

Abstract
Huntington's disease (HD) is a neurodegenerative disorder caused by expansion of a polyglutamine (polyQ) stretch within the Huntingtin (Htt) protein. Pathogenic Htt disrupts multiple neuronal processes, including gene expression, axonal trafficking, proteasome and mitochondrial activity, and intracellular vesicle trafficking. However, the primary pathogenic mechanism and subcellular site of action for mutant Htt are still unclear. Using a Drosophila HD model, we found that pathogenic Htt expression leads to a profound overgrowth of synaptic connections that correlates directly with the levels of Htt at nerve terminals. Branches of the same nerve containing different levels of Htt show distinct phenotypes, indicating that Htt acts locally to disrupt synaptic growth. The effects of pathogenic Htt on synaptic growth arise from defective synaptic endosomal trafficking, leading to expansion of a recycling endosomal signaling compartment containing Sorting Nexin 16 and a reduction in late endosomes containing Rab11. The disruption of endosomal compartments leads to elevated BMP signaling within nerve terminals, driving excessive synaptic growth. Blocking aberrant signaling from endosomes or reducing BMP activity ameliorates the severity of HD pathology and improves viability. Pathogenic Htt is present largely in a nonaggregated form at synapses, indicating that cytosolic forms of the protein are likely to be the toxic species that disrupt endosomal signaling. Our data indicate that pathogenic Htt acts locally at nerve terminals to alter trafficking between endosomal compartments, leading to defects in synaptic structure that correlate with pathogenesis and lethality in the Drosophila HD model.
SIGNIFICANCE STATEMENT Huntington's disease (HD) is the most commonly inherited polyglutamine expansion disorder, but how mutant Huntingtin (Htt) disrupts neuronal function is unclear. In particular, it is unknown within what subcellular compartment pathogenic Htt acts and whether the pathogenesis is associated with aggregated or more soluble forms of the protein. Using a Drosophila HD model, we find that nonaggregated pathogenic Htt acts locally at synaptic terminals to disrupt endosomal compartments, leading to aberrant wiring defects. Genetic manipulations to increase endosomal trafficking of synaptic growth receptors from signaling endosomes or to reduce BMP signaling reduce pathology in this HD model. These data indicate that pathogenic Htt can act locally within nerve terminals to disrupt synaptic endosomal signaling and induce neuropathology.
Keywords: BMP, Drosophila, endosome, Huntington's disease, polyglutamine, synapse
Introduction
Huntington's disease (HD) is an autosomal-dominant neurodegenerative disorder characterized by motor, cognitive, and emotional defects. The expansion of a polyglutamine tract (polyQ) within the Huntingtin (Htt) protein causes the disease (MacDonald et al., 1993; Bates, 2005). Numerous animal models of HD have been generated to examine how polyQ expansion leads to neurotoxicity. A diverse set of pathologies has been identified, including impaired vesicle trafficking and axonal transport, altered proteasomal degradation, mitochondrial dysfunction, and transcriptional dysregulation. At the molecular level, polyQ repeat expansion makes Htt prone to aggregation, a hallmark of the polyQ family of aggregation disorders. However, conflicting evidence regarding aggregation toxicity has been reported for different cell types (Saudou et al., 1998; Kim et al., 1999; Kuemmerle et al., 1999). In some cases, aggregation has neuroprotective roles, with soluble Htt identified as the toxic species (Arrasate et al., 2004). Indeed, several mouse (Hodgson et al., 1999) and Drosophila HD models (Romero et al., 2008) show neurodegeneration and HD-associated phenotypes without obvious aggregation. Despite efforts to identify the primary molecular pathology in HD, the pathogenic role of Htt and its normal cellular function are still uncertain.
One function for Htt that has emerged is the regulation of endosomal and vesicle trafficking. Htt is predominantly a cytoplasmic protein, but can be found in membrane fractions and on diverse vesicle compartments (DiFiglia et al., 1995). Many Htt binding partners function in vesicle endocytosis and trafficking, including PACSIN1, endophilin3, HIP1, and HAP1 (Wanker et al., 1997; Modregger et al., 2002; Li and Li, 2004). HIP1 has a conserved epsin N-terminal homology domain that interacts with membranes. An analogous protein, epsin, has been shown to modify membrane curvature directly (Ford et al., 2002). Htt and HIP1 also interact directly with α-adaptin and clathrin (Waelter et al., 2001), well known regulators of endocytosis. Together with other data, these studies indicate that Htt and its binding partners may regulate vesicle endocytosis and trafficking. It is currently unclear whether pathogenic Htt alters the normal function of the protein in vesicle trafficking and if altered membrane trafficking plays an important role in HD pathology.
In this study, we found that pathogenic Htt alters endosomal signaling in a Drosophila HD model. During classical receptor-mediated endocytosis, receptors are internalized via clathrin-dependent endocytosis and accumulate in sorting endosomes before being recycled back to the plasma membrane or degraded (Grant and Donaldson, 2009). Internalized receptors can transmit signals while residing in early endosomal compartments and additional trafficking to recycling endosomes or degradation is required to turn these pathways off (Sorkin et al., 2000; Miaczynska et al., 2004). At the Drosophila neuromuscular junction (NMJ), postsynaptic muscles release BMP ligands that drive synaptic growth and maturation by binding to receptors present in the presynaptic membrane (Harris and Littleton, 2015). Previous studies found that BMP receptors remain active after internalization into signaling endosomes (O'Connor-Giles et al., 2008; Rodal et al., 2011). Genetic manipulations that disrupt exit from signaling endosomes result in enhanced synaptic growth secondary to elevated BMP signaling. We find that pathogenic Htt interferes with endosomal recycling, leading to accumulation of early endosomal signaling compartments that trigger a robust overgrowth of synaptic connections. These changes result in elevated signaling from the BMP pathway. We observed enhanced levels of synaptic phosphorylated Mad (pMad), a downstream readout of BMP receptor activation, whereas nuclear pMad levels were unaltered. Manipulation of the small RAB GTPases, which regulate trafficking through endosomal compartments, indicate that promoting receptor recycling reduces synaptic overgrowth and lethality associated with pathogenic Htt expression. These changes in endosomal signaling occur secondary to local effects of pathogenic Htt at the synapse, rather than from cell-wide defects.
Materials and Methods
Fly stocks.
UAS-HttQ138-mRFP, UAS-HttQ15-mRFP, Snx16Δ1, Snx16Δ2 and UAS-Snx16-GFP were generated previously (Lee et al., 2004; Rodal et al., 2011; Weiss et al., 2012). The UAS-Rab7Q67L and UAS-Rab7T22N strains were obtained from Gregory Emery (Assaker et al., 2010). UAS-photoactivatable GFP lines were obtained from Richard Axel (Ruta et al., 2010). The remaining strains were obtained from the Bloomington Stock Center. All lines were raised at 25°C. Both male and female larvae were used unless noted.
Immunostaining and AZ counting.
Wandering third-instar larvae were dissected in Ca2+-free saline and fixed for 15 min in PBS solution with 4% paraformaldehyde. After a 1 h wash in PBST with 2% BSA and 2% NGS, samples were incubated overnight with primary antibody at 4°C and for 2–3 h with secondary antibodies at room temperature. Samples were washed and mounted in Vectashield (Vector Laboratories) for analysis. For comparative analysis, larvae from different genotypes were dissected and fixed together. After fixation, samples from different genotypes were cut to specific shapes and again transferred and incubated together in primary and secondary antibodies to ensure equal exposure to antigens. The following antibodies were used: Alexa Fluor 647-conjugated anti-HRP at 1:500 (Jackson ImmunoResearch); mouse anti-NC82 (BRP; RRID:AB_2314867) at 1:200 and mouse anti-Dlg (RRID:AB_2314321) at 1:500 (Developmental Studies Hybridoma Bank), guinea pig anti-Snx16 (gift from Avital Rodal laboratory) at 1:200; and guinea pig anti-pMad at 1:500 (a gift from Ed Laufer, Columbia University, NY). Species-specific Alexa Fluor-488 or Alexa Fluor-647 secondary antibody (Invitrogen RRID:AB_141882; RRID:AB_2576217; RRID:AB_142018) was used at 1:1000. To calculate the active zone (AZ) number, automated algorithms were created using Volocity version 3.3 software. Briefly, the software detects objects with levels of brightness >4 SDs above background within a preset radius of 0.5 μm. The total number of AZs was normalized by NMJ volume determined from the HRP-positive channel.
Western blot.
For detection of Htt levels, 10 third-instar larval brains were homogenized in 2× Laemmli sample buffer (Bio-Rad). After homogenization and removal of cell debris by centrifugation, protein lysates were analyzed by Western blotting. Then, 5 μl (1 brain) was loaded on a 10–15% SDS gel (Bio-Rad) and later transferred to nitrocellulose membrane. Htt levels were detected with anti-RFP (Abcam ab65562; RRID:AB_1142368) and anti-α-tubulin antibodies were used as a loading control (σ T5168; RRID:AB_477579). The primary antibody was applied at 4°C overnight and the secondary antibody (anti-rabbit and anti-mouse antibodies from LI-COR) for 1 h at room temperature. For detection of Snx16 levels from adults, flies were frozen in liquid nitrogen and heads were separated from bodies by vortexing. Ten heads were homogenized in 50 μl of 2× Laemmli sample buffer (Bio-Rad) and 5 μl (1 head) was loaded on a 10–15% SDS gel (Bio-Rad). Anti-Snx16 and anti-α-tubulin were used as primary antibodies, with anti-guinea pig and anti-mouse antibodies (LI-Cor RRID:AB_621843; RRID:AB_10706161) as a secondary. All antibodies were used at 1:5000.
Bouton counting and adult survivability rate determination.
NMJs from muscle 4 of segments A2–A3 were used for imaging and quantification of bouton number. Satellite boutons in HttQ138 often consisted of multiple long branches of small boutons <1 μm in diameter. Therefore, all boutons <1 μm that were not followed by larger boutons were quantified as satellite boutons. Statistical significance was calculated in Prism software (GraphPad) using Student's t tests when comparing two groups or ANOVA followed by Tukey's multiple-comparisons test when >2 groups were compared (in figures, **p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001). Error bars in figures represent SEM. Confocal images were obtained on an Axio Imager 2 from Zeiss equipped with a spinning-disk confocal head (CSU-X1; Yokagawa) and ImagEM X2 EM-CCD camera (Hammamatsu). Data analysis of fluorescence and NMJ volume was performed with Volocity software (PerkinElmer). For analysis of Snx16 and Rab intensities, 3D image stacks were used. NMJ volume was detected automatically by the software using the anti-HRP staining channel as a guide. Total Rab-positive NMJ fluorescence above background level was normalized by NMJ volume. When FYVE puncta were analyzed, a higher-intensity threshold was used that separates brighter larger endocytic structures from dimmer cytosolic component staining. Given that the GFP-labeled Rab lines have different intensities, each dataset is shown as a ratio of fluorescence change compared with control. For assaying adult survival, eight to 10 third-instar larvae of each genotype were transferred into fresh vials with food and the number of adults that eclosed was scored. Each data point represents the survivability of eight to10 flies.
FRAP experiments and photoconversion.
FRAP was used to analyze the mobility of Htt-RFP proteins. The brightest NMJ that had fluorescence similar to the fluorescence of HttQ138 aggregates was preselected. Using 100% excitation laser power, the axonal aggregates or an NMJ bouton were photobleached. Only boutons in the middle of the NMJ were used. Samples with complete bleaching of the desired region were analyzed. Fluorescence levels during recovery are presented as a percentage from the initial 100% fluorescence before bleaching and calculated for each time point. For photoconversion experiments, a FRAP module was used to initiate fluorescence conversion in a desired region. UAS-C3PA-GFP (photoactivatable GFP or PA-GFP; Ruta et al., 2010) was expressed together with HttQ138 and one terminal bouton of the NMJ was illuminated. PA-GFP exhibits low-level fluorescence when it is not activated, but fluorescence is dramatically enhanced after photoconversion with 488 nm high-energy light. Diffusion of photoactivated PA-GFP from the terminal bouton was followed over time to confirm the connection between individual NMJ branches.
Electron microscopy (EM).
Third-instar larvae expressing HttQ138 and HttQ15 were dissected in Ca2+-free solution and fixed in 1% glutaraldehyde, 4% formaldehyde, and 0.1 m sodium cacodylate for 2 h at room temperature and then 4°C overnight. After washing in 0.1 m sodium cacodylate and 0.1 m sucrose, samples were postfixed for 1 h in 1% osmium tetroxide, dehydrated through a graded series of ethanol and then acetone, and embedded in epoxy resin (Embed 812; Electron Microscopy Sciences). Thin sections (40–50 nm) were collected on Formvar/carbon-coated copper slot grids and contrasted with lead citrate. Sections were imaged at a 49,000× magnification at 80 kV on an electron microscope (Tecnai G2 Spirit; FEI) equipped with a charge-coupled device camera (Advanced Microscopy Techniques). Only type 1b boutons were analyzed.
Results
Pathogenic Htt alters synaptic morphology
We previously generated a transgenic Drosophila HD model expressing a 588 aa N-terminal fragment of human Htt tagged with mRFP and containing either a pathogenic polyQ tract of 138 repeats (HttQ138) or a nonpathogenic tract of 15 repeats (HttQ15) (Lee et al., 2004; Weiss et al., 2012). For this study, we used UAS lines with comparable expression levels of HttQ138 and HttQ15 (Schulte et al., 2011) driven with the pan-neuronal ElavC155 GAL4 driver. Expression of pathogenic HttQ138 causes strong cytoplasmic Htt aggregation in neurons of the ventral nerve cord, including in motor neuron axons, where it accumulates stochastically at synaptic terminals (Weiss et al., 2012). During our analysis of these lines, we observed that the morphology of larval NMJs of HttQ138-expressing lines was severely abnormal. We quantified synaptic growth at larval muscle 4 in abdominal segments 2 and 3, a well studied synaptic connection routinely used for synaptic growth analysis in Drosophila. We found that expression of HttQ138 causes severe NMJ overgrowth (Fig. 1A–C), accompanied by a large number of small synaptic boutons branching off from larger boutons and interbouton spaces. Such small boutons have been previously termed “satellite” boutons (Torroja et al., 1999) and are often observed in mutants in which endocytosis or endosomal trafficking is abnormal. The excess synaptic growth in HttQ138-expressing larvae is largely associated with these satellite boutons because they are immunopositive for the presynaptic AZ component BRP and the postsynaptic PSD-95 homolog, DLG. Larvae expressing HttQ15 had normal morphology, similar to control lines (ElavC155). Quantification of the number of satellite boutons at muscle 4 showed that HttQ138-expressing larvae had 28.7 ± 2.1 (n = 29, p ≤ 0.001) satellite boutons compared with only 3.1 ± 0.8 (n = 20) in HttQ15-expressing larvae. NMJs of control larvae were similar to HttQ15, with few satellite boutons (1.3 ± 0.3, n = 18, p > 0.05). The changes in satellite bouton number were not accompanied by alterations in muscle area (HttQ15: 77610 ± 3400 μm2 for HttQ15, n = 9; HttQ138: 67401 ± 3445 μm2, n = 11). AZ density was also unchanged (Fig. 1D), suggesting a more restricted defect in bouton budding as opposed to an overall change in synaptic maturation. Previous analysis of protein expression levels using adult flies showed that HttQ15 and HttQ138 lines have similar levels of expression (Schulte et al., 2011). We performed Western blot analysis on larvae and confirmed that Htt levels relative to tubulin (HttQ15: 0.98 ± 0.1, n = 9; HttQ138: 1.1 ± 0.1, n = 7) were also similar at the larval stage (Fig. 1E).
Figure 1.
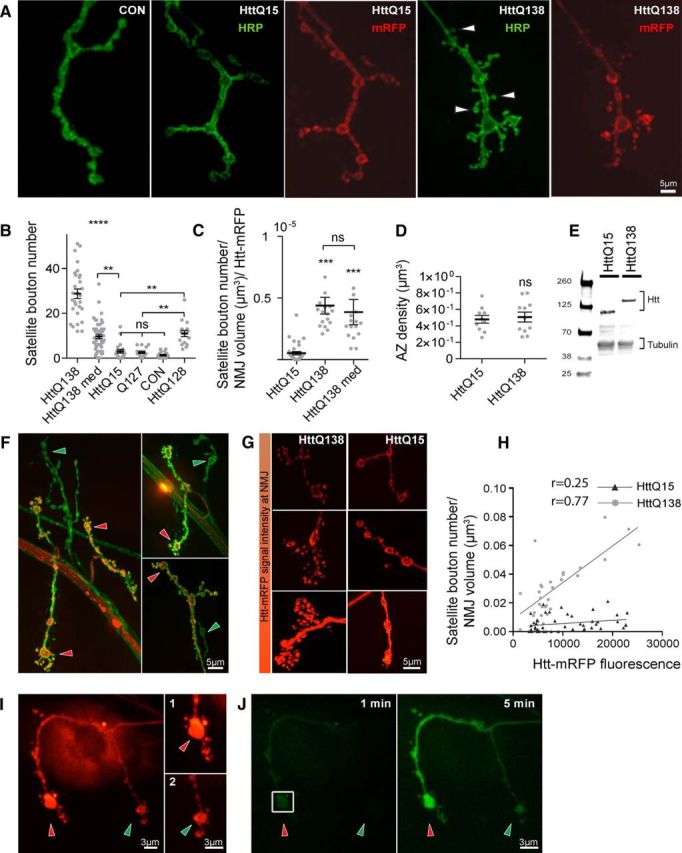
Expression of pathogenic HttQ138 induces overgrowth of NMJ synapses. A, Representative images of third instar NMJ 4 in control (ElavC155), ElavC155;UAS-HttQ15-mRFP and ElavC155;UAS-HttQ138-mRFP larvae stained with anti-HRP to label neuronal membranes. HttQ138 expression results in a robust overgrowth of smaller satellite boutons (arrowheads). B, Quantification of satellite bouton number at NMJ 4 in ElavC155;UAS-HttQ138-mRFP high expression, ElavC155;UAS-HttQ138-mRFP medium expression, ElavC155;UAS-HttQ15-mRFP, ElavC155;UAS-Q127, control (ElavC155), and ElavC155;UAS-HttQ128. C, Level of synaptic overgrowth induced by HttQ138, HttQ138med, and HttQ15 normalized by NMJ volume and Htt-RFP intensity. D, Expression of HttQ138 does not alter AZ density compared with HttQ15. E, Western blot data showing similar Htt expression levels in HttQ138 and HttQ15 larval extracts. F, NMJ 4 axon terminals expressing HttQ138-mRFP (red) stained with anti-HRP (green). HttQ138 accumulates stochastically in different nerve branches, resulting in synaptic overgrowth in those areas where HttQ138 has accumulated (red arrows). Branches lacking HttQ138 display more normal morphology (green arrows). G, Different levels of HttQ138 and HttQ15 accumulation (red) can be observed at distinct muscle 4 NMJs, resulting in increasing severity of synaptic overgrowth. H, Correlation between HttQ138 and HttQ15 expression at NMJ 4 synapses with the severity of satellite bouton overgrowth. I, Differential accumulation of HttQ138 can be seen at different branches of the same axon. The branch with more Htt expression (I1, red arrow) shows a more robust synaptic overgrowth phenotype compared with the branch with less HttQ138 accumulation (I2, green arrow). J, Diffusion of PA-GFP after photoactivation of the squared region of the NMJ shown in H. PA-GFP diffuses from the branch with more HttQ138 (red arrow) to the branch with lower HttQ138 accumulation, confirming that the branches arise from the same axon. Images were taken 1 min (left) and 5 min (right) after photoactivation. ANOVA followed by Tukey's multiple-comparisons tests was used for statistical analysis. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. Error bars indicate SEM.
Although we observed severe synaptic overgrowth in HttQ138-expressing lines, the phenotype was not uniform across all NMJs. Indeed, muscle 4 NMJs with mild overgrowth was associated with lower levels of HttQ138-mRFP accumulation detected optically (Fig. 1F, G). Although it is unknown why HttQ138 accumulation at nerve branches is stochastic, it may reflect previously identified axonal transport defects that occur in Drosophila HD models (Lee et al., 2004), leading to reduced accumulation at some synaptic terminal branches as opposed to others. Overall, we observed a positive correlation between mean HttQ138 fluorescence at the NMJ and the number of satellite boutons per NMJ volume (Pearson correlation coefficient r = 0.77, R2 = 0.59, n = 29, p ≤ 0.001; Fig. 1H). Different axonal branches located next to each other that accumulated distinct levels of HttQ138 had very distinct morphology, from normal (Fig. 1F, green arrows) to severely overgrown (Fig. 1F, red arrows). These results suggest that expression of pathogenic HttQ138 perturbs axonal branching and leads to NMJ overgrowth in a dose-dependent manner. HttQ15-expressing lines also had a nonuniform accumulation at terminals, although the heterogeneity for HttQ15 accumulation was less common than observed in HttQ138 lines (Fig. 1G). However, the correlation between HttQ15 accumulation and NMJ overgrowth was not significant (Pearson correlation coefficient r = 0.25, R2 = 0.06, n = 47, p ≤ 0.001; Fig. 1G,H). To further assay the dependence of HttQ138 levels on synaptic growth, we analyzed overgrowth in UAS lines that had ∼50% lower expression of HttQ138 (termed HttQ138 medium or HttQ138med; Schulte et al., 2011). Larvae expressing lower levels of HttQ138 had milder overgrowth (9.7 ± 1.0, n = 47, p ≤ 0.01), but still significantly higher than lines expressing nonpathogenic HttQ15 (Fig. 1B). To determine whether differences in NMJ overgrowth between HttQ138 and HttQ138 med were due to the different levels of Htt at the NMJ, we normalized overgrowth rate by NMJ volume and Htt-RFP levels. We found that both HttQ138 (4.4 ± 0.7e-006, n = 17) and HttQ138med (3.8 ± 1.0e-006, n = 16) induced overgrowth proportionally to the levels of Htt accumulation and significantly different from the HttQ15 line (0.5 ± 0.1e-006; n = 34; Fig. 1C).
In some cases, differential accumulation of HttQ138 was observed in NMJ branches that appeared to be part of the same axon from a single neuron, with differing severity of synaptic overgrowth observed (Fig. 1F,G). If these branches were from the same neuron, the data would argue that synaptic overgrowth is secondary to local synaptic alterations where pathogenic Htt accumulates, rather than from cell-wide defects such as global transcriptional dysregulation. Given that it is unclear from which cellular compartment Htt toxicity arises, it was important to establish whether synaptic growth alterations were secondary to local processes versus global effects of pathogenic Htt. To determine whether the same axon could show distinct phenotypes depending upon the local concentration of pathogenic Htt, we expressed HttQ138-mRFP along with PA-GFP (Ruta et al., 2010). At NMJs that had unequal accumulation of HttQ138 in different axonal branches (Fig. 1I), we photoactivated GFP in a bouton of one branch (Fig. 1G) and determined whether the PA-GFP signal was able to diffuse to the second branch. Indeed, we were able to observe PA-GFP signal propagation between branches with different HttQ138 levels (Fig. 1I,J). Therefore, HttQ138 accumulates at different levels within the same axon, where its local presence triggers synaptic overgrowth not only at the level of the axon, but also at the level of individual axonal branches. These observations suggest that more global cell-wide effects such as transcriptional dysregulation do not mediate the alterations in synaptic connectivity because one would expect such defects to be visible throughout the entire neuronal arbor.
Given that HD is associated with expansion of a polyQ stretch within the Htt protein, we investigated whether expression of a polyQ stretch alone would lead to NMJ overgrowth. We did not observe any overgrowth in larvae expressing a polyQ127 stretch alone (2.5 ± 0.5, n = 19, p ≤ 0.05; Fig. 1B). This was not due to the slightly smaller polyQ region (Q127 vs Q138 in Htt) because expression of a different Htt construct with a similar polyQ expansion length (HttQ128; Lee et al., 2004) still caused synaptic overgrowth (11.0 ± 1.4, n = 16, p ≤ 0.01; Fig. 1B). Therefore, synaptic overgrowth is observed after expression of pathogenic Htt polyQ, but not with an isolated polyQ expansion, suggesting that Htt-mediated protein interactions are likely involved with the pathogenesis.
Synaptic HttQ138 is largely present in a nonaggregated state
Aggregation of Htt with an expanded polyQ is considered a hallmark of HD. Because accumulation of HttQ138 causes overgrowth at the NMJ, we investigated whether Htt at NMJs exists predominantly in an aggregated form. HttQ138-mRFP appeared to be diffusely present at nerve terminals, rather than in the large, bright, immobile fluorescent puncta that are observed in axonal aggregates that we characterized previously (Weiss et al., 2012). To further address this question, we compared fluorescence recovery after photobleaching (FRAP) of HttQ138-mRFP and HttQ15-mRFP in synaptic boutons versus large, immobile axonal aggregates in HttQ138-mRFP. FRAP analysis of Htt in synaptic boutons showed a robust and immediate recovery, with continued increases in recovery during the 2 min imaging session. Recovery was similar between HttQ15 and HttQ138 line at most time points (Fig. 2B). In contrast, we observed no recovery of fluorescence in axonal HttQ138 aggregates during the same interval (Fig. 2A,B). The FRAP data argue that HttQ138 is highly mobile in synaptic terminals, suggesting that it is largely found in a nonaggregated form. Interestingly, small and actively moving Htt puncta were also observed in some boutons (Fig. 2C), similar to prior imaging analysis that we performed with labeled endosomal proteins (Rodal et al., 2011). Although these mobile structures were not present in every synaptic bouton, they were common in both HttQ138- and HttQ15-expressing larva. These data indicate that synaptic HttQ138 is largely found in the cytoplasm and on small highly mobile puncta, in contrast to the large, immobile aggregates observed in axons.
Figure 2.

HttQ138 accumulates at NMJ in nonaggregated form. A, Bleaching of a synaptic bouton with severe overgrowth (dotted region, top) from ElavC155;UAS-HttQ138-mRFP-expressing larvae. Fluorescence recovery was followed for 2 min in 12 s intervals, was observed within the first 12 s and continued throughout the imaging session. In contrast, bleaching of axonal aggregates from ElavC155;UAS-HttQ138-mRFP larvae (bottom, dotted area) showed no recovery of fluorescence during the 2 min imaging session. B, Quantification of fluorescence recovery in the bleached area in synaptic boutons of HttQ138 (gray circle line) and HttQ15 (black triangle line)-expressing larvae compared with axonal aggregates (gray square line) of HttQ138-mRFP-expressing larvae. C, More dynamic Htt-labeled puncta are also found in synaptic boutons from ElavC155;UAS-HttQ138-mRFP larvae, which resemble endocytic structures that transition between steady states and active movement.
Pathogenic Htt perturbs endosomal trafficking
Accumulation of pathogenic HttQ138 in axonal branches leads to NMJ overgrowth with excessive satellite boutons, a phenotype similar to that reported previously for a number of endocytosis and vesicle trafficking mutants, including Dap160, Rab11, and Nwk (Marie et al., 2004; Khodosh et al., 2006; O'Connor-Giles et al., 2008; Rodal et al., 2011). Given that prior studies have implicated Htt and Htt-binding proteins in endocytic trafficking (DiFiglia et al., 1995; Waelter et al., 2001), we investigated whether endosomal compartments might be perturbed after accumulation of pathogenic HttQ138 at presynaptic terminals. Distinct endosomal compartments can be identified by the presence of specific Rab proteins (Zerial and McBride, 2001; Grant and Donaldson, 2009; Jean and Kiger, 2012, 2012). Using fluorescently labeled Rabs, we visualized endosomal compartments and examined their changes in the presence of pathogenic HttQ138 versus HttQ15 lines. Specifically, we used GFP-labeled Rab5 and FYVE to visualize early endosomal compartments, Rab11 to label recycling endosomes, and Rab7 and Spinster (Spin) to label late endosomal and lysosomal compartments (Ullrich et al., 1996; Bucci et al., 2000, 2000; Nakano et al., 2001; Stein et al., 2003; Dermaut et al., 2005). These markers were coexpressed with HttQ138 or HttQ15 using ElavC155. Rab5 labeling was slightly diminished in HttQ138-expressing larvae (relative fluorescence intensity of 0.8 ± 0.1, n = 27, p ≤ 0.05) compared with HttQ15 (1.0 ± 0.0, n = 26) (Fig. 3A,B). FYVE-positive staining was not statistically altered in HttQ138 boutons (0.8 ± 0.1, n = 53, p > 0.05) compared with HttQ15 (1.0 ± 0.1, n = 54). Given that endosomal compartments can be better visualized in live animals compared with fixed tissue (Rodal et al., 2011), we also imaged FYVE-GFP puncta at synaptic terminals in live dissected larvae. Prior work indicated that FYVE-positive endosomes are associated with receptor trafficking and accumulate transferrin (Gillooly et al., 2000). Imaging of FYVE-positive endosomes revealed that HttQ138 induced a significant increase in staining within puncta (1.8 ± 0.3-fold increase, n = 18, p ≤ 0.05). Analysis of the later recycling endosomal compartment labeled by Rab11 indicated that the recycling endosomal compartment was reduced in the presence of HttQ138 (0.8 ± 0.0, n = 50, p ≤ 0.001; Fig. 3A,B). In contrast, late endosomal and lysosomal compartments positive for Rab7 and Spin were largely unaffected by HttQ138 expression (Fig. 3A,B). Rab7 positive fluorescence distributed uniformly at NMJs, with some rare larger components occasionally observed. When fluorescence of these brighter puncta was quantified, we observed that HttQ138 lines had a higher accumulation of Rab7-positive puncta compared with controls (2.3 ± 0.2-fold, n = 11, p ≤ 0.01; Fig. 3A,B). These findings suggest that HttQ138 expression alters compartments that participate in endosomal trafficking and recycling at the synapse.
Figure 3.
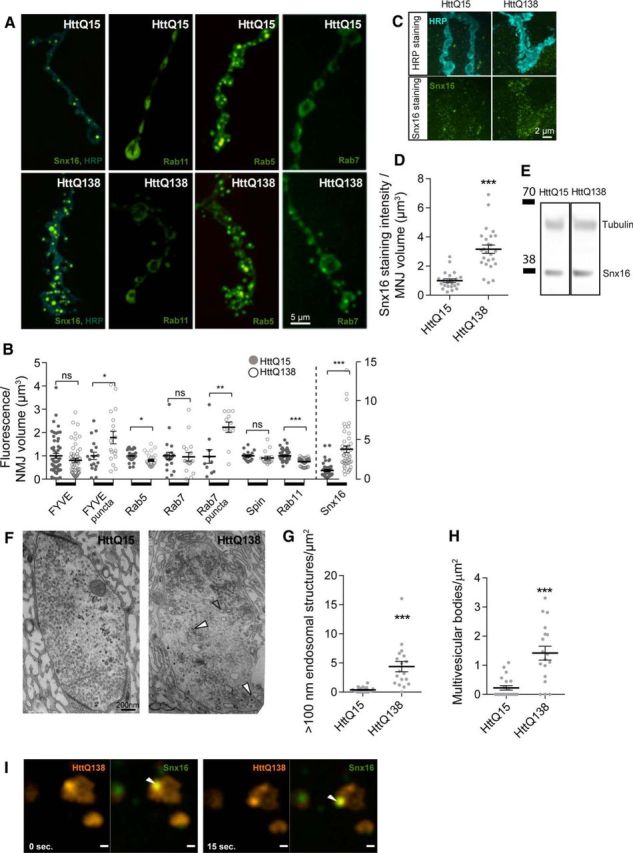
HttQ138 alter endosomal trafficking and leads to abnormal changes in a key endocytic compartments. A, Representative images of third-instar NMJ 4 showing distribution of ElavC155-driven UAS-Snx16-GFP, UAS-Rab11-GFP, UAS-Rab5-GFP, and UAS-Rab7-GFP proteins on a background of control ElavC155;UAS-HttQ15-mRFP or pathogenic ElavC155;UAS-HttQ138-mRFP lines. Anti-HRP staining was used to visualize neuronal membranes (blue). Strong accumulation of Snx16-positive compartments was observed in ElavC155;UAS-HttQ138-mRFP/UAS-Snx16-GFP larvae. The levels of UAS-Rab11-GFP and UAS-Rab5-GFP were reduced after expression of pathogenic UAS-HttQ138-mRFP (ElavC155;UAS-HttQ138-mRFP/UAS-Rab11-GFP, ElavC155;UAS-HttQ138-mRFP/UAS-Rab5-GFP and ElavC155;UAS-HttQ138-mRFP/UAS-Rab7-GFP). B, Quantification of GFP fluorescence with ElavC155-driven expression of UAS-FYVE-GFP, UAS-Rab5-GFP, UAS-Rab7-GFP, UAS-spin-GFP, UAS-Rab11-GFP, and UAS-Snx16-GFP, together with UAS-HttQ15-mRFP (filled circles) or UAS-HttQ138-mRFP (empty circles). Note the different scale (right y-axis) used for Snx16 levels (separated by dotted line). C, ElavC155;UAS-HttQ15-mRFP and ElavC155;UAS-HttQ138-mRFP lines were stained with anti-HRP (top) and anti-Snx16 (bottom). D, Quantification of Snx16-positive staining. Snx16 levels were normalized to NMJ volume determined by HRP labeling. E, Western blot analysis of Snx16 levels from head extracts of HttQ15- and HttQ138-expressing larvae. F, Representative ultramicrographs of ElavC155;UAS-HttQ15-mRFP- and ElavC155;UAS-HttQ138-mRFP-expressing boutons. Accumulation of large vacuolar structures (empty arrows) and multivesicular bodies (filled arrows) were observed in HttQ138-expressing boutons. G, Quantification of large vacuolar structures that are larger than 100 nm in diameter in HttQ15- and HttQ138-expressing larvae. H, Quantification of multivesicular bodies. Structures with inclusion of smaller vesicles inside (white filled arrow) were counted and normalized by bouton area. I, Dynamics of HttQ138 (red) and Snx16 (green) puncta in ElavC155;UAS-HttQ138-mRFP/UAS-Snx16-GFP larval NMJ boutons imaged from single confocal planes. The merged image is on the right, with colocalized puncta marked with a white arrow. Snx16 puncta colocalize with HttQ138 puncta and move together. Student's t test was used for statistical analysis. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. Error bars indicate SEM.
Growth factor ligand/receptor signaling complexes that regulate synapse expansion traffic through distinct endosomal stages. To visualize more directly the growth factor receptor-containing compartments, we imaged transgenic larvae expressing GFP-tagged Sorting Nexin 16 (Snx16; Hanson and Hong, 2003; Choi et al., 2004; Rodal et al., 2011). Snx16 regulates trafficking of BMP and Wnt receptors in Drosophila presynaptic terminals (Hanson and Hong, 2003), defining an early signaling endosome that is critical for growth factor signaling. Coexpression of Snx16-GFP and HttQ138 resulted in the abnormal accumulation of this sorting endosomal compartment compared with HttQ15 (Fig. 3A,B). Overall, Snx16-GFP fluorescence levels increased 3.8 ± 0.4-fold (n = 43, p ≤ 0.001) at synaptic terminals in HttQ138 animals compared with HttQ15. The size of individual Snx16 synaptic puncta also increased from 0.1 ± 0.02 μm3 (n = 20) in HttQ15 terminals to 0.3 ± 0.1 μm3 (n = 12, p ≤ 0.001) in HttQ138. To confirm the increase in the synaptic Snx16 compartment, we performed immunochemistry using antisera to the protein (Rodal et al., 2011). We could barely detect Snx16-positive staining in nonpathogenic HttQ15 lines (Fig. 3C), but could clearly detect Snx16 in HttQ138 lines. We observed a 3.2 ± 0.3-fold increase (n = 25, p ≤ 0.001) in Snx16 immunostaining in HttQ138 synapses compared with control HttQ15 synapses (Fig. 3C,D). We also investigated whether Snx16 protein levels change after HttQ138 expression. Western analysis from adult head extracts indicated that Snx16 protein levels were not upregulated (HttQ15: 1.1 ± 0.4, n = 3; HttQ138: 0.9 ± 0.2, n = 3; Fig. 3E), suggesting altered synaptic distribution rather than changes in overall protein levels.
As noted above, we often observed Htt puncta in some boutons (Fig. 2C). These Htt-positive puncta did not show obvious colocalization with any of the visualized Rab-positive compartments in our experiments. However, in many cases, they were positioned next to the Snx16-labeled compartment. We hypothesized that these compartments may interact transiently, which has been observed previously for endosomal compartments containing the Snx family (Merino-Trigo et al., 2004; van Weering et al., 2010; Rodal et al., 2011). Therefore, we performed live imaging of these actively moving compartments and observed multiple fusion and separation events between the HttQ138-mRFP- and Snx16-GFP-labeled compartments. Some puncta were positive for HttQ138 and Snx16 simultaneously and could be seen actively moving together (Fig. 3I). These data indicate that Htt is likely to interact transiently with the Snx16 early endosomal signaling compartment and may alter cargo transport out of this compartment, leading to an increase in its size and a decrease in compartments labeled by Rab5 and Rab11.
Expression of pathogenic Htt leads to an increase in large internal synaptic membrane compartments visualized by EM
To determine whether the expansion of the Snx16 endosomal signaling compartment could be observed by transmission electron microcopy (EM), we performed EM analysis of synaptic terminals from larvae expressing HttQ138 or HttQ15. Synaptic boutons expressing HttQ15 were filled with uniform synaptic vesicles and occasionally had 100–200 nm long endocytic-like structures (Fig. 3F). In contrast, boutons expressing pathogenic HttQ138 accumulated large vesicular structures and multivesicular bodies (Fig. 3F–H). Quantification of synaptic vesicular structures larger than 100 nm in diameter revealed that HttQ138 flies had 4.4 ± 1.0 (n = 18, p ≤ 0.001) large vesicular structures per square micrometer, whereas HttQ15 lines only had 0.4 ± 0.1 (n = 21; Fig. 3G). There was also a significant enhancement in the number of synaptic multivesicular bodies in HttQ138 (1.4 ± 0.2/μm2, n = 18, p ≤ 0.001) compared with HttQ15 (0.2 ± 0.1/μm2, n = 21; Fig. 3H). Therefore, both immunocytochemistry and EM indicate that pathogenic Htt disrupts endosomal trafficking and leads to aberrant endosomal compartments not observed after expression of nonpathogenic Htt.
Synaptic BMP signaling is enhanced by pathogenic Htt
Given the increase in Snx16 immunostaining and the accumulation of large membrane compartments observed by EM, we investigated what signaling systems might be disrupted by HttQ138 that result in the overgrowth of synaptic terminals. Among the well characterized signaling pathways required for NMJ growth and maturation in Drosophila are the BMP and Jun-N-terminal kinase (JNK) cascades (Collins and DiAntonio, 2007). BMP signaling is crucial for normal synaptic growth and homeostasis, with enhanced synaptic BMP signaling leading to excessive satellite bouton formation (McCabe et al., 2003; Goold and Davis, 2007). The Drosophila BMP ortholog Glass bottom boat (Gbb) binds to a heterotetrameric receptor complex containing Wishful thinking (Wit), Thickveins (Tkv), and Saxophone (Sax). Activation of these receptors leads to increased accumulation of the pMad transcription factor in motor neuron nuclei and synaptic terminals (McCabe et al., 2003; Dudu et al., 2006). We used genetic interaction studies to determine whether components of this signaling pathway altered the ability of HttQ138 to induce synaptic overgrowth. Expressing HttQ138 in the Gbb-null background (gbb1/gbb2) completely suppressed NMJ overgrowth observed with HttQ138 alone (Fig. 4A). The number of satellite boutons decreased from 28.7 ± 2.1 (n = 29) in HttQ138 larvae to 1.1 ± 0.3 (n = 18, p ≤ 0.001) when HttQ138 was expressed in the Gbb-null background. Although Gbb is crucial for NMJ growth and Gbb null mutants display synaptic undergrowth, the absence of a single copy of the gene does not affect NMJ morphology on its own (Nahm et al., 2010b; Kamimura et al., 2013). However, reduction of Gbb levels in heterozygous flies (gbb1/+) substantially suppressed HttQ138-induced overgrowth (Fig. 4A,B), indicating that BMP signaling regulates HttQ138-mediated synaptic overgrowth. Because we observed elevated levels of Snx16-positive compartments in HttQ138 flies and those compartments were previously associated with BMP trafficking (Rodal et al., 2011), we investigated whether downregulation of Snx16 could suppress synaptic overgrowth after HttQ138 expression. Indeed, we observed a strong reduction in satellite bouton number in Snx16Δ1/Snx16Δ2-null mutant larvae expressing HttQ138 (11.7 ± 2.1, n = 17, p ≤ 0.001) versus HttQ138 alone (28.7 ± 2.1, n = 29) (Fig. 4B). To confirm that changes in overgrowth were not due to reduced HttQ138 levels, we analyzed Htt-mRFP fluorescence at the NMJ. HttQ138 fluorescence was unchanged for the Snx16Δ1/Snx16Δ2;HttQ138 and gbb1/+;HttQ138 lines and increased in the gbb1/ gbb2;HttQ138 background (Fig. 4C). In addition, these genotypes did not have any difference in satellite bouton number compared with controls when tested alone without HttQ138 (gbb1/gbb2: 0.8 ± 0.3, n = 12; gbb1/+: 1.0 ± 0.3, n = 15; and Snx16Δ1/Snx16Δ2: 0.7 ± 0.3, n = 15). These data indicate that hyperactivation of the BMP signaling pathway regulated by Snx16 is a key driver of synaptic overgrowth after expression of HttQ138.
Figure 4.
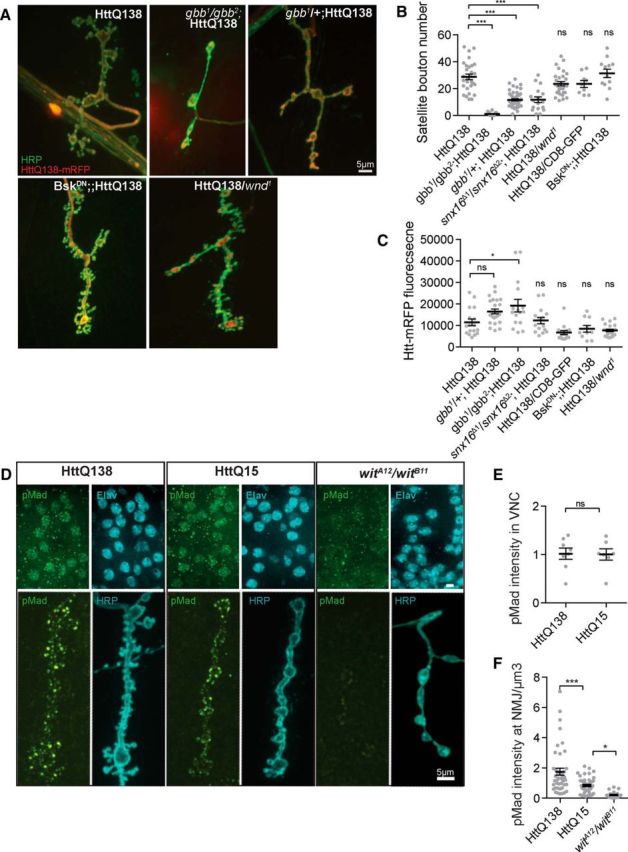
HttQ13Q-induced synaptic overgrowth requires BMP signaling. A, Representative images of HttQ138-dependent synaptic overgrowth in genetic backgrounds altering BMP and JNK signaling. ElavC155;UAS-HttQ138-mRFP caused severe synaptic overgrowth that was completely suppressed in ElavC155;gbb1/gbb2;UAS-HttQ138-mRFP and partially suppressed in ElavC155;gbb1/+;UAS-HttQ138-mRFP larvae. Expression of DN Bsk did not reduce HttQ138-induced overgrowth. ElavC155/UAS-BskDN;UAS-HttQ138-mRFP (female larvae) had overgrowth similar to ElavC155;UAS-HttQ138-mRFP alone. Strong overgrowth was still observed when the levels of Wnd were reduced 50% reduced in ElavC155;UAS-HttQ138-mRFP/wnd1 strains. B, Quantification of satellite bouton number in the indicated genotypes. Removal of Snx16 in ElavC155;snx16Δ1/ snx16Δ2;UAS-HttQ138-mRFP rescues HttQ138-induced synaptic overgrowth. C, Synaptic HttQ138-mRFP levels detected in the indicated genetic backgrounds shown in B. D, Representative images showing pMad nuclear and NMJ immunostaining. Synaptic and nuclear pMad staining is lost in wit mutants (witA12/witB12, right panel), whereas synaptic pMad is elevated in HttQ138-expressing larvae. E, Quantification of pMad fluorescent labeling in nuclei of ventral nerve cord neurons. F, Quantification of pMad labeling at the NMJ. ANOVA followed by Tukey's multiple-comparisons test was used for statistical analysis. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. Error bars indicate SEM.
In addition to BMP signaling, the Drosophila JNK (DJNK, also known as Basket) is a key regulator of synaptic growth. JNK level is regulated by upstream components of a mitogen-activated protein kinase (MAPK) cascade that includes the Wallenda (Wnd) protein (Collins et al., 2006). To analyze the potential function of JNK signaling in HttQ138-mediated synaptic overgrowth, we inhibited JNK activation in HttQ138 larvae by expressing a dominant-negative (DN) form of Drosophila JNK (BskDN) with ElavC155. To avoid possible effects from titration of the GAL4 driver, we compared lines expressing UAS-BskDN and UAS-HttQ138 with lines expressing UAS-CD8-GFP and UAS-HttQ138 (Fig. 4A,B). We observed no ability of JNK inhibition to reduce synaptic overgrowth after HttQ138 expression. The number of satellite boutons was similar after HttQ138 expression in the CD8-GFP coexpression line (23.6 ± 1.6, n = 29) and the BskDN coexpression line (31.3 ± 3.2, n = 12, p ≤ 0.05). To further assay whether JNK signaling was required for synaptic overgrowth in HttQ138 larvae, we tested whether reducing the levels of Wnd would alter synaptic overgrowth. Larvae expressing HttQ138 and missing one copy of Wnd had 28.7 ± 2.1 (n = 29, p ≤ 0.05) satellite boutons, similar to HttQ138; CD8-GFP larvae, which had 23.6 ± 1.6 (n = 29). Loss of Wnd did not alter HttQ138 fluorescence at NMJs (Fig. 4C). In addition, these genotypes did not alter satellite bouton number on their own (Wnd1/+: 0.7 ± 0.3 n = 14; ElavC155/BskDN female larvae: 3.0 ± 0.4 n = 15). We conclude that HttQ138 does not alter the JNK synaptic growth pathway.
Activation of BMP signaling leads to phosphorylation and accumulation of pMad in motor neuron nuclei (nuclear pMad; McCabe et al., 2003; Dudu et al., 2006) and at NMJ synapses (synaptic pMad; Higashi-Kovtun et al., 2010; Sulkowski et al., 2014). Using pMad-specific antibodies, we tested whether pMad levels were increased after expression of HttQ138. No changes in nuclear pMad were found after expression of pathogenic Htt (Fig. 4D,E). Specific pMad staining was not detected in mutants lacking the BMP receptor (wit1/wit2), confirming the specificity of the pMad antisera (Fig. 4D). In contrast to nuclear pMad, synaptic pMad fluorescence was elevated in HttQ138-expressing larvae (1.8 ± 0.2, normalized to NMJ volume in cubic micrometers, n = 44, p ≤ 0.001) compared with HttQ15 (1.0 ± 0.2, n = 45) and BMP receptor mutants (wit1/wit2, 0.2 ± 0.1, n = 15), in which staining was strongly diminished (Fig. 4D,F). These findings indicate that elevated BMP signaling within nerve terminals leads to enhanced synaptic growth after HttQ138 accumulation at synapses.
HttQ138 expression within motor neurons is sufficient to induce synaptic overgrowth and lethality
The changes in endosomal compartments observed in HttQ138 larvae are associated with peripheral nervous system synapses at the NMJ. We next investigated whether disruptions of NMJ biology induced by HttQ138 contributed to the lethality observed after pan-neuronal expression of HttQ138. To test this directly, we expressed HttQ138 in motor neurons with the C164-GAL4 and OK371-GAL4 motor-neuron-restricted GAL4 drivers. Expression of HttQ138 with these drivers resulted in substantially overgrown NMJs (Fig. 5A,B). To determine whether motor neuron HttQ138 expression alone can trigger pupal lethality, we scored the number of third-instar larvae of each genotype that emerged as adults (Fig. 5C). Pan-neuronal expression of pathogenic HttQ138 caused robust pupal lethality, with only a few rare adults able to emerge from pupal cases (survival rate of 6.2 ± 2.6%, n = 8). With C164-GAL4-driven HttQ138 expression, 51.6 ± 3.1% (n = 8, p ≤ 0.001) of larvae emerged as adults. OK371-GAL4-driven expression caused stronger lethality, with only 39.1 ± 5.3% (n = 8, p ≤ 0.01) of larvae surviving to adulthood (Fig. 5C).
Figure 5.

Pathology induced by motor neuron HttQ138 expression. Expression of HttQ138 in motor neurons causes overgrowth similar to that observed after pan-neuronal expression. A, Representative images of muscle 4 NMJ overgrowth in ElavC155;UAS-HttQ138-mRFP, C164-GAL4;UAS-HttQ138-mRFP, OK371;UAS-HttQ138-mRFP, Mef2-GAL4;UAS-HttQ138-mRFP, and ElavC155;UAS-HttQ15-mRFP larvae. B, Quantification of satellite bouton number for each genotype. C, Quantification of the percentage of third-instar larval survival to adulthood for ElavC155;UAS-HttQ138-mRFP, C164-GAL4;UAS-HttQ138-mRFP, OK371;UAS-HttQ138-mRFP, Mef2-GAL4;UAS-HttQ138-mRFP, ElavC155;UAS-HttQ15-mRFP, MD-GAL4;UAS-HttQ138-mRFP, and OK107-GAL4;UAS-HttQ138-mRFP. ANOVA followed by Tukey's multiple-comparisons test was used for statistical analysis. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. Error bars indicate SEM.
We also examined the effects of HttQ138 expression in two other essential neuronal populations, mushroom body neurons (Shin and DiAntonio, 2011; Bates et al., 2014) and peripheral sensory neurons (Song et al., 2007). In contrast to motor neurons, expression of HttQ138 with the pan-mushroom body driver OK107 did not cause lethality; all larvae produced healthy adults with 100 ± 0.0% survivability (n = 9; Fig. 5C). Likewise, expression in multiple dendritic neurons generated larvae that were able to survive to adults at a 97.4 ± 1.7% (n = 9) rate (Fig. 5C). Similarly high levels of viability, without any obvious changes in NMJ morphology, were observed when HttQ138 was expressed postsynaptically, using the Mef2-Gal4 muscle driver (Fig. 5B,C). Although we do not exclude roles for other cell populations in HttQ138 pathology, HttQ138-induced changes in motor neurons are sufficient to induce synaptic overgrowth and contribute to lethality, identifying this cell population as being critically sensitive to pathogenic Htt expression in the Drosophila HD model.
Manipulation of the Snx16 endosomal compartment results in alterations in synaptic growth
Based on the disruptions of endosomal compartments and BMP signaling, we hypothesized that the NMJ overgrowth in HttQ138-expressing larvae was secondary to enhanced synaptic growth signals emanating from activated receptors trapped in early endosomal signaling compartments. Because the most dramatic change observed was a robust expansion of the Snx16 endosomal compartment, we investigated whether disruptions of Rab family members alone would alter the size and distribution of Snx16 positive puncta. Prior studies have shown Rab5 overexpression increases endocytic compartments in Drosophila (Grbovic et al., 2003; Wucherpfennig et al., 2003). In contrast, overexpression of Rab7 has been shown to enlarge late endosomal compartments (Bucci et al., 2000), with compartment dispersion observed after expression of DN Rab7 (Dodson et al., 2012). We overexpressed Rab5, Rab7, and Rab11 in constitutively active (CA) or DN forms, together with Snx16-GFP (Fig. 6A,B). As a control, we expressed Snx16-GFP with UAS-myrRFP to account for possible GAL4 titration. We observed that expression of Rab7CA (Rab7Q67L) expanded the Snx16-positive compartment (relative fluorescence normalized to NMJ volume was 4.2 ± 0.5, n = 20, p ≤ 0.001 vs control Snx16-GFP; myrRFP larvae, 1.0 ± 0.1, n = 32). Overexpressing Rab7DN (Rab7T22N) caused a substantial reduction in the Snx16-positive compartment (0.2 ± 0.0, n = 50, p ≤ 0.01), whereas expression of Rab11CA (Rab11Q70L) led to a mild reduction (0.4 ± 0.3, n = 66, p ≤ 0.1). Expression of Rab11DN (Rab11S25N) did not affect the distribution of Snx16 puncta (0.8 ± 0.1, n = 28, p > 0.05), although it resulted in a more diffuse Snx16-GFP pattern (Fig. 6A). Rab5CA (Rab5Q88L) expression strongly altered the formation of Snx16 puncta, with only 1–2 enlarged puncta observed per NMJ instead of the normal pattern of multiple puncta distributed along the synaptic arbor (relative fluorescence intensity was 0.2 ± 0.0, n = 30, p ≤ 0.05; Fig. 6A,B). Expression of Rab5DN (Rab5S43N) together with Snx16-GFP caused early larval lethality and was excluded from analysis. These findings indicate that the Snx16 endosomal compartment is sensitive to changes in multiple routes of endocytic trafficking after disruption of synaptic Rab proteins.
Figure 6.
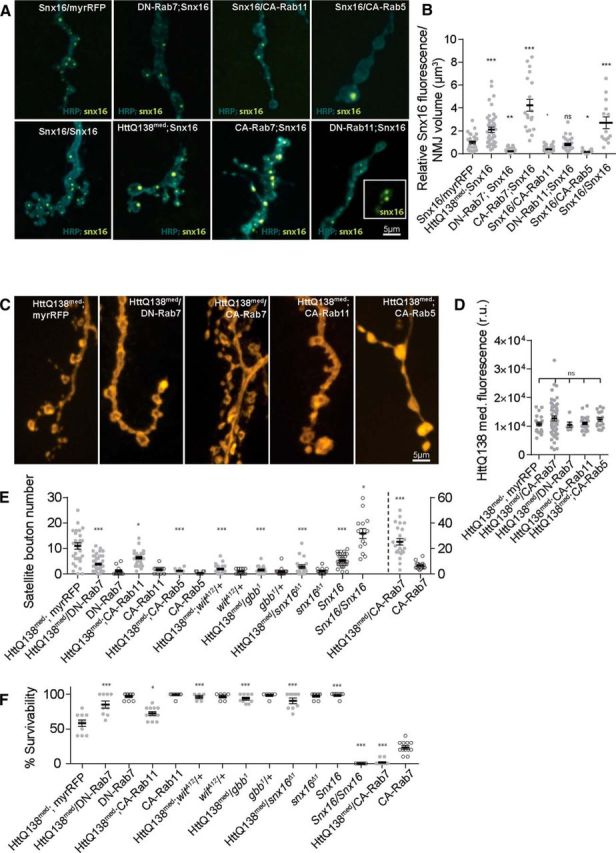
Manipulations of endosomal and BMP trafficking alter Snx16 compartments and interact synergistically with HttQ138. A, Representative images showing changes in Snx16 distribution in ElavC155;UAS-Snx16-GFP/UAS-myrRFP control lines (A1) compared with manipulations of the indicated Rab proteins (A2: ElavC155;UAS-Rab7DN;UAS-Snx16-GFP, A3: ElavC155;UAS-Snx16-GFP/UAS-Rab11CA, A4: ElavC155;UAS-Snx16-GFP/UAS-Rab5CA, A5: ElavC155;UAS-Snx16-GFP/UAS-Snx16-GFP, A6: ElavC155;UAS-HttQ138-RFPmed/+; UAS-Snx16-GFP/+, A7: ElavC155;UAS-Rab7CA/+;UAS-Snx16-GFP/+, and A8: ElavC155;UAS-Rab11DN;UAS-Snx16-GFP). B, Quantification of Snx16-GFP fluorescence levels in the indicated genotypes. C, Representative images of synaptic growth in ElavC155;UAS-HttQ138-RFPmed/+;UAS-myrRFP/+ controls (C1) versus ElavC155;UAS-HttQ138-RFPmed;UAS-Rabs (C2: ElavC155;UAS-HttQ138-RFPmed/UAS-Rab7DN, C3: ElavC155;UAS-HttQ138-RFPmed/ UAS-Rab7CA, C4: ElavC155;UAS-HttQ138-RFPmed/+;UAS-Rab11CA/+, and C5: ElavC155;UAS-HttQ138-RFPmed/+;UAS-Rab5CA/+). D, Quantification of UAS-HttQ138-mRFPmed intensity from the genotypes shown in C. E, Quantification of satellite bouton number in the indicated genotypes from ElavC155-driven expression of the indicated transgenes. F, Quantification of the percentage of third-instar larvae surviving to adults in the indicated genotypes as above. ANOVA followed by Tukey's multiple-comparisons test was used for statistical analysis. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. Error bars indicate SEM.
HttQ138 induced toxicity can be modified by Rab proteins that alter the Snx16 compartment
As noted above, the size and distribution of the Snx16-positive compartment can be altered by expression of CA or DN Rabs. To determine whether manipulations of the Snx16 compartment by Rabs might interact synergistically with pathogenic HttQ138 to alter NMJ overgrowth and larval viability, we expressed CA and DN versions of Rab5, Rab7, and Rab11 with HttQ138. For these genetic interaction experiments, we took advantage of a lower-expressing HttQ138-mRFP line (HttQ138med) to avoid saturation of the overgrowth pathway. As a control for GAL4 titration, we used animals expressing UAS-myrRFP together with HttQ138med for comparisons. ElavC155;UAS-HttQ138-mRFPmed;UAS-myrRFP larvae displayed mild NMJ overgrowth (11.0 ± 1.3, n = 32; Fig. 6C,E) and were partially viable to adulthood (58.0 ± 4.7% third-instar larvae surviving to adults, n = 10; Fig. 6F). Expression of Rab5CA, Rab7DN, and Rab11CA caused a downregulation of the Snx16 compartment, as noted above (Fig. 6A,B). Expression of Rab7DN or Rab11CA with HttQ138med caused a suppression of HttQ138-induced synaptic overgrowth (HttQ138med;Rab7DN = 3.7 ± 0.4 satellite boutons, n = 53, p ≤ 0.001; HttQ138med;Rab11CA = 6.4 ± 0.5, n = 31, p ≤ 0.05; Fig. 6C,E). Expression of Rab5CA produced dramatic NMJ undergrowth on its own, phenotypically resembling the NMJs of gbb-null mutants that lack BMP signaling. Expression of Rab5CA was able to suppress Htt-induced overgrowth completely (1.2 ± 0.3, n = 9, p ≤ 0.001), with the NMJ underdeveloped compared with controls. Rab5CA-expressing larvae were lethal at late larval stages on their own or when combined with HttQ138med. Lines expressing Rab7DN and Rab11CA had 96.7 ± 1.7% (n = 9) and 99.0 ± 1.0% (n = 10) survivability, respectively (Fig. 6F). Rab7DN and Rab11CA rescued the lethality associated with HttQ138med expression alone when coexpressed with pathogenic Htt (84.9 ± 5.1%, n = 9, p ≤ 0.001 and 72.3 ± 2.7%, n = 12, p ≤ 0.05, respectively; Fig. 6F). These data indicate that blocking synaptic overgrowth by reducing signaling from the Snx16 compartment can rescue both the synaptic overgrowth and the lethality associated with HttQ138 expression.
In contrast to the rescue observed with manipulations of Rab proteins that decreased the Snx16 compartment and synaptic overgrowth, we observed the opposite effects with manipulations that expanded the Snx16 compartment. Rab7CA expression enlarged the Snx16 compartment (Fig. 6A,B). When expressed together with HttQ138med, Rab7CA further enhanced NMJ overgrowth (25.3 ± 2.6, n = 23, p ≤ 0.001) compared with either manipulation alone (Fig. 6E). Expression of Rab7CA alone is partially viable (23 ± 0.0% survival from third instar to adult, n = 12, p ≤ 0.001), although adult escapers are unable to inflate their wings, similar to defects observed in HttQ138med surviving adults. Coexpression of Rab7CA with HttQ138med results in synergistic lethality (1.4 ± 0.9% survival, n = 14, p ≤ 0.001; Fig. 6E). Expression of the different Rab5, Rab7, and Rab11 lines did not alter HttQ138 expression or localization at synapses (Fig. 6D), indicating that the synergistic alterations in synaptic growth and viability are not secondary to defective HttQ138 trafficking or expression.
These results support the hypothesis that HttQ138 promotes synaptic growth and lethality in Drosophila by downregulating trafficking from early endosomal signaling compartments and upregulating local BMP signaling. To further test this model, we altered BMP signaling directly in the HttQ138med background. We observed similar synergistic interactions to those observed with Rab manipulations. Expression of HttQ138med in heterozygotes with a 50% reduction in Gbb or Wit resulted in suppression of synaptic overgrowth and an increase in adult viability (gbb1/+;HttQ138med, 1.4 ± 0.4 satellite boutons, n = 16, p ≤ 0.001, 93.6 ± 2.1% survival, n = 9, p ≤ 0.001; Wit1/+;HttQ138med, 1.9 ± 0.4 satellite boutons, n = 23, p ≤ 0.001, 96.0 ± 0.0% survival, n = 6, p ≤ 0.001; Fig. 6E,F). These data indicate that enhanced BMP signaling underlies the synaptic overgrowth and contributes to lethality in the Drosophila HD model.
The observation that HttQ138 induced changes in the formation of Snx16-positive compartments are correlated with NMJ overgrowth and viability suggests that Snx16 overexpression alone might be sufficient to induce pathology in the absence of HttQ138. Indeed, we found that expression of two copies of the Snx16 transgene caused an increase in the number of Snx16 puncta (Fig. 6A,B). Fluorescent levels of Snx16 increased 2.7 ± 0.5-fold (n = 14, p ≤ 0.001). Although overexpression of a single copy of Snx16 induced a mild increase in satellite bouton number (5.5 ± 0.4, n = 31), expression of 2 copies of Snx16 induced pronounced overgrowth of satellite boutons (16.1 ± 1.9, n = 14). This overgrowth was slightly higher (p ≤ 0.01) than that observed in the HttQ138 med line (Fig. 6A,E). Although Drosophila expressing a single copy of Snx16 are fully viable, with a 100 ± 0.0% survival rate (n = 9), expression of 2 copies of the Snx16 transgene caused complete adult lethality (Fig. 6F). These data indicate that Snx16 levels are crucial for controlling normal NMJ overgrowth and that upregulation of the protein can perturb endosomal signaling and induce lethality.
Discussion
In this study, we found that synaptic pathology and lethality are linked to Htt-mediated perturbation of endosomal compartments in a Drosophila HD model. Pathogenic Htt accumulation at nerve terminals results in expansion of an early endosomal synaptic signaling compartment and leads to enhanced BMP signaling and synaptic overgrowth. Synaptic HttQ138 is present largely in a nonaggregated from at the NMJ, suggesting that abnormal interactions mediated by soluble Htt are responsible for the disruption of endosomal signaling. Although aggregated versions of pathogenic Htt have been found along axons and can disrupt axonal transport (Lee et al., 2004; Weiss et al., 2012), our data indicate that the synaptic overgrowth defect is not downstream of Htt aggregation. Indeed, we found that axon branches of the same neuron that accumulate different levels of pathogenic Htt have striking differences in synaptic morphology, ruling out cell-wide effects as a cause of the local NMJ overgrowth. We hypothesize that nonaggregated forms of pathogenic Htt block the exit and subsequent termination of BMP receptor signaling complexes locally by binding to and interfering with the activity of key regulators of membrane trafficking present at synapses.
Multiple genetic interaction experiments indicated that Htt-induced alterations in receptor trafficking cause pathological changes that contribute to Drosophila pharate lethality in this HD model. Using both immunocytochemistry and EM, we found that HttQ138 expression results in a robust expansion in the size and number of early synaptic signaling endosomes labeled by Snx16. Manipulations that reduce the accumulation of the Snx16 sorting endosomal compartment reduce synaptic overgrowth and increase survivability of HttQ138-expressing larvae, including expression of Rab11CA and Rab7DN. Expression of Rab7CA causes the opposite effect, enhancing synaptic growth defects and increasing lethality induced by HttQ138. Previous work at the Drosophila NMJ has implicated Rab11 and the Snx16-binding protein Nwk as key mediators of sorting from early to late endosomes (Rodal et al., 2011). Indeed, manipulations of Rab11 function have been reported to alter pathology in other HD models (Richards et al., 2011; Steinert et al., 2012, 2012). Rab7 is associated with late endocytic structures and lysosomes and has been shown to be crucial for receptor trafficking to lysosomes (Bucci et al., 2000; Miaczynska et al., 2004). Therefore, manipulations of Rab7 may alter endosomal function through distinct mechanisms involving trafficking to lysosomes.
In addition to Rab modulation of Htt-mediated pathology, our genetic interaction studies indicate that BMP signaling through the Gbb pathway is altered by defective endosomal traffic and contributes to toxicity. Htt-mediated synaptic overgrowth and larval lethality were alleviated by multiple factors that downregulate BMP signaling, including reducing the levels of the ligand Gbb, the receptor Wit, and the endosomal trafficking regulator Snx16. Although BMP signaling is a key target for dysregulation, HttQ138 only results in a local accumulation of the BMP effector pMad at synapses without changes in nuclear pMad. Therefore, nuclear pMad signaling events are not likely to be as strongly affected by HttQ138. Specific and local changes in synaptic pMad levels at the NMJ have been reported in a number of other Drosophila mutants, including importin-β11 (Higashi-Kovtun et al., 2010) and Neto mutants (Sulkowski et al., 2014). More directly related to endocytic trafficking, mutations in the α2-adaptin subunit of the AP-2 complex also lead to increased synaptic growth, with satellite bouton formation and altered synaptic BMP signaling (Choudhury et al., 2016). Although BMP signaling is closely linked to synaptic development and growth in Drosophila, changes in synaptic structure have also been found in HD patients and other mammalian HD models. Extensive dendritic branching with increased number and size of dendritic spines are observed in striatal spiny neurons in HD patients (Ferrante et al., 1991). In addition, changes in dendrite structure and function have been observed in many mammalian models of neurodegenerative disorders (Herms and Dorostkar, 2016), indicating that alterations in synapse formation and stability may extend beyond the Drosophila HD model.
Although multiple mechanisms linking abnormal endosomal trafficking of growth factors to the formation of satellite boutons are established in Drosophila (Marie et al., 2004; O'Connor-Giles et al., 2008; Rodal et al., 2011), the connection between NMJ overgrowth and lethality is less clear. Although other neuronal defects are likely present in the Drosophila HD model and may contribute to neuropathology, our genetic interaction experiments support a direct link between altered endosomal trafficking and lethality. Several other Drosophila synaptic mutants that block membrane trafficking and show synaptic overgrowth are also lethal during larval development, often displaying locomotion defects (Marie et al., 2004; Khodosh et al., 2006; Koh et al., 2007; Sulkowski et al., 2014). In contrast, mutations in dCIP4 and Nwk, which also have NMJ overgrowth due to elevated BMP signaling, are viable (Nahm et al., 2010a). More studies are required to determine why certain disruptions of synaptic endosomal trafficking are tolerated, whereas others result in lethality.
What is the mechanism by which Htt disrupts endosomal trafficking? Given the host of endocytic proteins that interact with Htt, an attractive model is that either enhanced or reduced binding to a key effector caused by polyQ expansion within Htt leads to the pathology. Such an effector could be a protein or a specific lipid. Pathogenic Htt has been shown to have enhanced binding to phosphatidylinositol 3,4,5-triphosphate [PI(3,4,5)P3] compared with wild-type Htt (Kegel et al., 2009). The levels of PI(3,4,5)P3 are generally low in cells, but can increase rapidly upon growth factor stimulation. Pathogenic Htt can also associate with other phospholipids not recognized by wild-type Htt (Kegel et al., 2009), providing additional avenues for disruptions in endosomal trafficking that require unique lipid signatures to regulate the flow of cargo. Indeed, Htt is recruited to the cell surface upon growth-factor-dependent increases in the phospholipids PI(3,4)P2 and PI(3,4,5)P3 (Kegel et al., 2009).
In terms of protein targets, a number of studies have shown that polyQ-expanded Htt interacts with many of the same proteins as wild-type Htt, but with distinct affinities (Harjes and Wanker, 2003; Kegel et al., 2009). For example, many Htt-interacting partners that contain SH3 domains bind more tightly to mutant Htt (Li and Li, 2004). Htt also interacts with the epidermal growth factor receptor Grb2 and its intracellular partner RasGap and these associations are regulated by epidermal growth factor stimulation (Liu et al., 1997). Therefore, Htt could have more direct interaction with signaling receptors themselves that alter trafficking. Htt has also been shown to regulate other steps in membrane trafficking, including macroautophagy (Rui et al., 2015) and early endosomal trafficking during cellular stress (Nath et al., 2015). These activities represent additional avenues for mutant Htt to block trafficking by altering the activity of wild-type Htt interactions normally required for endosomal function.
In conclusion, these data support a model whereby pathogenic nonaggregated Htt alters early steps of receptor trafficking within the early endosomal system, prolonging signaling from intracellular compartments at the synapse. Abnormal endosomal function causes changes in synaptic morphology that contribute to the lethality caused by mutant Htt expression. Therefore, downregulation of synaptic endosomal signaling might provide a promising potential therapeutic target for HD pathology.
Footnotes
This work was supported by the JBP Foundation and the National Institutes of Health (Grants NS40296 and MH104536). We thank Avi Rodal for helpful suggestions, the Bloomington Stock Center for Drosophila strains, and Ed Laufer for generously providing antibodies.
The authors declare no competing financial interests.
References
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431:805–810. 10.1038/nature02998 [DOI] [PubMed] [Google Scholar]
- Assaker G, Ramel D, Wculek SK, González-Gaitán M, Emery G (2010) Spatial restriction of receptor tyrosine kinase activity through a polarized endocytic cycle controls border cell migration. Proc Natl Acad Sci U S A 107:22558–22563. 10.1073/pnas.1010795108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates GP. (2005) History of genetic disease: the molecular genetics of Huntington disease: a history. Nat Rev Genet 6:766–773. 10.1038/nrm1738 [DOI] [PubMed] [Google Scholar]
- Bates KE, Sung C, Hilson L, Robinow S (2014) Unfulfilled interacting genes display branch-specific roles in the development of mushroom body axons in Drosophila melanogaster. G3 (Bethesda) 4:693–706. 10.1534/g3.113.009829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B (2000) Rab7: a key to lysosome biogenesis. Mol Biol Cell 11:467–480. 10.1091/mbc.11.2.467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Hong WP, Kim MJ, Kim JH, Ryu SH, Suh PG (2004) Sorting nexin 16 regulates EGF receptor trafficking by phosphatidylinositol-3-phosphate interaction with the Phox domain. J Cell Sci 117:4209–4218. 10.1242/jcs.01233 [DOI] [PubMed] [Google Scholar]
- Choudhury SD, Mushtaq Z, Reddy-Alla S, Balakrishnan SS, Thakur RS, Krishnan KS, Raghu P, Ramaswami M, Kumar V (2016) α2-Adaptin facilitates basal synaptic transmission and is required for regenerating endo-exo cycling pool under high-frequency nerve stimulation in Drosophila. Genetics 203:369–385. 10.1534/genetics.115.183863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CA, DiAntonio A (2007) Synaptic development: insights from Drosophila. Curr Opin Neurobiol 17:35–42. 10.1016/j.conb.2007.01.001 [DOI] [PubMed] [Google Scholar]
- Collins CA, Wairkar YP, Johnson SL, DiAntonio A (2006) Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron 51:57–69. 10.1016/j.neuron.2006.05.026 [DOI] [PubMed] [Google Scholar]
- Dermaut B, Norga KK, Kania A, Verstreken P, Pan H, Zhou Y, Callaerts P, Bellen HJ (2005) Aberrant lysosomal carbohydrate storage accompanies endocytic defects and neurodegeneration in Drosophila benchwarmer. J Cell Biol 170:127–139. 10.1083/jcb.200412001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, Martin E, Vonsattel JP, Carraway R, Reeves SA (1995) Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron 14:1075–1081. 10.1016/0896-6273(95)90346-1 [DOI] [PubMed] [Google Scholar]
- Dodson MW, Zhang T, Jiang C, Chen S, Guo M (2012) Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum Mol Genet 21:1350–1363. 10.1093/hmg/ddr573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudu V, Bittig T, Entchev E, Kicheva A, Jülicher F, González-Gaitán M (2006) Postsynaptic mad signaling at the Drosophila neuromuscular junction. Curr Biol 16:625–635. 10.1016/j.cub.2006.02.061 [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Richardson EP Jr (1991) Proliferative and degenerative changes in striatal spiny neurons in Huntington's disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J Neurosci 11:3877–3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford MG, Mills IG, Peter BJ, Vallis Y, Praefcke GJ, Evans PR, McMahon HT (2002) Curvature of clathrin-coated pits driven by epsin. Nature 419:361–366. 10.1038/nature01020 [DOI] [PubMed] [Google Scholar]
- Gillooly DJ, Morrow IC, Lindsay M, Gould R, Bryant NJ, Gaullier JM, Parton RG, Stenmark H (2000) Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J 19:4577–4588. 10.1093/emboj/19.17.4577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold CP, Davis GW (2007) The BMP ligand Gbb gates the expression of synaptic homeostasis independent of synaptic growth control. Neuron 56:109–123. 10.1016/j.neuron.2007.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BD, Donaldson JG (2009) Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol 10:597–608. 10.1038/nrm2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grbovic OM, Mathews PM, Jiang Y, Schmidt SD, Dinakar R, Summers-Terio NB, Ceresa BP, Nixon RA, Cataldo AM (2003) Rab5-stimulated up-regulation of the endocytic pathway increases intracellular beta-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Abeta production. J Biol Chem 278:31261–31268. 10.1074/jbc.M304122200 [DOI] [PubMed] [Google Scholar]
- Hanson BJ, Hong W (2003a) Evidence for a role of SNX16 in regulating traffic between the early and later endosomal compartments. J Biol Chem 278:34617–34630. 10.1074/jbc.M300143200 [DOI] [PubMed] [Google Scholar]
- Harjes P, Wanker EE (2003) The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci 28:425–433. 10.1016/S0968-0004(03)00168-3 [DOI] [PubMed] [Google Scholar]
- Harris KP, Littleton JT (2015) Transmission, development, and plasticity of synapses. Genetics 201:345–375. 10.1534/genetics.115.176529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herms J, Dorostkar MM (2016) Dendritic spine pathology in neurodegenerative diseases. Annu Rev Pathol 11:221–250. 10.1146/annurev-pathol-012615-044216 [DOI] [PubMed] [Google Scholar]
- Higashi-Kovtun ME, Mosca TJ, Dickman DK, Meinertzhagen IA, Schwarz TL (2010) Importin-$\beta$11 regulates synaptic phosphorylated mothers against decapentaplegic, and thereby influences synaptic development and function at the Drosophila neuromuscular junction. J Neurosci 30:5253–5268. 10.1523/JNEUROSCI.3739-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, **Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR (1999) A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 23:181–192. 10.1016/S0896-6273(00)80764-3 [DOI] [PubMed] [Google Scholar]
- Jean S, Kiger AA (2012) Coordination between RAB GTPase and phosphoinositide regulation and functions. Nat Rev Mol Cell Biol 13:463–470. 10.1038/nrm3379 [DOI] [PubMed] [Google Scholar]
- Kamimura K, Ueno K, Nakagawa J, Hamada R, Saitoe M, Maeda N (2013) Perlecan regulates bidirectional Wnt signaling at the Drosophila neuromuscular junction. J Cell Biol 200:219–233. 10.1083/jcb.201207036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kegel KB, Sapp E, Alexander J, Valencia A, Reeves P, Li X, Masso N, Sobin L, Aronin N, DiFiglia M (2009) Polyglutamine expansion in huntingtin alters its interaction with phospholipids. J Neurochem 110:1585–1597. 10.1111/j.1471-4159.2009.06255.x [DOI] [PubMed] [Google Scholar]
- Khodosh R, Augsburger A, Schwarz TL, Garrity PA (2006) Bchs, a BEACH domain protein, antagonizes Rab11 in synapse morphogenesis and other developmental events. Development 133:4655–4665. 10.1242/dev.02650 [DOI] [PubMed] [Google Scholar]
- Kim M, Lee HS, LaForet G, McIntyre C, Martin EJ, Chang P, Kim TW, Williams M, Reddy PH, Tagle D, Boyce FM, Won L, Heller A, Aronin N, DiFiglia M (1999) Mutant huntingtin expression in clonal striatal cells: dissociation of inclusion formation and neuronal survival by caspase inhibition. J Neurosci 19:964–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh TW, Korolchuk VI, Wairkar YP, Jiao W, Evergren E, Pan H, Zhou Y, Venken KJ, Shupliakov O, Robinson IM, O'Kane CJ, Bellen HJ (2007) Eps15 and Dap160 control synaptic vesicle membrane retrieval and synapse development. J Cell Biol 178:309–322. 10.1083/jcb.200701030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ (1999) Huntington aggregates may not predict neuronal death in Huntington's disease. Ann Neurol 46:842–849. [PubMed] [Google Scholar]
- Lee WC, Yoshihara M, Littleton JT (2004) Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington's disease. Proc Natl Acad Sci U S A 101:3224–3229. 10.1073/pnas.0400243101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SH, Li XJ (2004) Huntingtin-protein interactions and the pathogenesis of Huntington's disease. Trends Genet 20:146–154. 10.1016/j.tig.2004.01.008 [DOI] [PubMed] [Google Scholar]
- Liu YF, Deth RC, Devys D (1997) SH3 domain-dependent association of huntingtin with epidermal growth factor receptor signaling complexes. J Biol Chem 272:8121–8124. 10.1074/jbc.272.13.8121 [DOI] [PubMed] [Google Scholar]
- MacDonald ME, et al.; The Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72:971–983. 10.1016/0092-8674(93)90585-E [DOI] [PubMed] [Google Scholar]
- Marie B, Sweeney ST, Poskanzer KE, Roos J, Kelly RB, Davis GW (2004) Dap160/intersectin scaffolds the periactive zone to achieve high-fidelity endocytosis and normal synaptic growth. Neuron 43:207–219. 10.1016/j.neuron.2004.07.001 [DOI] [PubMed] [Google Scholar]
- McCabe BD, Marqués G, Haghighi AP, Fetter RD, Crotty ML, Haerry TE, Goodman CS, O'Connor MB (2003) The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron 39:241–254. 10.1016/S0896-6273(03)00426-4 [DOI] [PubMed] [Google Scholar]
- Merino-Trigo A, Kerr MC, Houghton F, Lindberg A, Mitchell C, Teasdale RD, Gleeson PA (2004) Sorting nexin 5 is localized to a subdomain of the early endosomes and is recruited to the plasma membrane following EGF stimulation. J Cell Sci 117:6413–6424. 10.1242/jcs.01561 [DOI] [PubMed] [Google Scholar]
- Miaczynska M, Pelkmans L, Zerial M (2004) Not just a sink: endosomes in control of signal transduction. Curr Opin Cell Biol 16:400–406. 10.1016/j.ceb.2004.06.005 [DOI] [PubMed] [Google Scholar]
- Modregger J, DiProspero NA, Charles V, Tagle DA, Plomann M (2002) PACSIN 1 interacts with huntingtin and is absent from synaptic varicosities in presymptomatic Huntington's disease brains. Hum Mol Genet 11:2547–2558. 10.1093/hmg/11.21.2547 [DOI] [PubMed] [Google Scholar]
- Nahm M, Kim S, Paik SK, Lee M, Lee S, Lee ZH, Kim J, Lee D, Bae YC, Lee S (2010a) dCIP4 (Drosophila Cdc42-interacting protein 4) restrains synaptic growth by inhibiting the secretion of the retrograde Glass bottom boat signal. J Neurosci 30:8138–8150. 10.1523/JNEUROSCI.0256-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahm M, Long AA, Paik SK, Kim S, Bae YC, Broadie K, Lee S (2010b) The Cdc42-selective GAP rich regulates postsynaptic development and retrograde BMP transsynaptic signaling. J Cell Biol 191:661–675. 10.1083/jcb.201007086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano Y, Fujitani K, Kurihara J, Ragan J, Usui-Aoki K, Shimoda L, Lukacsovich T, Suzuki K, Sezaki M, Sano Y, Ueda R, Awano W, Kaneda M, Umeda M, Yamamoto D (2001) Mutations in the novel membrane protein Spinster interfere with programmed cell death and cause neural degeneration in Drosophila melanogaster. Mol Cell Biol 21:3775–3788. 10.1128/MCB.21.11.3775-3788.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath S, Munsie LN, Truant R (2015) A huntingtin-mediated fast stress response halting endosomal trafficking is defective in Huntington's disease. Hum Mol Genet 24:450–462. 10.1093/hmg/ddu460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor-Giles KM, Ho LL, Ganetzky B (2008) Nervous wreck interacts with thickveins and the endocytic machinery to attenuate retrograde BMP signaling during synaptic growth. Neuron 58:507–518. 10.1016/j.neuron.2008.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards P, Didszun C, Campesan S, Simpson A, Horley B, Young KW, Glynn P, Cain K, Kyriacou CP, Giorgini F, Nicotera P (2011) Dendritic spine loss and neurodegeneration is rescued by Rab11 in models of Huntington's disease. Cell Death Differ 18:191–200. 10.1038/cdd.2010.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodal AA, Blunk AD, Akbergenova Y, Jorquera RA, Buhl LK, Littleton JT (2011) A presynaptic endosomal trafficking pathway controls synaptic growth signaling. J Cell Biol 193:201–217. 10.1083/jcb.201009052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero E, Cha GH, Verstreken P, Ly CV, Hughes RE, Bellen HJ, Botas J (2008) Suppression of neurodegeneration and increased neurotransmission caused by expanded full-length huntingtin accumulating in the cytoplasm. Neuron 57:27–40. 10.1016/j.neuron.2007.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui YN, Xu Z, Patel B, Cuervo AM, Zhang S (2015) HTT/Huntingtin in selective autophagy and Huntington disease: a foe or a friend within? Autophagy 11:858–860. 10.1080/15548627.2015.1039219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruta V, Datta SR, Vasconcelos ML, Freeland J, Looger LL, Axel R (2010) A dimorphic pheromone circuit in Drosophila from sensory input to descending output. Nature 468:686–690. 10.1038/nature09554 [DOI] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME (1998) Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95:55–66. 10.1016/S0092-8674(00)81782-1 [DOI] [PubMed] [Google Scholar]
- Schulte J, Sepp KJ, Wu C, Hong P, Littleton JT (2011) High-content chemical and RNAi screens for suppressors of neurotoxicity in a Huntington's disease model. PLoS One 6:e23841. 10.1371/journal.pone.0023841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, DiAntonio A (2011) Highwire regulates guidance of sister axons in the Drosophila mushroom body. J Neurosci 31:17689–17700. 10.1523/JNEUROSCI.3902-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Onishi M, Jan LY, Jan YN (2007) Peripheral multidendritic sensory neurons are necessary for rhythmic locomotion behavior in Drosophila larvae. Proc Natl Acad Sci U S A 104:5199–5204. 10.1073/pnas.0700895104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkin A, McClure M, Huang F, Carter R (2000) Interaction of EGF receptor and grb2 in living cells visualized by fluorescence resonance energy transfer (FRET) microscopy. Curr Biol 10:1395–1398. 10.1016/S0960-9822(00)00785-5 [DOI] [PubMed] [Google Scholar]
- Stein MP, Dong J, Wandinger-Ness A (2003) Rab proteins and endocytic trafficking: potential targets for therapeutic intervention. Adv Drug Deliv Rev 55:1421–1437. 10.1016/j.addr.2003.07.009 [DOI] [PubMed] [Google Scholar]
- Steinert JR, Campesan S, Richards P, Kyriacou CP, Forsythe ID, Giorgini F (2012) Rab11 rescues synaptic dysfunction and behavioural deficits in a Drosophila model of Huntington's disease. Hum Mol Genet 21:2912–2922. 10.1093/hmg/dds117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulkowski M, Kim YJ, Serpe M (2014) Postsynaptic glutamate receptors regulate local BMP signaling at the Drosophila neuromuscular junction. Development 141:436–447. 10.1242/dev.097758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torroja L, Packard M, Gorczyca M, White K, Budnik V (1999) The Drosophila beta-amyloid precursor protein homolog promotes synapse differentiation at the neuromuscular junction. J Neurosci 19:7793–7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich O, Reinsch S, Urbé S, Zerial M, Parton RG (1996) Rab11 regulates recycling through the pericentriolar recycling endosome. J Cell Biol 135:913–924. 10.1083/jcb.135.4.913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Weering JR, Verkade P, Cullen PJ (2010) SNX-BAR proteins in phosphoinositide-mediated, tubular-based endosomal sorting. Semin Cell Dev Biol 21:371–380. 10.1016/j.semcdb.2009.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waelter S, Scherzinger E, Hasenbank R, Nordhoff E, Lurz R, Goehler H, Gauss C, Sathasivam K, Bates GP, Lehrach H, Wanker EE (2001) The huntingtin interacting protein HIP1 is a clathrin and alpha-adaptin-binding protein involved in receptor-mediated endocytosis. Hum Mol Genet 10:1807–1817. 10.1093/hmg/10.17.1807 [DOI] [PubMed] [Google Scholar]
- Wanker EE, Rovira C, Scherzinger E, Hasenbank R, Wälter S, Tait D, Colicelli J, Lehrach H (1997) HIP-I: a huntingtin interacting protein isolated by the yeast two-hybrid system. Hum Mol Genet 6:487–495. 10.1093/hmg/6.3.487 [DOI] [PubMed] [Google Scholar]
- Weiss KR, Kimura Y, Lee WC, Littleton JT (2012) Huntingtin aggregation kinetics and their pathological role in a Drosophila Huntington's disease model. Genetics 190:581–600. 10.1534/genetics.111.133710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wucherpfennig T, Wilsch-Bräuninger M, González-Gaitán M (2003) Role of Drosophila Rab5 during endosomal trafficking at the synapse and evoked neurotransmitter release. J Cell Biol 161:609–624. 10.1083/jcb.200211087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerial M, McBride H (2001) Rab proteins as membrane organizers. Nat Rev Mol Cell Biol 2:107–117. 10.1038/35052055 [DOI] [PubMed] [Google Scholar]