. 10.1016/j.jneumeth.2015.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Yu S, Duncan T, Li Y, Liu P, Gene E, Cortes-Pena Y, Qian H, Dong L, Redmond TM (2015) Mouse model of human RPE65 P25L hypomorph resembles wild type under normal light rearing but is fully resistant to acute light damage. Hum Mol Genet 24:4417–4428. 10.1093/hmg/ddv178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JL, Tian DS, Li ZW, Qu WS, Zhan Y, Xie MJ, Yu ZY, Wang W, Wu G (2010) Tamoxifen alleviates irradiation-induced brain injury by attenuating microglial inflammatory response in vitro and in vivo. Brain Res 1316:101–111. 10.1016/j.brainres.2009.12.055 [DOI] [PubMed] [Google Scholar]
- Lonard DM, Smith CL (2002) Molecular perspectives on selective estrogen receptor modulators (SERMs): progress in understanding their tissue-specific agonist and antagonist actions. Steroids 67:15–24. 10.1016/S0039-128X(01)00133-7 [DOI] [PubMed] [Google Scholar]
- Ma W, Coon S, Zhao L, Fariss RN, Wong WT (2013) A2E accumulation influences retinal microglial activation and complement regulation. Neurobiol Aging 34:943–960. 10.1016/j.neurobiolaging.2012.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell DP, Connor CE, Wijayaratne A, Chang CY, Norris JD (2002) Definition of the molecular and cellular mechanisms underlying the tissue-selective agonist/antagonist activities of selective estrogen receptor modulators. Recent Prog Horm Res 57:295–316. 10.1210/rp.57.1.295 [DOI] [PubMed] [Google Scholar]
- McLaughlin ME, Sandberg MA, Berson EL, Dryja TP (1993) Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat Genet 4:130–134. 10.1038/ng0693-130 [DOI] [PubMed] [Google Scholar]
- Méndez-Luna D, Martínez-Archundia M, Maroun RC, Ceballos-Reyes G, Fragoso-Vázquez MJ, González-Juárez DE, Correa-Basurto J (2015) Deciphering the GPER/GPR30-agonist and antagonists interactions using molecular modeling studies, molecular dynamics, and docking simulations. J Biomol Struct Dyn 33:2161–2172. 10.1080/07391102.2014.994102 [DOI] [PubMed] [Google Scholar]
- Nayfield SG, Gorin MB (1996) Tamoxifen-associated eye disease: a review. J Clin Oncol 14:1018–1026. [DOI] [PubMed] [Google Scholar]
- Ni YQ, Xu GZ, Hu WZ, Shi L, Qin YW, Da CD (2008) Neuroprotective effects of naloxone against light-induced photoreceptor degeneration through inhibiting retinal microglial activation. Invest Ophthalmol Vis Sci 49:2589–2598. 10.1167/iovs.07-1173 [DOI] [PubMed] [Google Scholar]
- Noureddin BN, Seoud M, Bashshur Z, Salem Z, Shamseddin A, Khalil A (1999) Ocular toxicity in low-dose tamoxifen: a prospective study. Eye 13:729–733. 10.1038/eye.1999.217 [DOI] [PubMed] [Google Scholar]
- Ogueta SB, Schwartz SD, Yamashita CK, Farber DB (1999) Estrogen receptor in the human eye: influence of gender and age on gene expression. Invest Ophthalmol Vis Sci 40:1906–1911. [PubMed] [Google Scholar]
- Onishi A, Peng GH, Poth EM, Lee DA, Chen J, Alexis U, de Melo J, Chen S, Blackshaw S (2010) The orphan nuclear hormone receptor ERRbeta controls rod photoreceptor survival. Proc Natl Acad Sci U S A 107:11579–11584. 10.1073/pnas.1000102107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, Hempstead BL, Littman DR, Gan WB (2013) Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155:1596–1609. 10.1016/j.cell.2013.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis NA, Petris C, Briassoulis E, Klouvas G, Psilas C, Rempapis J, Petroutsos G (1992) Clear evidence that long-term, low-dose tamoxifen treatment can induce ocular toxicity: a prospective study of 63 patients. Cancer 69:2961–2964. 10.1002/1097-0142(19920615)69:12%3C2961::AID-CNCR2820691215%3E3.0.CO%3B2-W [DOI] [PubMed] [Google Scholar]
- Peng B, Xiao J, Wang K, So KF, Tipoe GL, Lin B (2014) Suppression of microglial activation is neuroprotective in a mouse model of human retinitis pigmentosa. J Neurosci 34:8139–8150. 10.1523/JNEUROSCI.5200-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennesi ME, Michaels KV, Magee SS, Maricle A, Davin SP, Garg AK, Gale MJ, Tu DC, Wen Y, Erker LR, Francis PJ (2012) Long-term characterization of retinal degeneration in rd1 and rd10 mice using spectral domain optical coherence tomography. Invest Ophthalmol Vis Sci 53:4644–4656. 10.1167/iovs.12-9611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs BL, Hartmann LC (2003) Selective estrogen-receptor modulators–mechanisms of action and application to clinical practice. N Engl J Med 348:618–629. 10.1056/NEJMra022219 [DOI] [PubMed] [Google Scholar]
- Roque RS, Imperial CJ, Caldwell RB (1996) Microglial cells invade the outer retina as photoreceptors degenerate in Royal College of Surgeons rats. Invest Ophthalmol Vis Sci 37:196–203. [PubMed] [Google Scholar]
- Russo R, Varano GP, Adornetto A, Nucci C, Corasaniti MT, Bagetta G, Morrone LA (2016) Retinal ganglion cell death in glaucoma: exploring the role of neuroinflammation. Eur J Pharmacol 787:134–142. 10.1016/j.ejphar.2016.03.064 [DOI] [PubMed] [Google Scholar]
- Salomão SR, Watanabe SE, Berezovsky A, Motono M (2007) Multifocal electroretinography, color discrimination and ocular toxicity in tamoxifen use. Curr Eye Res 32:345–352. 10.1080/02713680701229638 [DOI] [PubMed] [Google Scholar]
- Schaufelberger SA, Rosselli M, Barchiesi F, Gillespie DG, Jackson EK, Dubey RK (2016) 2-Methoxyestradiol, an endogenous 17beta-estradiol metabolite, inhibits microglial proliferation and activation via an estrogen receptor-independent mechanism. Am J Physiol Endocrinol Metab 310:E313–E322. 10.1152/ajpendo.00418.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz R, Sobotka M, Caramoy A, Stempfl T, Moehle C, Langmann T (2015) Minocycline counter-regulates pro-inflammatory microglia responses in the retina and protects from degeneration. J Neuroinflammation 12:209. 10.1186/s12974-015-0431-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suuronen T, Nuutinen T, Huuskonen J, Ojala J, Thornell A, Salminen A (2005) Anti-inflammatory effect of selective estrogen receptor modulators (SERMs) in microglial cells. Inflamm Res 54:194–203. 10.1007/s00011-005-1343-z [DOI] [PubMed] [Google Scholar]
- Tapia-Gonzalez S, Carrero P, Pernia O, Garcia-Segura LM, Diz-Chaves Y (2008) Selective oestrogen receptor (ER) modulators reduce microglia reactivity in vivo after peripheral inflammation: potential role of microglial ERs. J Endocrinol 198:219–230. 10.1677/JOE-07-0294 [DOI] [PubMed] [Google Scholar]
- Toler SM. (2004) Oxidative stress plays an important role in the pathogenesis of drug-induced retinopathy. Exp Biol Med (Maywood) 229:607–615. [DOI] [PubMed] [Google Scholar]
- Tremblay GB, Bergeron D, Giguere V (2001a) 4-Hydroxytamoxifen is an isoform-specific inhibitor of orphan estrogen-receptor-related (ERR) nuclear receptors beta and gamma. Endocrinology 142:4572–4575. 10.1210/endo.142.10.8528 [DOI] [PubMed] [Google Scholar]
- Tremblay GB, Kunath T, Bergeron D, Lapointe L, Champigny C, Bader JA, Rossant J, Giguère V (2001b) Diethylstilbestrol regulates trophoblast stem cell differentiation as a ligand of orphan nuclear receptor ERR beta. Genes Dev 15:833–838. 10.1101/gad.873401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CL, Wu HM, Lin CY, Lin YJ, Chao A, Wang TH, Hsueh S, Lai CH, Wang HS (2013) Estradiol and tamoxifen induce cell migration through GPR30 and activation of focal adhesion kinase (FAK) in endometrial cancers with low or without nuclear estrogen receptor alpha (ERalpha). PLoS One 8:e72999. 10.1371/journal.pone.0072999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa A, Vegeto E, Poletti A, Maggi A (2016) Estrogens, neuroinflammation and neurodegeneration. Endocr Rev 37:372–402. 10.1210/er.2016-1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivacqua A, Bonofiglio D, Recchia AG, Musti AM, Picard D, Andò S, Maggiolini M (2006a) The G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17beta-estradiol and hydroxytamoxifen in endometrial cancer cells. Mol Endocrinol 20:631–646. 10.1210/me.2005-0280 [DOI] [PubMed] [Google Scholar]
- Vivacqua A, Bonofiglio D, Albanito L, Madeo A, Rago V, Carpino A, Musti AM, Picard D, Andò S, Maggiolini M (2006b) 17beta-estradiol, genistein, and 4-hydroxytamoxifen induce the proliferation of thyroid cancer cells through the g protein-coupled receptor GPR30. Mol Pharmacol 70:1414–1423. 10.1124/mol.106.026344 [DOI] [PubMed] [Google Scholar]
- Wang X, Zhao L, Zhang J, Fariss RN, Ma W, Kretschmer F, Wang M, Qian HH, Badea TC, Diamond JS, Gan WB, Roger JE, Wong WT (2016) Requirement for microglia for the maintenance of synaptic function and integrity in the mature retina. J Neurosci 36:2827–2842. 10.1523/JNEUROSCI.3575-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe SE, Berezovsky A, Motono M, Sacai PY, Pereira JM, Sallum JM, Gebrim LH, Salomão SR (2010) Retinal function in patients treated with tamoxifen. Doc Ophthalmol 120:137–143. 10.1007/s10633-009-9203-8 [DOI] [PubMed] [Google Scholar]
- Wert KJ, Lin JH, Tsang SH (2014) General pathophysiology in retinal degeneration. Dev Ophthalmol 53:33–43. 10.1159/000357294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S (2013) Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38:79–91. 10.1016/j.immuni.2012.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida N, Ikeda Y, Notomi S, Ishikawa K, Murakami Y, Hisatomi T, Enaida H, Ishibashi T (2013a) Laboratory evidence of sustained chronic inflammatory reaction in retinitis pigmentosa. Ophthalmology 120:e5–e12. 10.1016/j.ophtha.2012.07.008 [DOI] [PubMed] [Google Scholar]
- Yoshida N, Ikeda Y, Notomi S, Ishikawa K, Murakami Y, Hisatomi T, Enaida H, Ishibashi T (2013b) Clinical evidence of sustained chronic inflammatory reaction in retinitis pigmentosa. Ophthalmology 120:100–105. 10.1016/j.ophtha.2012.07.006 [DOI] [PubMed] [Google Scholar]
- Yu Y, Chen H, Su SB (2015) Neuroinflammatory responses in diabetic retinopathy. J Neuroinflammation 12:141. 10.1186/s12974-015-0368-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabel MK, Zhao L, Zhang Y, Gonzalez SR, Ma W, Wang X, Fariss RN, Wong WT (2016) Microglial phagocytosis and activation underlying photoreceptor degeneration is regulated by CX3CL1-CX3CR1 signaling in a mouse model of retinitis pigmentosa. Glia 64:1479–1491. 10.1002/glia.23016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Lei B, Lam TT, Yang F, Sinha D, Tso MO (2004) Neuroprotection of photoreceptors by minocycline in light-induced retinal degeneration. Invest Ophthalmol Vis Sci 45:2753–2759. 10.1167/iovs.03-1344 [DOI] [PubMed] [Google Scholar]
- Zhang H, Xie M, Schools GP, Feustel PF, Wang W, Lei T, Kimelberg HK, Zhou M (2009) Tamoxifen mediated estrogen receptor activation protects against early impairment of hippocampal neuron excitability in an oxygen/glucose deprivation brain slice ischemia model. Brain Res 1247:196–211. 10.1016/j.brainres.2008.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Milatovic D, Aschner M, Feustel PJ, Kimelberg HK (2007) Neuroprotection by tamoxifen in focal cerebral ischemia is not mediated by an agonist action at estrogen receptors but is associated with antioxidant activity. Exp Neurol 204:819–827. 10.1016/j.expneurol.2007.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Zabel MK, Wang X, Ma W, Shah P, Fariss RN, Qian H, Parkhurst CN, Gan WB, Wong WT (2015) Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol Med 7:1179–1197. 10.15252/emmm.201505298 [DOI] [PMC free article] [PubMed] [Google Scholar]