

Abstract
Enormously growing genomic datasets present a new challenge on missing data imputation, a notoriously resource-demanding task. Haplotype imputation requires ethnicity-matched references. However, to date, haplotype references are not available for the majority of populations in the world. We explored to use existing unphased genotype datasets as references; if it succeeds, it will cover almost all of the populations in the world. The results showed that our HiFi software successfully yields 99.43% accuracy with unphased genotype references. Our method provides a cost-effective solution to breakthrough the bottleneck of limited reference availability for haplotype imputation in the big data era.
Keywords: Imputation, big data, references
1. Introduction
As the number and size of big genomic datasets enormously grow, people will regularly encounter a limitation on missing information, and this problem may be exacerbated continuously. Imputation has rapidly become one of the most useful strategies for dealing with missing values (Browning, 2008). However, solving the missing-value problem with small errors is a notoriously resource-demanding task; the challenge is how to make good use of the available data as the references to obtain high imputation accuracy.
Haplotype imputation needs large population-matched references. The phase information is lost in the data from high-throughput genotyping and sequencing platforms, but this piece of lost information is critically important for research and clinics. Currently the reference datasets are obtained from the HapMap project and the 1,000 Genomes Project (KGP) because the haplotypes inferred from trios are as accurate as experimentally determined molecular haplotypes except at those triple heterozygous sites (Ma et al., 2010). However, only a small number of ethnicities are available with trio haplotypes from these two projects. When a reference panel from one ethnicity is used to impute variation in a sample of another ethnicity, the quality of imputation results will be significantly reduced (Huang et al., 2009; Rao et al., 2013) although using a pooled reference panel with all available ethnicities can give acceptable results (Chambers et al., 2008; Huang et al., 2009). To date, high-density single-nucleotide polymorphism (SNP) genotyping and next-generation sequencing have generated a large number of unphased genotype datasets for many ethnic populations in the world. These unphased genotypes can be statistically phased into haplotypes. If we can use these statistically configured haplotypes and can overcome their well-known “switch error” issue in the imputation, the scarcity issue of reference haplotypes with known phases matched to most of ethnic populations in the world will be fundamentally solved.
2. Method
2.1 Reference of molecular haplotypes
All haplotype data were downloaded from HapMap, CEU (CEPH, U.S. Utah residents with ancestry from northern and western Europe). The reference haplotypes contain 116,415 SNPs, corresponding to the entire set of SNPs on chromosome-1 in the HapMap datasets. The Caucasian Reference Haplotype Panel was composed of 176 haplotypes of 88 HapMap CEU individuals (CEPH, U.S. Utah residents with ancestry from northern and western Europe). Each trio consists of 3 individuals (two parents and one child). The offspring haplotypes were inferred with parental genotypes except those triple heterozygous loci. To avoid redundancy, only the haplotypes of the parents are included in the HapMap dataset, the children’s haplotypes are not included in the final phased files. The haplotypes of an individual that was being imputed or the parents’ haplotypes of an individual that was being imputed were always temporarily removed from the reference panel.
2.2 Reference of statistically configured haplotypes
To generate the statistical haplotype reference panel, we erased the phase information from those trio haplotypes downloaded from HapMap, and then used the software Beagle version 3.3.2 to resolve the haplotypes from the unphased genotypes.
2.3 Target samples to be imputed
Our imputation software HiFi needs three input data, a reference panel, a genotype dataset, and a low-resolution haplotype dataset (Rao et al., 2013). To create the seed haplotype and genotype input, we randomly selected 6 Caucasians (NA11919, NA12144, NA12248, NA12341, NA12749, NA12763) from the HapMap CEU population. These 6 individuals are the children of the HapMap trios, so that their accurate haplotypes have been known except those triple heterozygous loci, so that we can compare imputed results with their known data. We then randomly blinded the allele phases at 70% loci of entire SNP set. The blinded loci included both homozygous and heterozygous to mimic the 30% resolution in actual haplotyping experiments. To create the seed genotype input, we randomly blinded the genotypes at 0%, 10%, 30%, or 50% loci (loci% with missing values).
2.4 HiFi imputation
The algorithm of HiFi was described by Rao et al.(Rao et al., 2013). Briefly, HiFi exhaustively seeks non-ambiguous matches to an individual’s seed haplotypes and genotypes among a panel of reference haplotypes along a sliding window. Once HiFi identifies a single non-equivocal match in a window, it will use the identified reference haplotypes to impute the phases at all loci within this window. If HiFi finds multiple matches or no match, it will adjust the window size and repeat the search automatically until a single match is found (Rao et al., 2013). HiFi imputes the sample genotypes one person by one person instead of imputing all samples together (Rao et al., 2013). HiFi output two haplotypes for each individual instead of dosage (a continuous random variable between 0 and 2) (Rao et al., 2013).
We have investigated the effects of potential impact factors on the accuracy of HiFi and developed a scoring system as an accuracy prediction metrics. This quality scoring system had been implemented into the HiFi software; the scores will be output together with the pair of haplotypes. Each imputed site will receive a 0–1 score. The higher scores, the higher quality of imputation calls (Rao et al., 2013). The performance of this quality score system had been shown previously (Rao et al., 2013).
HiFi was executed on a laptop computer [intel i7, 2.9 Ghz, 15.7 G usable RAM, 64-bit system, Win7] to impute those missing values in the datasets created above. Two reference panels were used in the imputation, both were the HapMap CEU haplotypes; however, one is composed of molecular haplotypes (trio haplotypes) downloaded from the HapMap database; the other is composed of statistically resolved haplotypes inferred from the unphased genotypes with Beagle (Fig. 1). Two reference panels have an identical number of individual haplotypes (reference size) on identical CEU individuals. When a haplotype is used as an imputation target sample haplotype, the haplotypes of this individual or his/her parents were always temporarily removed from the reference panel during the analysis. We then used HiFi to retrieve the allele phases at those blinded loci. For each case, we use 174 haplotypes from 87 individual as reference haplotypes. The phasing distance was 249-Mb, composed of 116,415 SNPs, spanning across the entire chromosome-1. Availability and implementation: http://www.4dgenome.com/software/hifi.html.
Figure 1.
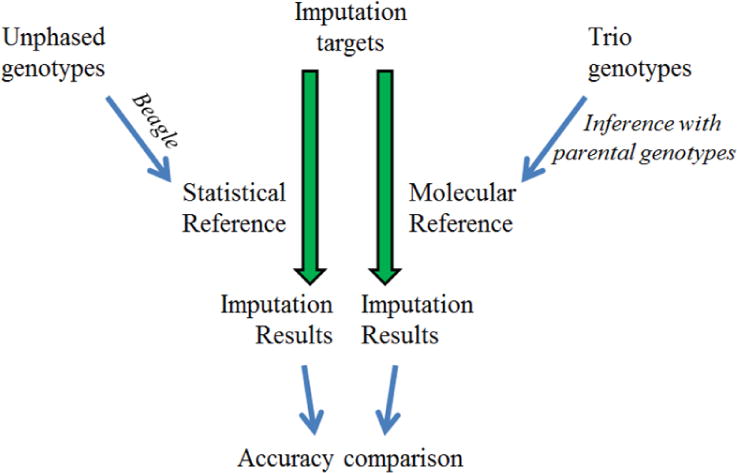
The flowchart of this study.
2.5 Measurement of accuracy
The children’s haplotypes of each trio can be determined accurately and unambiguously with trio genotypes according to Mendelian Laws of Inheritance except those triple heterozygous loci (Hodge et al., 1999; Howie et al., 2009). It has been shown that the haplotypes yielded with trio genotypes are consistent with the haplotypes determined experimentally (Ma et al., 2010; Fan et al., 2011; Kitzman et al., 2011; Yang et al., 2011) and are a widely accepted method for accuracy evaluation for phasing methods. In this study, the accuracy of the imputation results was evaluated by the concordance rate between the imputation outputs and the haplotypes inferred from trio genotypes on the same individual. We used a stringent criterion in accuracy evaluation, in which only the loci that were correct on both allele calls and allelic phases on both alleles were reported as “correct”. If a locus received only one correct allele imputation among two alleles at a locus, or both allele calls are correct but with wrong phases, this locus will be treated as an error.
In the metrics for accuracy measurement, the denominator is either the total number of heterozygous loci or the total number of the SNP loci in the reference panel. During the comparisons, all triple-heterozygous SNPs, phase-known heterozygous SNPs in the haplotype input, and all symmetric SNP loci (A/T SNPs and C/G SNPs), were excluded. Because homozygous loci are already phase-known, the accuracy with the number of imputed heterozygous SNP as the denominator can reflect the technical capacity of a method for data imputation; and the accuracy reading with the number of all SNPs in a dataset provides the information on the overall accuracy and reliability for potential applications when they choose the imputation method.
3. Results
3.1 The accuracy of imputation results
With the molecular haplotypes in the reference panel, the imputation accuracy is 99.49 ± 0.05%; with the statistically resolved haplotypes in the reference panel, the imputation accuracy is 99.43 ± 0.05% (Table 1). The accuracy of imputation results is very consistent across different individuals included in this study (Table 2). This result demonstrates that these two different reference panels can yield imputation output with similar accuracy. It is well-known that statistically resolved haplotypes are very accurate with a short-range but suffer from switch errors in the chromosomal-range. As shown in the Figure 2A, there are many large segments in the statistically resolved haplotypes in which all adjacent heterozygous SNPs are incorrectly phased. However, this statistically resolved reference panel can still lead to a high accuracy in the subsequent data imputation.
Table 1.
A comparison of haplotype imputation accuracy with molecular reference haplotype reference panel and statistically-resolved haplotype reference panel.
| Imputation Accuracy | Among all SNPs | Among heterozygous SNPs | ||
|---|---|---|---|---|
| Reference Panel | Molecular haplotypes | Statistical haplotypes | Molecular haplotypes | Statistical haplotypes |
| Data missing % | ||||
| 0% | 99.49±0.05 | 99.43±0.05 | 98.11±0.19 | 97.87±0.17 |
| 10% | 99.39±0.06 | 99.31±0.05 | 97.72±0.22 | 97.42±021 |
| 30% | 99.09±0.07 | 99.02±0.04 | 96.61±0.25 | 96.32±0.13 |
| 50% | 98.61±0.08 | 98.49±0.06 | 94.81±0.32 | 94.38±0.23 |
Table 2.
The imputation accuracies on each individual sample.
| Samples
|
NA11919 | NA12144 | NA12248 | NA12341 | NA12749 | NA12763 | Mean |
|---|---|---|---|---|---|---|---|
| References | |||||||
| Denominator = all homozygous &heterozygous SNPs, haplotype missing = 70%, genotype missing = 0% | |||||||
| Molecular panel | 99.52 | 99.51 | 99.51 | 99.44 | 99.42 | 99.56 | 99.49 |
| Statistical panel | 99.46 | 99.41 | 99.48 | 99.37 | 99.39 | 99.47 | 99.43 |
|
| |||||||
| Denominator = imputed heterozygous SNPs, haplotype missing = 70%, genotype missing = 0% | |||||||
| Molecular panel | 98.22 | 98.19 | 98.15 | 97.9 | 97.86 | 98.33 | 98.11 |
| Statistical panel | 98 | 97.82 | 98.04 | 97.63 | 97.75 | 98 | 97.87 |
|
| |||||||
| Denominator = all homozygous &heterozygous SNPs, haplotype missing = 70%, genotype missing = 10% | |||||||
| Molecular panel | 99.4 | 99.38 | 99.47 | 99.39 | 99.28 | 99.41 | 99.39 |
| Statistical panel | 99.33 | 99.37 | 99.35 | 99.27 | 99.23 | 99.3 | 99.31 |
|
| |||||||
| Denominator = imputed heterozygous SNPs, haplotype missing = 70%, genotype missing = 10% | |||||||
| Molecular panel | 97.78 | 97.70 | 98.03 | 97.7 | 97.34 | 97.75 | 97.72 |
| Statistical panel | 97.55 | 97.68 | 97.57 | 97.25 | 97.15 | 97.33 | 97.42 |
|
| |||||||
| Denominator = all homozygous &heterozygous SNPs, haplotype missing = 70%, genotype missing = 30% | |||||||
| Molecular panel | 99.17 | 99.07 | 99.17 | 99.04 | 99.01 | 99.09 | 99.09 |
| Statistical panel | 99 | 99.04 | 99.01 | 98.99 | 98.97 | 99.08 | 99.02 |
|
| |||||||
| Denominator = imputed heterozygous SNPs, haplotype missing = 70%, genotype missing = 30% | |||||||
| Molecular panel | 96.93 | 96.58 | 96.89 | 96.39 | 96.33 | 96.56 | 96.61 |
| Statistical panel | 96.3 | 96.44 | 96.27 | 96.19 | 96.2 | 96.51 | 96.32 |
|
| |||||||
| Denominator = all homozygous &heterozygous SNPs, haplotype missing = 70%, genotype missing = 50% | |||||||
| Molecular panel | 98.73 | 98.66 | 98.6 | 98.58 | 98.5 | 98.56 | 98.61 |
| Statistical panel | 98.52 | 98.55 | 98.55 | 98.44 | 98.4 | 98.5 | 98.49 |
|
| |||||||
| Denominator = imputed heterozygous SNPs, haplotype missing = 70%, genotype missing = 50% | |||||||
| Molecular panel | 95.33 | 95.05 | 94.74 | 94.67 | 94.48 | 94.57 | 94.81 |
| Statistical panel | 94.54 | 94.64 | 94.56 | 94.14 | 94.09 | 94.32 | 94.38 |
Figure 2. The distribution of imputation errors.
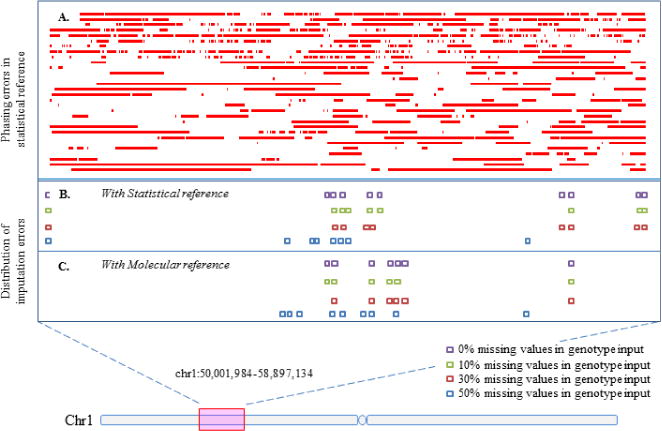
Only the imputation errors in one sample haplotype (NA11919) are shown in an 8.9-Mbp genomic region of chromosome 1. (A) The errors in the reference haplotypes resolved by Beagle. Each horizontal line represents a haplotype in the reference panel, and each horizontal bar represents an erroneous region in which all heterozygous SNPs are phased incorrectly. The results show that the statistically resolved haplotypes suffer from large “switched” stretches of incorrect phases. We calculated the switch error rate among these reference haplotypes, which is 99.59% ± 0.06%. (B) The distribution of imputation errors with statistical haplotype panel. Each square dot represents an imputation error. (C) The distribution of imputation errors with molecular haplotypes.
We further investigated the potential difference in the error distribution between data imputation with a statistical haplotype reference panel and imputation with a molecular haplotype reference panel. We found that the error distribution is very similar between the two imputation results (Fig. 2B and 2C). The errors tend to cluster together in both imputation results. This result indicates that these errors may be caused by a lack of coverage of haplotype diversity at some loci rather than the long-range switch errors.
Moreover, since big data is never big enough, we examined if our imputation method can still generate highly accurate results with a high rate of missing values in the input files. We found that even when the missing rate was as high as 50% in the genotype input data file, HiFi can still provide highly accurate imputation results with either molecular haplotype reference panel (98.61 ± 0.08% or statistical haplotype reference panel (98.49 ± 0.06%).
3.2 Computational speed
A challenge in imputation methods is how to be computationally efficient. If a method is computationally intensive and slow, it cannot be broadly applicable to genome analysis in the big data era. We examined the runtime of our imputation software HiFi on a laptop [intel i7, 2.9 Ghz, 15.7 G usable RAM, 64-bit system, Window7]. It took about 9.7 seconds to impute the human chromosome 1, and about 1.8 minutes to complete the imputation of a human genome, which is estimated to be able to impute 5,000 human individual genomes in less than a week on a laptop.
4. Discussion
With the rapid development of high-throughput technologies, the incoming flood of large-scale sequence data presents new challenges on how to deal with lost information. First, allele phase - a piece of fundamental information essential for personalized medicine - is not reported in big sequence data. Second, allele content may often be incomplete at many loci, especially when combining the data generated from different experimental platforms. However, haplotypes are essential for understanding haploinsufficiency, recessive functional variants, dosage compensation, parent-of-origin imprinting effects, drug response, disease susceptibility (Bansal et al., 2011; Fan et al., 2011). Deterministic haplotypes can greatly increase the power of genome-wide association studies (Browning, 2008), and facilitate the population genetics (Conrad et al., 2006; Green et al., 2010), deciphering cis-interactions in gene regulation (Tycko, 2010) and compound heterozygosity (McLaughlin et al., 2010; Ng et al., 2010). The data imputation of phase information is likely to become increasingly important.
The forthcoming era of genome-wide studies presents two new challenges to haplotype imputation. First, the size of datasets is about to increase dramatically, in terms of both numbers of loci and numbers of individuals. Second, to date, the number of references for the imputation is only limited to a small number of ethnicities in the world. The experimental phasing approach is still expensive at present and has not generated any large dataset yet that can be used for imputation as references. An alternative approach to obtain references is to deduce the personal haplotypes from trios (Hodge et al., 1999). However, The International HapMap Project and The 1,000 Genomes Projects have only performed SNP genotyping on a small number of human populations (Fig. 3). It is known that a mismatch of reference panel and imputation target samples on ethnicities may yield false positive results (Campbell et al., 2005). To yield the most accurate imputation results without available specifically ethnicity-matched reference panels, one strategy is to use the pooled panels from at least two populations can give acceptable results (Chambers et al., 2008; Huang et al., 2009). Indeed, we have compared the accuracy of HiFi results among matched references, unmatched references, and pooled references, and the results showed that pooled reference panel could yield the results similar to the matched reference panel (Rao et al., 2013). However, for many phasing algorithm, this approach needs to identify the “optimal” recipe for mixing the reference panels for each individual (Huang et al., 2009), which may be problematic for individuals with uncertain ethnic background and individuals with ethnically mosaic background. The other strategy is to use internal reference panels to avoid the problem of a substantial mismatch in ancestral background between the study population and the reference population (Fridley et al., 2010; Jewett et al., 2012; Zhang et al., 2013). To efficiently select an internal panel, an idea of generating “the most diverse reference panel” by phylogenetic diversity from mathematical phylogenetics and comparative genomics was proposed, which has been shown to be able to substantially improve the imputation accuracy compared to randomly selected reference panels (Zhang et al., 2013). Now there is a need to expand the number and representation of ethnicities in the haplotype references for imputation.
Figure 3. A comparison of availability between statistical references vs. molecular references in the world populations.
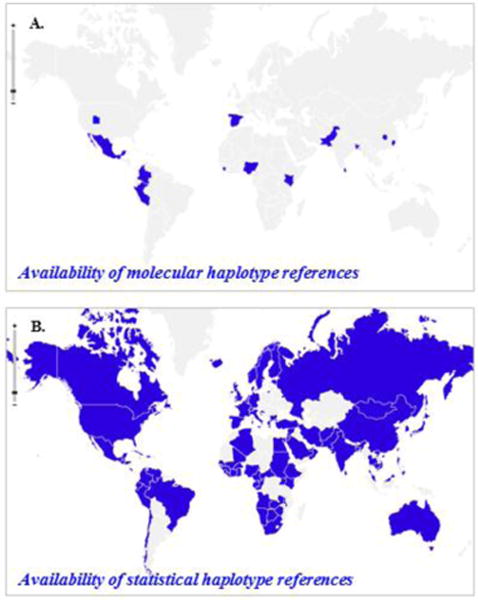
The molecular haplotypes are composed of trio haplotypes retrieved from the HapMap project and the 1,000 Genomes Project (KGP). The statistical references are composed of statistically resolved haplotype from unphased genotypes obtained from genotyping and next-generation sequencing platforms. The data is mainly retrieved form dbGaP, African Genome Variation Project, Human Genome Diversity Project (HGDP), AADM and GALA. The map was generated with “openheatmap” software.
The big data era demands new methods to be capable to smartly make good use of the available data. In the past few years, enormous amounts of unphased genotype data are being generated by genome-wide SNP microarrays and whole-genome sequencing tools. Haplotype information can be retrieved from unphased genotype data using statistical inference (Browning and Browning, 2011), which can produce reliable haplotypes for moderately long stretches of a chromosome. However, these statistically resolved haplotypes are notorious for their significant ‘switching error’ inaccuracies, where chromosomal segments are accurately phased but their connections to each other are incorrect along the entire chromosomes. Such errors can occur many times over different chromosomes. If our method can overcome these significant ‘switching error’ inaccuracies and leverage these existing pieces of information, we will provide a cost-effective method for quickly expanding the number and representations of ethnicities in the reference panels for imputation (Fig. 3).
In order to examine the imputation performance of this reference panel created from statistically resolved haplotypes, we compared its imputation accuracy and error distribution with the imputation results using the molecular haplotype references. We found that the reference panel composed of statistically resolved haplotypes can yield the high accuracy similar to the reference panel composed of molecular haplotypes (Table 1). The error distribution is also similar between two imputation results (Fig. 2B and 2C), indicating that these errors may be caused by the local haplotypes rather than the long-range switch errors. The HiFi algorithm exhaustively seeks a unique non-equivocal solution for each individual haplotype instead of assigning the haplotypes with the most likelihood (Rao et al., 2013); it is likely that those incorrectly phased “switched” segments are much longer than the non-equivocal haplotype stretches for the imputation target sites, so that our imputation with HiFi can overcome the weakness of the references composed of statistically resolved haplotypes.
Regarding phasing implementation, Beagle has been widely used in practice because of its ease to implement and convenience in accepting both genotypes from unrelated persons and related persons. It has also been found that trio-based phasing has much better accuracy than phasing based on unrelated persons. We have shown the results of comparison of HiFi performance between using references inferred by Beagle statistical phasing and references obtained by Mendelian-inheritance-based phasing of trio genotypes (Table 1). Furthermore, we compared the HiFi performances with three different implementations (Li et al., 2015): (A) Beagle statistical phasing of unrelated persons and Mendelian-inheritance-based phasing of trios, and then pool the results together; (B) Beagle statistical phasing of pooled unrelated persons and trios, but presume all as unrelated; and (C) Beagle statistical phasing of pooled unrelated persons and trios, and specifying the family structure in the input. Our results showed that the performance of these three implementations are similar on accuracy, in which the accuracy of implementation-B is slightly but consistently higher than A and C (see Supplementary Table S1 in reference (Li et al., 2015)). It will be interesting to know why implementation-B gave a consistently higher accuracy; we believe that this implementation may enable our imputation to take the advantages from both statistical-based algorithm in the reference generation and our non-statistical-based algorithm of using the references. This result may provide some ideas about choosing the optimal phasing pipeline for each user.
We compared the performance between HiFi and three standard imputation software tools (MACH, IMPUTE2 and BEAGLE) (Browning and Browning, 2007; Howie et al., 2009; Li et al., 2010). As the results, HiFi performed better on haplotype imputation accuracy (99.5% - 84.3% - 81.8% - 86.9%, HiFi – MACH – IMPUTE2 – BEAGLE) (see Supplementary Table S2 in reference (Li et al., 2015)), and speed (9 – 1698 – 173 – 239 seconds, HiFi – MACH – IMPUTE2 – BEAGLE) (see Supplementary Table S4 in reference (Li et al., 2015)), whereas the other three software performed better on genotype imputation accuracy (99.73% - 99.98% - 99.99% - 99.80%, HiFi – MACH – IMPUTE2 – BEAGLE) when the missing value rates were 70% on haplotypes and 10% on genotypes (see Supplementary Table S3 in reference (Li et al., 2015)), in which MACH and IMPUTE2 performed the best on genotype imputation. Even if HiFi performs worse than standard imputation software, it still has the benefits of quick running. Compared with MACH, IMPUTE2 and BEAGLE, HiFi requires three input: a reference panel consisting of a set of haplotypes at all sites, a genotype dataset consisting genotypes of a set of samples at genotyped sites, and a low-resolution haplotype dataset consisting a set of haplotypes from same samples in the genotype dataset (Rao et al., 2013); whereas those three major phasing software needs only two input datasets. On one hand, this is the advantage of these software over HiFi; on the other hand, our result demonstrated that a low-resolution of haplotype seeds generated from experimental phasing pipeline could substantially improve the imputation accuracy.
In conclusion, the existing unphased genotype datasets can be used for high-accuracy (99.43%) haplotype imputation using the HiFi software. It can finish a whole-genome imputation of a human individual within 1.8 minutes on a laptop computer. It can yield a 98.5% imputation accuracy even when 50% values are missing in the dataset. The big data era demands new methods that are computationally efficient and that make good use of the available data. Our method provides a cost-effective solution for the issue of lack of population-matched reference panels in data imputation.
Highlights.
The accuracy is as high as 99.43%.
It can finish a whole-genome imputation within 2 minutes on a laptop computer.
The availability issue of ethnicity-matched references is solved.
Acknowledgments
This work was supported by National Institutes of Health (R21HG006173, R43HG007621, HL117929, MD007602, MD005964, RR003034, U54MD07588); and the American Heart Association grant (09GRNT2300003).
Abbreviations list
- KGP
1,000 Genomes Project
- SNP
single-nucleotide polymorphism
- CEU, CEPH
U.S. Utah residents with ancestry from northern and western Europe
- HGDP
Human Genome Diversity Project
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: none declared
References
- Bansal V, Tewhey R, Topol EJ, Schork NJ. The next phase in human genetics. Nat Biotechnol. 2011;29:38–9. doi: 10.1038/nbt.1757. [DOI] [PubMed] [Google Scholar]
- Browning SR. Missing data imputation and haplotype phase inference for genome-wide association studies. Hum Genet. 2008;124:439–50. doi: 10.1007/s00439-008-0568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81:1084–97. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR, Browning BL. Haplotype phasing: existing methods and new developments. Nat Rev Genet. 2011;12:703–14. doi: 10.1038/nrg3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell CD, Ogburn EL, Lunetta KL, Lyon HN, Freedman ML, Groop LC, Altshuler D, Ardlie KG, Hirschhorn JN. Demonstrating stratification in a European American population. Nat Genet. 2005;37:868–72. doi: 10.1038/ng1607. [DOI] [PubMed] [Google Scholar]
- Chambers JC, Elliott P, Zabaneh D, Zhang W, Li Y, Froguel P, Balding D, Scott J, Kooner JS. Common genetic variation near MC4R is associated with waist circumference and insulin resistance. Nat Genet. 2008;40:716–8. doi: 10.1038/ng.156. [DOI] [PubMed] [Google Scholar]
- Conrad DF, Jakobsson M, Coop G, Wen X, Wall JD, Rosenberg NA, Pritchard JK. A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nat Genet. 2006;38:1251–60. doi: 10.1038/ng1911. [DOI] [PubMed] [Google Scholar]
- Fan HC, Wang J, Potanina A, Quake SR. Whole-genome molecular haplotyping of single cells. Nat Biotechnol. 2011;29:51–7. doi: 10.1038/nbt.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridley BL, Jenkins G, Deyo-Svendsen ME, Hebbring S, Freimuth R. Utilizing genotype imputation for the augmentation of sequence data. PLoS One. 2010;5:e11018. doi: 10.1371/journal.pone.0011018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH, Hansen NF, Durand EY, Malaspinas AS, Jensen JD, Marques-Bonet T, Alkan C, Prufer K, Meyer M, Burbano HA, Good JM, Schultz R, Aximu-Petri A, Butthof A, Hober B, Hoffner B, Siegemund M, Weihmann A, Nusbaum C, Lander ES, Russ C, Novod N, Affourtit J, Egholm M, Verna C, Rudan P, Brajkovic D, Kucan Z, Gusic I, Doronichev VB, Golovanova LV, Lalueza-Fox C, de la Rasilla M, Fortea J, Rosas A, Schmitz RW, Johnson PL, Eichler EE, Falush D, Birney E, Mullikin JC, Slatkin M, Nielsen R, Kelso J, Lachmann M, Reich D, Paabo S. A draft sequence of the Neandertal genome. Science. 2010;328:710–22. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge SE, Boehnke M, Spence MA. Loss of information due to ambiguous haplotyping of SNPs. Nat Genet. 1999;21:360–1. doi: 10.1038/7687. [DOI] [PubMed] [Google Scholar]
- Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Li Y, Singleton AB, Hardy JA, Abecasis G, Rosenberg NA, Scheet P. Genotype-imputation accuracy across worldwide human populations. Am J Hum Genet. 2009;84:235–50. doi: 10.1016/j.ajhg.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett EM, Zawistowski M, Rosenberg NA, Zollner S. A coalescent model for genotype imputation. Genetics. 2012;191:1239–55. doi: 10.1534/genetics.111.137984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzman JO, Mackenzie AP, Adey A, Hiatt JB, Patwardhan RP, Sudmant PH, Ng SB, Alkan C, Qiu R, Eichler EE, Shendure J. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat Biotechnol. 2011;29:59–63. doi: 10.1038/nbt.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu W, He S, Ma L, Song Q. Data supporting the high-accuracy haplotype imputation using unphased genotype data as the references Data in Brief. 2015 doi: 10.1016/j.dib.2016.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–34. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Xiao Y, Huang H, Wang Q, Rao W, Feng Y, Zhang K, Song Q. Direct determination of molecular haplotypes by chromosome microdissection. Nat Methods. 2010;7:299–301. doi: 10.1038/nmeth.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin HM, Sakaguchi R, Liu C, Igarashi T, Pehlivan D, Chu K, Iyer R, Cruz P, Cherukuri PF, Hansen NF, Mullikin JC, Program NCS, Biesecker LG, Wilson TE, Ionasescu V, Nicholson G, Searby C, Talbot K, Vance JM, Zuchner S, Szigeti K, Lupski JR, Hou YM, Green ED, Antonellis A. Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am J Hum Genet. 2010;87:560–6. doi: 10.1016/j.ajhg.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, Shendure J, Bamshad MJ. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42:30–5. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao W, Ma Y, Ma L, Zhao J, Li Q, Gu W, Zhang K, Bond VC, Song Q. High-resolution whole-genome haplotyping using limited seed data. Nat Methods. 2013;10:6–7. doi: 10.1038/nmeth.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko B. Allele-specific DNA methylation: beyond imprinting. Hum Mol Genet. 2010;19:R210–20. doi: 10.1093/hmg/ddq376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Chen X, Wong WH. Completely phased genome sequencing through chromosome sorting. Proc Natl Acad Sci U S A. 2011;108:12–7. doi: 10.1073/pnas.1016725108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Zhan X, Rosenberg NA, Zollner S. Genotype imputation reference panel selection using maximal phylogenetic diversity. Genetics. 2013;195:319–30. doi: 10.1534/genetics.113.154591. [DOI] [PMC free article] [PubMed] [Google Scholar]