

Abstract
Tn is a carbohydrate antigen uniquely exposed on tumor mucins and thus, an ideal target for immunotherapy. However, it has been difficult to elicit protective antibody responses against Tn antigen and other tumor associated carbohydrate antigens. Our study demonstrates this can be attributed to PD-1 immuno-inhibition. Our data show a major role for PD-1 in suppressing mucin- and Tn-specific B-cell activation, expansion, and antibody production important for protection against Tn-bearing tumor cells. These Tn/mucin-specific B cells belong to the innate-like B-1b cell subset typically responsible for T cell–independent antibody responses. Interestingly, PD-1–mediated regulation is B cell–intrinsic and CD4+ cells play a key role in supporting Tn/mucin-specific B cell antibody production in the context of PD-1 deficiency. Mucin-reactive antibodies produced in the absence of PD-1 inhibition largely belong to the IgM subclass and elicit potent antitumor effects via a complement-dependent mechanism. The identification of this role for PD-1 in regulating B cell–dependent antitumor immunity to Tn antigen highlights an opportunity to develop new therapeutic strategies targeting tumor associated carbohydrate antigens.
Introduction
Tumor-associated carbohydrate antigens (TACAs), including Tn (Thomsen-nouvelle/CD175) antigen, represent ideal targets for the antitumor response, as these antigens are masked on glycoproteins and glycolipids of normal cells (1). Tn antigen, composed of an N-acetylgalactosamine (GalNAc) sugar linked to a serine or threonine on the peptide backbone of mucins, and its sialylated derivate (sTn), are abnormally exposed on tumor mucins due to hypoglycosylation. Tn antigen has been reported on up to 70–90% of adenocarcinomas (1,2), although discrepancies in overall frequencies among certain malignancies exist due to differences in Tn Ab specificities (3). Tn and sTn expression by several types of tumors is associated with metastatic potential and poor prognosis (1). Patients who produce Ab against TACAs and mucins, either naturally or in response to vaccination, may have improved survival outcomes (4–9). However, eliciting protective antibody responses against these carbohydrate antigens has proven challenging.
The regulation of B-cell antibody responses to carbohydrate antigens, especially TACAs, is not completely understood. The PD-1 immunoinhibitory receptor plays a critical role in suppressing IgG responses to classical T cell–independent type 2 (TI-2) carbohydrate antigens, including pneumococcal polysaccharides, but nonetheless promotes IgG responses to T cell–dependent (TD) antigens (10–15). PD-1 is a B7/CD28 superfamily receptor expressed on activated lymphoid and myeloid cells (16). Upon engagement of its ligands, PD-L1 and PD-L2, PD-1 negatively regulates critical signaling events and thereby often dampens immune responses. Intense interest in exploiting the PD-1:PD-L regulatory axis for treatment of cancer stems from several preclinical and clinical studies demonstrating the potent effects this pathway has on suppressing T cell antitumor responses (17–24). However, a role for effects on other immune cell types has not been fully explored.
In this study, we examined a role for PD-1 in regulating the humoral immune response to mucin and Tn and its potential impact on protection against Tn+ mucin–bearing tumors. Our results show PD-1 is a major suppressor of the humoral immune response to mucin bearing high amounts of Tn (25) and to the production of cross-reactive Abs against tumor cells expressing Tn+ mucin. Furthermore, we demonstrate Tn-specific B cells largely belong to the B-1b lymphocyte subset, are nonresponsive to mucin immunization in wild-type mice, but become responsive under conditions of PD-1 deficiency. This regulation is of great physiologic relevance as B cell–intrinsic PD-1 expression suppresses the protective B cell–dependent antitumor response elicited by immunization. Remarkably, generation of high concentrations of mucin- and Tn-specific antibody in the context of PD-1 deficiency requires CD4+ cells and thereby reveals an unexpected role for PD-1 in regulating immunity to these unique antigens. In summary, our results demonstrate a critical role for B cell–intrinsic PD-1 expression in regulating protective B-cell responses to Tn-bearing tumors.
Materials and Methods
Mice
Wild-type C57BL/6 and μMT mice were from Jackson Laboratories. PD-1−/− (10) mice were on a C57BL/6 background (permission obtained from Dr. Tasuku Honjo). Studies were approved by Wake Forest’s Animal Care and Use Committee.
Immunizations and ELISAs
Bovine submaxillary mucin (Sigma, Millipore) was desialyated (dBSM) as described (26) and treated with endotoxin-removal resin. Bacteriophage Qβ virus-like particles bearing Tn (Qβ-Tn) were generated as described (27). Mice were immunized intraperitoneally (i.p.) with 5 μg Qβ-Tn, 100 μg dBSM (Sigma), or 250 μg dBSM plus 10μg LPS (Escherichia coli O111:B4, Sigma) in 200 μl PBS. CD4 depleting (GK1.5) and control (LTF-2) antibodies were from BioXcell (inVivoMAb).
ELISAs were as described (28) using Nunc Maxisorp plates coated with 10 μg/ml dBSM in 0.1M borate buffered saline and pre-blocked with TBS-BSA prior to incubation with sera. To detect dBSM-specific Abs, alkaline phosphatase-conjugated polyclonal goat anti-mouse IgM and IgG Abs (Southern Biotechnology) diluted in TBS-BSA and pNPP (Sigma) were used. ELISA values are reported as relative absorbance units (AU; OD405nm reading for serum samples minus OD405nm reading from wells with serum omitted).
Tumor challenge
TA3-Ha cells were obtained from Dr. Richard Lo-Man (Pasteur Institute, Paris, France) in 2010. This stock was tested for rodent pathogens (IMPACT IV testing, IDEXX-RADIL). One pooled ascites frozen stock was used for all subsequent challenge experiments. Cells were expanded for several days prior to injection. Mice developing ascites with signs of distress (lethargy, dehydration, reduced/impaired movement, reduced grooming, labored breathing, etc.) were humanely euthanized.
Cell transfers and cobra venom factor administration
Naïve spleen and peritoneal B cells were purified using negative depletion as described (11,13). B cells from immune mice were purified using EasySep untouched mouse B-cell purification (Stem Cell Technologies) with biotinylated F4/80 antibody included. Cobra venom factor (Millipore) was administered i.p. (20 μg/mouse) one day prior to tumor challenge and on days 1, 3, 5, 7, 9, and 11.
Flow cytometry
TA3-Ha cells, E0771 cells, and Jurkat cells (1 × 106/ml) were stained with diluted sera (1:10–1:50) in PBS containing 2% calf serum for 30 minutes at RT and washed. Goat anti-IgM-FITC and anti-IgG-PE (Southern Biotechnology Associates, Inc.) were used to detect bound Ab. For antigen-specific analysis, cells were pre-incubated with 0.5 μg/ml Fc block and stained with 18 μg/ml dBSM-AlexaFluor488 or 2.5 μg/ml Tn-BSA-AlexaFluor647, and mAbs conjugated to fluorochromes or biotin: CD5 (53-7.3), CD80(16-10A1), CD86(GL-1), CD11b(M1/70), CD138(281-2) all from Biolegend, CD21/35 (7E9) from eBioscience, and CD19(1D3), PD-1(J43) from BD Biosciences, and corresponding isotype controls. Biotin-conjugated mAbs were detected using streptavidin-fluorochrome conjugates. Cells were analyzed using a FACSCanto II cytometer (Becton Dickinson).
Statistical analysis
Data are shown as means ± SEM with differences assessed using unpaired Student’s t test. Differences in Kaplan-Meier survival curves were assessed using the Log Rank or Gehan-Wilcoxon tests.
Results
PD-1−/− mice produce Abs that cross-react with Tn+ mucin-expressing tumors
Desialylated ovine and bovine submaxillary gland mucins (dBSM) have been used to study Ab responses to T, Tn, and sTn in both mice and humans due to their display of natural glycan clusters mimicking TACAs found on tumor-derived mucins (8,25,26,29,30). In contrast to weak IgM and IgG responses to dBSM in WT mice, PD-1−/− mice produced robust dBSM-specific IgM and IgG responses following boosting (Fig. 1A). Moreover, sera from dBSM-immunized PD-1−/− mice exhibited significant IgM, and to a lesser extent IgG, reactivity with TA3-Ha cells—a mucinous Tn-expressing mammary tumor line ((26,31); Fig. 1B–C). Free GalNAc, but not glucose, inhibited IgM binding, indicating a portion of dBSM-elicited IgM in PD-1−/− mice was Tn-reactive (Fig. 1D). Free GalNAc had no measurable effect on WT sera binding (percent reduction in MFI: WT, 2.6%; PD-1−/−, 31%). We did not detect differences between WT and PD-1−/− d35 immune sera reactivity with a Tn-negative mammary carcinoma line, E0771 (Fig. 1E). Sera from dBSM-immune PD-1−/−, but not WT, mice also showed significant reactivity with Jurkat cells, a human T-cell leukemia line with high Tn expression (Fig. 1F–G; (1)). GalNAc inhibited binding, suggesting reactivity was due to Tn Ab (Fig. 1H). Consistent with dBSM results, PD-1−/− mice immunized with Qβ-Tn, a bacteriophage displaying Tn (27), had significantly more IgM and IgG reactive with TA3-Ha cells (Fig. 1I). Thus, PD-1−/− mice produce significantly more Abs that are cross-reactive with Tn/mucin-bearing tumor cells following immunization with Qβ-Tn and dBSM, which could be attributed in part to increased Tn-specific Ab production.
Figure 1. PD-1−/− mice produce increased dBSM- and Tn+ tumor mucin-specific Ab following dBSM and Qβ-Tn immunization.
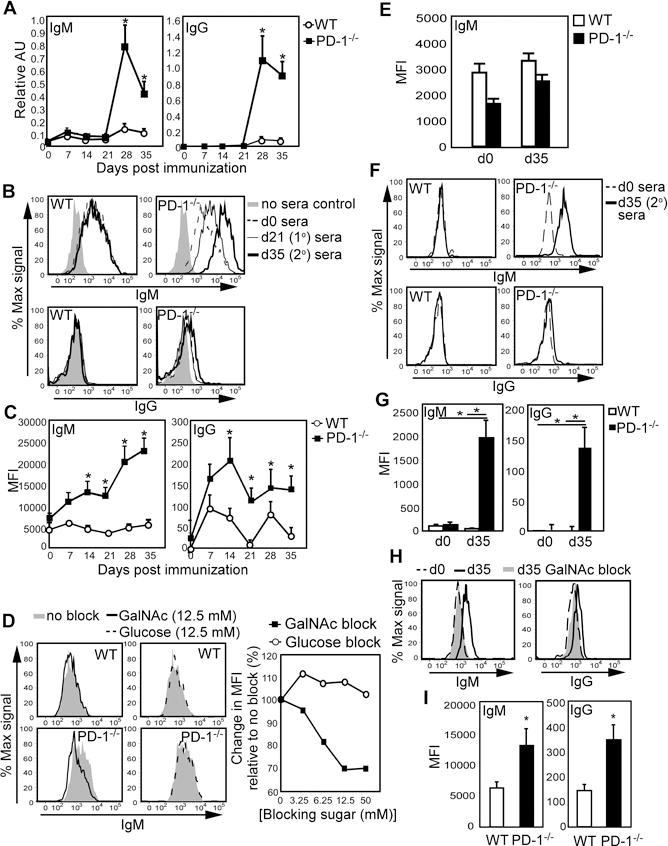
A–H) WT and PD-1−/− mice were immunized with 100μg dBSM on d0 and 21. A) Mean(± SEM) dBSM-specific serum IgM and IgG concentrations. B–C) Serum IgM and IgG reactivity with TA3-Ha cells. Representative staining (B) and average MFI (±SEM) values (C). Results representative of 4 independent experiments. D) GalNAc but not glucose inhibits IgM from dBSM-immunized PD-1−/− mice (d28) from binding TA3-Ha cells. E) Serum IgM (mean MFI ±SEM) reactivity with E0771 cells. F–H) Immune (d35; n=7–8 mice/group) and naive serum IgM and IgG reactivity with Jurkat cells. Representative staining (F) and mean MFI (±SEM) values (G). H) GalNAc(50 mM) inhibits PD-1−/− serum (d35) from binding to Jurkat cells. I) Mice were immunized with 5 μg Qβ-Tn on d0 and d21. Serum IgM reactivity (d35) with TA3-Ha cells shown as mean MFI(±SEM; n≥6/group). Asterisks (*) indicate significant differences (p <0.05) between mean values.
Protection against an aggressive Tn-bearing tumor in PD-1−/− mice by immunization
TA3-Ha cells are used as an aggressive model of peritoneal carcinomatosis (26,32–34). TA3-Ha cells (104) injected i.p. into naïve WT C57BL/6 mice induced visible ascites between days 8–10. Mice rapidly succumbed to tumor burden and overwhelming ascites development by d14 (Fig. 2A). Naïve WT, naïve PD-1−/−, and immune WT mice showed similar susceptibility to TA3-Ha challenge, with all mice succumbing to tumors. However, dBSM and Qβ-Tn immunization provided PD-1−/− mice with significant protection as shown by increased median time to death and ~35% overall survival (Fig. 2A–B).
Figure 2. DBSM immunization elicits protection against a Tn-bearing tumor in PD-1−/− but not WT mice.
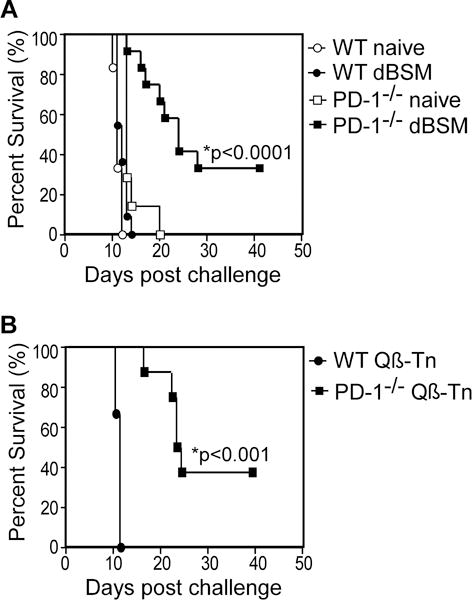
WT and PD-1−/− mice were immunized with dBSM (A) or Qβ-Tn (B) on d0 and d21. Mice were given 104 TA3-Ha cells i.p. on d42 (A) or d49 (B). Significant differences in survival were assessed by Log-rank analysis (A, n=6–7 naive mice/group and 11–12 immune mice/group; B, n=6–8 mice/group).
Adjuvant further enhances mucin-elicited B cell-dependent tumor protection in PD-1−/− mice
We investigated whether LPS would enhance responses to dBSM, due to its potent activity in promoting B-cell activation and Ab production. PD-1−/− mice produced significantly higher dBSM- and TA3-Ha-specific IgM concentrations relative to WT mice in response to primary dBSM+LPS immunization (Fig. 3A–B). Despite negligible increases in IgG reactivity with TA3-Ha cells (not shown), immune PD-1−/− mice were nonetheless protected during tumor challenge. Staining with tumor-reactive Helix pomatia agglutinin (HPA) (35) indicated that > 70% of blood cells (> 107 cells/ml) in naïve WT and PD-1−/− mice, as well as immunized WT mice, consisted of HPA+FSChi(TA3-Ha) cells 10d post-challenge (Fig. 3C). In contrast, HPA+FSChi cell numbers were at background in immunized PD-1−/− mice, suggesting tumor cells had not disseminated to the blood. Significant differences in HPA− (leukocyte) cell numbers among naïve and immune groups were not detected (Fig. 3C). Consistent with these findings, > 90% of naïve WT, naïve PD-1−/−, and immune WT mice succumbed to the tumor (Fig. 3D). In contrast, the majority of immune PD-1−/− mice survived. Immunization with dBSM did not increase survival in B cell–deficient μMT mice lacking PD-1 (μMT/PD-1−/−) (Fig. 3E). Thus, primary dBSM+LPS immunization significantly prevented tumor dissemination and promoted survival in PD-1−/−, but not WT, mice, and this effect depended on B cells.
Figure 3. Primary dBSM + LPS immunization protects PD-1−/− mice against TA3-Ha challenge and requires B cells.
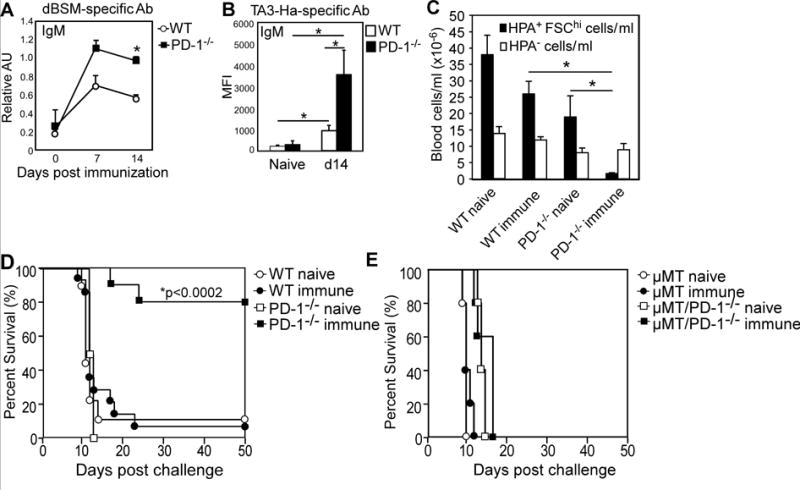
WT and PD-1−/− mice were immunized with 250 μg dBSM plus 10 μg LPS. A–B) Serum IgM reactivity against dBSM (A) and TA3-Ha cells (B). C–D) Mice were challenged with 104 TA3-Ha cells on d21. Ten days post-challenge, tumor cells in blood were detected using HPA staining and FSC (FSChiHPA+) (C). D) Differences in survival were assessed by Log-rank analysis (n=4–9 naive and 10–14 immune mice/group). Results representative of 3 independent challenge experiments. E) Survival in μMT mice and μMT/PD-1−/− mice immunized and challenged with TA3-Ha cells as above (n=5 mice/genotype). Asterisks(*) indicate significant differences (p <0.05) between mean (±SEM) values for WT and PD-1−/− mice.
PD-1 suppressed dBSM- and Tn-specific B-1 B cell responses against dBSM
We assessed changes in dBSM-specific B cell populations in WT and PD-1−/− mice following dBSM immunization. Representative gating strategies for dBSM-specific B cells are shown in Fig. 4A for peritoneal cavity and Supplemental Fig. S1A for spleen. Peritoneal and splenic dBSM-binding B-cell numbers were lower in naïve PD-1−/− mice relative to WT mice (Fig. 4B). However, peritoneal and splenic dBSM-specific B-cell numbers were significantly increased 5 days following dBSM boosting in PD-1−/−, but not WT mice (Fig. 4B). Overall, dBSM-specific FSChiCD86+ B-cell frequencies and numbers in peritoneal cavity (Fig. 4C) and spleen (Fig. 4D) increased > 2.5-fold in immune PD-1−/− mice relative to naïve PD-1−/−, immune WT, and naïve WT mice (Fig. 4C–D and Fig. S1B). Activated dBSM-specific peritoneal B cells were CD19hi, CD11b+(94±1%), and CD5neg(74±7%), indicating most belonged to the B-1b cell subset (Supplemental Fig. S1C). Thus, dBSM-specific B cells were selectively expanded and activated following dBSM immunization in PD-1−/−, but not WT, mice.
Figure 4. PD-1 suppresses activation and expansion of dBSM- and Tn-specific B-1b cells and B cells from immune PD-1−/− mice transfer antitumor protection.
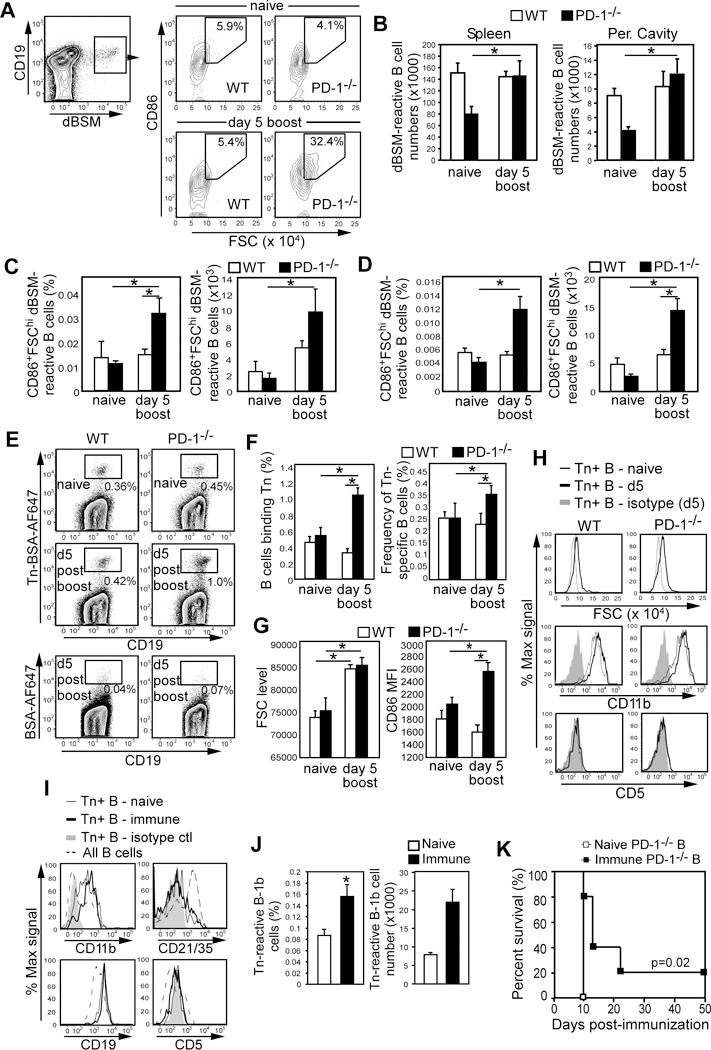
A–D) Analysis of dBSM-specific B cells in peritoneal cavities and spleens. A) Representative gating for peritoneal dBSM+CD19+ B cells with subgating on CD86+FSChi cells for naïve and immune (d5 post dBSM boost) mice. B) Mean numbers (±SEM) of dBSM-binding B cells. C–D) Mean frequencies and numbers (±SEM) of CD86+FSChi dBSM-binding B cells in peritoneal cavities (C) and spleens (D). E–K) Analysis of Tn-specific peritoneal B cells. E) Representative gating of Tn-BSA-binding and BSA control-binding CD19+ peritoneal B cells. F) Mean frequencies of B cells binding Tn(left panel) and frequencies of Tn-specific B cells among peritoneal leukocytes (right panel). G) Mean (±SEM) FSC and CD86 MFI values for Tn-specific peritoneal B cells. H) Phenotype of Tn-specific peritoneal B cells. Shaded histograms represent isotype controls. For panels A–H, WT and PD-1−/− mice were immunized with dBSM on d0, 21, and 176. Ag-specific B cells were analyzed on d181 (d5 post boost). In A–G, asterisks(*) indicate significant differences in mean (± SEM) values (p <0.05; n=4–8 mice/group). I–J) PD-1−/− mice were immunized with dBSM on d0 and 21 and challenged with TA3-Ha cells on d42. On d125, Tn-specific peritoneal B cell phenotype (I) and numbers (J) were assessed (n=3–4 mice/group). K) Spleen B (3 × 107 i.v.) and peritoneal B (8 × 106 i.p.) cells from dBSM-immune PD-1−/− mice that survived TA3-Ha challenge were transferred into naïve WT mice. Two days later, mice were challenged with 2 × 104 TA3-Ha cells (n= 5/group).
We analyzed Tn-specific B cells in WT and PD-1−/− mice 5 days after dBSM boosting in a similar manner. The frequencies of peritoneal B cell specific for Tn were significantly increased in PD-1−/− (~2-fold) 5 days after dBSM boosting, but remained unchanged in WT mice (Fig. 4E–F). Consistent with this, the overall frequencies of Tn-specific peritoneal B cells among total leukocytes were significantly increased in immune PD-1−/− mice relative to naïve PD-1−/− and immune WT mice (Fig. 4F, right). FSC was increased for Tn-specific peritoneal B cells from both immune WT and PD-1−/− mice relative to naïve mice (Fig. 4G–H). However, CD86 expression was only increased on Tn-specific peritoneal B cells from immune PD-1−/− mice (Fig. 4G). Moreover, in immune PD-1−/−, but not WT, mice, an increased frequency of Tn-specific peritoneal B cells expressed the plasmablast marker, CD138 (Supplementary Fig. S1D). Tn-specific peritoneal B cells were CD11b+ and CD5–, suggesting they also belonged to the B-1b cell subset (Fig. 4H). CD11b+CD5− Tn-specific B cells were also present in the spleens of some immune PD-1−/− mice (data not shown). Thus, dBSM immunization resulted in significant activation and expansion of Tn-specific B-1b cells and increased Tn-specific plasmablast numbers in peritoneal cavities of PD-1−/−, but not WT, mice.
An expanded pool of Tn-specific memory B cells in PD-1−/− mice transfers tumor protection
Twelve weeks post-tumor challenge, Tn-specific peritoneal B-1b cells remained CD19hiCD11b+CD5negCD21/35lo/neg and were significantly increased over naïve mice (Fig. 4I–J). Tn-specific spleen B-cell frequencies in survivor mice were also significantly increased over naive mice and nonspecific BSA-binding B-cell frequencies (Supplemental Fig. 1E). In particular, the frequency of Tn-specific spleen CD21/35lo/neg B cells in PD-1−/− tumor survivors was significantly increased (2.5-fold; Supplemental Fig. S1F), suggesting Tn-specific B-1 cells had expanded and/or trafficked to the spleen. Thus, the expanded pool of Tn-specific B cells resulting from immunization and/or tumor challenge persists and is predominantly composed of B-1b cells. This expanded “memory-like” population along with other tumor-reactive cells may contribute to long-term protection. Consistent with this notion, immune PD-1−/− survivors survived a secondary TA3-Ha challenge (Supplemental Fig. S2). Moreover, transfers of spleen and peritoneal B cells from PD-1−/− survivors into WT naïve mice prolonged survival following TA3-Ha challenge, whereas all mice receiving naïve PD-1−/− cells were deceased on day 10 (Fig. 4K).
Secondary mucin- and Tn-specific antibody responses in PD-1−/− mice required CD4+ cells
Mucins have characteristics of TI antigens—given their high carbohydrate content, repeating antigenic epitopes, and predominant elicitation of IgM—but have the potential to elicit T-cell help, given their peptide content. We therefore examined whether CD4+ cells played a role in the augmented dBSM-specific and TA3-Ha cross-reactive antibody response in PD-1−/− mice. CD4+ cell–depletion during the primary response had no measurable effect on IgM or IgG responses to dBSM or TA3-Ha cells (Fig. 5A–B). However, CD4+ cell–depletion significantly blunted secondary dBSM and cross-reactive TA3-Ha antibody responses in PD-1−/− mice (Fig. 5A–B). Free GalNAc moderately reduced primary immune (d20) sera binding (by 10–20%) to TA3-Ha cells (Fig. 5C). Blocking was similar between primary sera from control and CD4+ cell–depleted mice. By contrast, free GalNAc reduced binding of sera from control dBSM-boosted mice (d37) to TA3-Ha cells by 60%, whereas sera from dBSM-boosted mice that had received CD4-depleting antibody was only moderately blocked, as observed for primary sera (Fig. 5C). Similar results were obtained using Tn-expressing Jurkat cells (Fig. 5D). Not unexpectedly, mice depleted of CD4+ cells during primary and secondary dBSM immunization all succumbed to TA3-Ha challenge despite a return of CD4+ cells by the time of tumor challenge, whereas mice that had received control Ab exhibited significantly increased survival (Fig. 5E). Thus, in PD-1−/− mice, CD4+ cells are required for optimal secondary antibody responses to dBSM, including the production of Abs that cross-react with TA3-Ha cells and Tn antigen.
Figure 5. Secondary mucin- and Tn-specific antibody responses in PD-1−/− mice requires CD4+ cells.
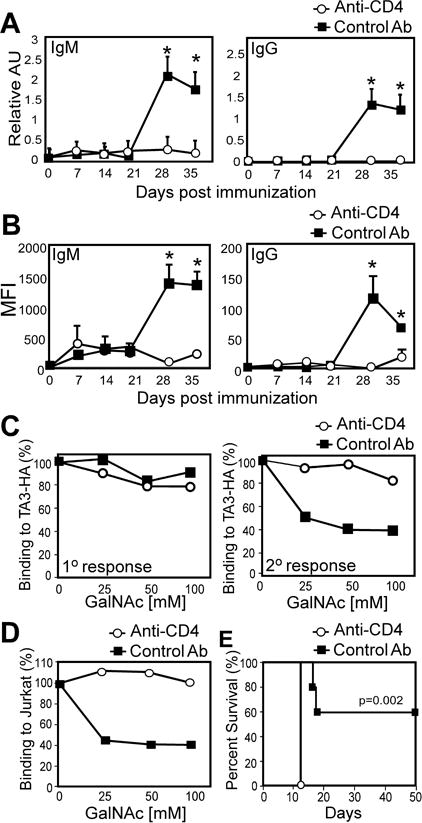
A–E) PD-1−/− mice were immunized with 100μg dBSM on d0 and 21. Mice received GK1.5 (anti-CD4) or control rat IgG2b i.p. on days −3 (200 μg), 0 (200 μg), and +3 (100 μg) of primary and secondary immunization. A–B) dBSM-reactive (A) and TA3-Ha-reactive (B) serum IgM and IgG. C) GalNAc inhibition of dBSM-elicited IgM binding to TA3-Ha cells. MFI values for GalNAc-incubated samples were compared to PBS-only samples (pooled sera (d20 [1/10 dilution]; left panel and d37 [1/50 dilution]; right panel) to determine percent blocking. (D) Inhibition of IgM (d37; 1:25 dilution) binding to Jurkat cells by GalNAc. (E) Survival of dBSM-immunized PD-1−/− mice following TA3-Ha challenge on d42 (n=5 mice/group). Asterisks indicate significant differences between GK1.5- and control Ab-treated mice (p<0.05; n=5/group).
B cell–intrinsic PD-1 suppresses dBSM-specific Ab responses and weakens tumor protection
DBSM-specific B-1b cells upregulated PD-1 following dBSM immunization (Fig. 6A). PD-1 expression was not increased on nonspecific B-1 cells. We therefore assessed the role of B cell–intrinsic and nonintrinsic PD-1 expression in regulating dBSM antibody responses. Transfers of splenic and peritoneal B cells from naïve PD-1−/− mice into μMT mice yielded significantly higher primary and secondary IgM responses to dBSM relative to mice reconstituted with WT B cells (Fig. 6B). In contrast, μMT mice and μMT/PD-1−/− mice that had received WT B cells showed no significant differences in dBSM-specific Ab responses (Fig. 6C), suggesting a limited role for B cell-extrinsic PD-1 expression in regulating the Ab response. Transfers of splenic PD1−/− B cells alone into μMT mice also yielded increased dBSM-specific IgM responses relative to WT B cells (Fig. 6D) and significantly prolonged survival in response to TA3-Ha challenge relative to mice reconstituted with WT B cells (Fig. 6E). Not unexpectedly, all mice eventually succumbed to the tumor likely due to the limited amount of TA3-Ha-reactive antibody produced in reconstituted mice. Total serum IgM concentrations in μMT mice reconstituted with PD-1−/− B cells were not increased over that in mice reconstituted with WT B cells (WT: 164 ± 14 μg/ml; PD-1−/−: 75 ± 17 μg/ml, d10). Thus, B cell–intrinsic PD-1 expression regulates the protective mucin-specific Ab response.
Figure 6. PD-1 regulation of dBSM and Tn-specific antibody responses is B cell–intrinsic.
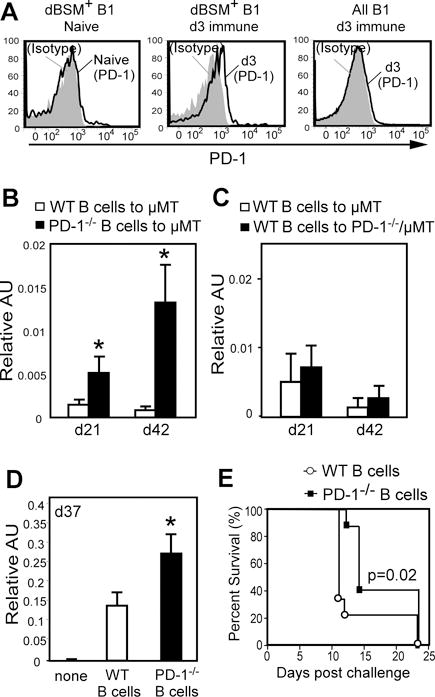
A) PD-1 expression on peritoneal dBSM-specific B cells from naïve and dBSM-immunized (d3) WT mice (left and middle histograms) and all B-1 cells in immune mice (right histogram). (B–C) Purified spleen B cells (1 × 107) and peritoneal B cells (1 × 106) from naïve WT and PD-1−/− mice were transferred i.v. and i.p., respectively, into μMT or PD-1−/−μMT mice. Ten days later, mice were immunized with 100μg dBSM (d0 and d21). Mean dBSM-specific IgM concentrations (± SEM) are indicated (n=7–8/group). (D–E) Purified spleen B cells (2 × 107) from naïve WT and PD-1−/− mice were transferred i.v. into μMT mice. Mice were immunized as above. (D) Mean (± SEM) dBSM-specific IgM concentrations at d37 (n=7–8/group). (E) μMT recipient mice were challenged with 2 × 104 TA3-Ha cells on d42. Differences in survival factoring for time-to-death were assessed by Gehan-Breslow-Wilcoxon test (p=0.02; n=8–9/group). Significant differences between Ab concentrations in reconstituted mice in B and D are indicated (*, p<0.05).
Antibody from dBSM-immunized PD-1−/− mice elicits protection in the absence of T cells
To determine whether dBSM-elicited Abs in PD-1−/− mice contributed to increased survival following TA3-Ha challenge, we performed passive serum transfers. Relative to immune WT sera, immune sera from PD-1−/− mice provided significantly increased protection during TA3-Ha challenge (Fig. 7A). The total tumor burden was significantly reduced in moribund mice that had received PD-1−/− sera, suggesting enhanced antitumor immunity in these mice (Fig. 7B). T cells were not essential for therapeutic dBSM-elicited antibody-mediated protection, as T cell–deficient nude mice administered sera from dBSM-immunized PD-1−/− mice had significantly reduced TA3-Ha cell frequencies and numbers in the circulation 10d post-challenge (Fig. 7C) and had significantly prolonged and improved survival (Fig. 7D). Thus, passive transfer of dBSM-elicited antibody from PD-1−/− mice significantly limited tumor dissemination and improved survival in the absence of T cells.
Figure 7. Protection against TA3-Ha growth and dissemination in PD-1−/− mice depends on antibody and complement.
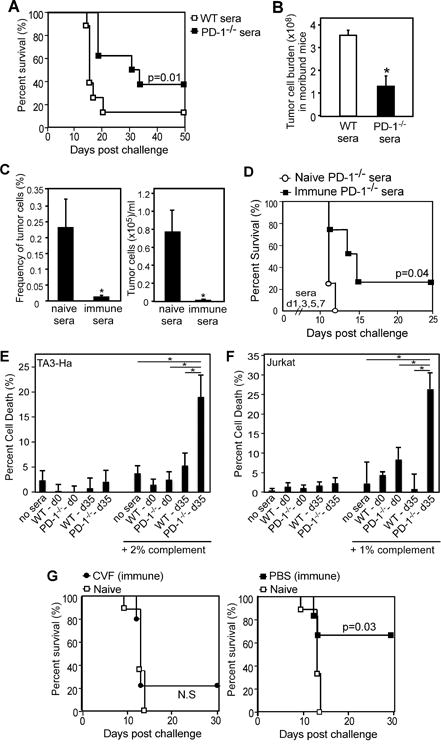
A–B) Transfer of serum from dBSM-immunized WT or PD-1−/− mice into naïve mice. Mice were challenged with TA3-Ha cells and received 30μl sera on d0 and 100 μl sera on d1, 3, and 6 i.p. A) Pooled survival results (n=4/group) represent 2 independent challenge experiments (n=8/group). B) Mean TA3-Ha cell numbers (±SEM) in ascites of moribund mice. (C–D) Naïve nude mice were challenged with 1,000 TA3-Ha cells on d0 and received 100 μl pooled sera from dBSM-immune or naïve PD-1−/− mice on d1, 3, 5, and 7 (i.p.). Nude mice received 1mg cyclophosphamide on d1 i.p. (n=4/group). Mean frequencies and numbers of TA3-Ha cells (HPA+CD138+CD19−CD11b−) present in blood 10 days post-challenge (C) and survival results (n=4/group; D). E–F) TA3-Ha (E) or Jurkat (F) cells were incubated with naïve (d0) or dBSM-immune (d35) serum (1/25 dilution) from individual WT or PD-1−/− mice (n=3–4/group) with or without complement (1–2%). 7AAD staining was analyzed after 3 hours. Results representative of 2 independent experiments. G) Effect of CVF administration on survival during TA3-Ha challenge in dBSM-immune PD-1−/− mice versus survival in naïve PD-1−/− mice(n=5-9 mice/group; Log-rank analysis). In B, C, E, and F, significant differences between groups are indicated (*, p<0.05).
Complement is required for dBSM-elicited Ab-mediated killing of tumor cells
To investigate whether complement was involved in Ab-mediated protection, we performed CDC assays. Incubation of naïve or dBSM-immunized mouse sera with TA3-Ha or Jurkat cells had no effect on cell viability relative to no sera control (Fig. 7E–F). However, when complement was present, sera from dBSM-immunized PD-1−/− mice elicited significant killing of TA3-Ha and Jurkat cells relative to immune WT, naïve PD-1−/−, and naïve WT sera (Fig. 7E–F). We therefore assessed the effect of depleting complement in vivo using the complement activator, cobra venom factor (CVF). DBSM immunization-induced protection in PD-1−/− mice was completely lost with CVF treatment, as survival in immune CVF-treated mice (in contrast to PBS control mice), no longer differed from that of naïve mice (Fig. 7G). Thus, dBSM-elicited Ab-mediated protection in PD-1−/− mice is complement dependent.
Discussion
Tn- and sialyl Tn-based vaccines have shown efficacy in mice and a fraction of patients in clinical trials(8,36–38). Despite the promise of these vaccines, inducing optimal concentrations of protective Abs against these glycan antigens displayed on natural mucins remains a challenge. Our data demonstrating PD-1 plays a major role in limiting protective Ab responses to these and other TACAs thereby reveals a tangible opportunity for improving protective responses in patients. Specifically, we report several clinically relevant findings, the most important of which include: 1) PD-1 suppresses mucin- and Tn-specific Ab responses that provide complement-dependent protection in a peritoneal carcinomatosis model of tumor growth and dissemination, 2) PD-1 suppresses activation, expansion, and plasmablast differentiation of mucin/Tn-reactive peritoneal B-1b cells, and suppresses anti-mucin responses via B cell–intrinsic expression, and 3) CD4+ cells play a key role in promoting mucin/Tn-reactive Ab responses in the context of PD-1 deficiency, but are not required for these Abs to elicit protection against tumor growth and dissemination. Thus, our novel study reveals a critical role for PD-1 in suppressing B cell–dependent anti-Tn/mucin Ab responses that provide protection against tumors.
The TA3-Ha model of peritoneal cavity tumor growth and ascites development (peritoneal carcinomatosis) with dissemination into the circulation is an ideal model system to study effects of Tn-specific B cell responses (26,32–34). In this tumor model, mucin-reactive Abs produced by PD-1−/− mice contributed to tumor killing through complement activation, as evidenced by their efficacy in eliciting CDC against both TA3-Ha cells and Tn+ Jurkat cells in vitro, and the requirement for complement in immune-mediated protection in vivo. These effects may be attributed to the high levels of mucin-reactive IgM produced in PD-1−/− mice. Mucin-elicited IgG may also contribute via antibody-dependent cellular cytotoxicity and antibodies may function by altering mucin signaling and/or adhesion properties important for limiting metastases. Indeed, the decreased dissemination observed in immune PD-1−/− mice and in serum transfer experiments supports the latter (39). Finally, although mucin/Tn-specific Ab contributed to protection, it is possible B cells may regulate additional aspects of antitumor immunity, such as antigen presentation (40–42) and/or cytokine production. Future studies will address these possibilities.
The identification of B-1 cells as a major B-cell subset harboring mucin- and Tn-specific B cells has important implications for therapeutic strategies activating immune responses to TACA-bearing tumors. We have identified a B-1b counterpart responsive to TI-2 antigens in nonhuman primates(12,43) and a recirculating B-1–like population has also been described in humans (44). In both WT and PD-1−/− mice, dBSM- and Tn-specific peritoneal B cells were predominantly B-1b cells. Interestingly, the frequency of Tn-reactive B cells was enriched in the peritoneal cavity relative to spleen. This has been noted for B-1 cells with other self-antigen specificities (45). Evidence of selective activation, expansion, and differentiation of these antigen-binding cells in immunized PD-1−/− mice, but not WT mice, clearly supports the notion that the PD-1 regulatory axis controls B-1 responses to mucins. PD-1 suppresses humoral responses to classical TI-2 antigens (10,11,13). PD-1 is transiently upregulated by antigen-specific B-1b cells (11,13,43) and thereby contributes to suppression of antigen-specific B cell division, isotype switching, and Ab production during TI-2 responses (11,13). However, dBSM shares functional characteristics of both TI-2 (multivalent carbohydrate display) and TD (protein backbone) antigens. Thus, it is possible that conjugating Tn to an immunogenic protein carrier could recruit B-2 cells into the response. Future work will address this possibility.
Our identification of B cell–intrinsic PD-1-mediated suppression of humoral responses to dBSM and Tn involving CD4+ cells represents a unique finding, as previous studies have suggested a positive regulatory role for PD-1 in TD Ab responses based on evidence for PD-1-mediated support of T follicular helper cell function (14,15). The poor immunogenicity of dBSM in WT mice suggests there may be limited T cell help available, and this along with PD-1 suppression likely contributes to poor activation of mucin/Tn-specific B cells. Mucins also bind receptors on professional antigen presenting cells and elicit generalized suppression, which can contribute to tumor immune evasion (46–48). Inefficient processing and presentation of heavily glycosylated mucins to T cells by dendritic cells may also contribute to poor immunogenicity (49). Thus, T-cell activation by mucin-derived peptide, and hence, provision of T-cell help, may rely more heavily on antigen presentation by activated mucin-specific B cells expressing appropriate T-cell costimulatory signals—the upregulation of which are suppressed by PD-1. Nonetheless, it remains entirely possible that the CD4+ help involved in supporting Tn-specific antibody responses in our study represents a unconventional (non-cognate) type of T/NKT cell help. Support for this is based on a previous study showing a TCR-nonspecific CD86/CD80-dependent form of T-cell help promotes polysaccharide (phosphorylcholine)-specific antibody responses against bacteria (50)—a response attributed to innate-like B1 cells and marginal zone B cells. Future work is necessary to identify the precise mechanisms by which CD4+ cells support mucin/Tn-specific antibody responses and the extent to which PDL1 and PDL2 expressed by these and other cells contribute to PD-1–mediated regulation.
In summary, our results demonstrate that PD-1 suppresses B-cell functions critical for optimal antitumor immunity to TACA-bearing tumor mucins. Therapeutics targeting PD-1:PD-1 ligand interactions have shown tremendous clinical results (17,18). Although T-cell modulation represents a major mechanism by which these therapies elicit their effects, this study raises the exciting possibility that PD-1–directed therapies may also support B-cell responses to TACAs. Our preliminary results using PD-1 mAb blockade in our model system support this possibility (unpublished observations, KMH and MAH). An understanding of the impact this regulatory pathway has on human B cells may ultimately lead to the development of additional strategies specifically aimed at eliciting effective antitumor B-cell responses.
Supplementary Material
Acknowledgments
The authors would like to thank MG Finn(Georgia Tech) for assistance in Qβ-Tn production.
This work was supported by an American Cancer Society research scholar grant (RSG-12-170-01-LIB) and NIAID/NIH R01AI118876 awarded to KMH, and R01CA149451 awarded to XH. Shared resources support was provided by NCI-CCSG grant P30CA012197. MAH was supported by NIH/NCI F31CA183567. KMH holds a patent for PD-1 blockade in pneumococcal vaccines.
Footnotes
The authors have no other conflicts to disclose.
References
- 1.Ju T, Otto VI, Cummings RD. The Tn antigen-structural simplicity and biological complexity. Angewandte Chemie. 2011;50:1770–91. doi: 10.1002/anie.201002313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Springer GF. T and Tn, general carcinoma autoantigens. Science. 1984;224:1198–206. doi: 10.1126/science.6729450. [DOI] [PubMed] [Google Scholar]
- 3.Li Q, Anver MR, Butcher DO, Gildersleeve JC. Resolving conflicting data on expression of the Tn antigen and implications for clinical trials with cancer vaccines. Molecular cancer therapeutics. 2009;8:971–9. doi: 10.1158/1535-7163.MCT-08-0934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pashov A, Monzavi-Karbassi B, Kieber-Emmons T. Immune surveillance and immunotherapy: lessons from carbohydrate mimotopes. Vaccine. 2009;27:3405–15. doi: 10.1016/j.vaccine.2009.01.074. [DOI] [PubMed] [Google Scholar]
- 5.Monzavi-Karbassi B, Pashov A, Kieber-Emmons T. Tumor-Associated Glycans and Immune Surveillance. Vaccines. 2013;1:174–203. doi: 10.3390/vaccines1020174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell CT, Gulley JL, Oyelaran O, Hodge JW, Schlom J, Gildersleeve JC. Serum antibodies to blood group A predict survival on PROSTVAC-VF. Clin Cancer Res. 2013;19:1290–9. doi: 10.1158/1078-0432.CCR-12-2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dalziel M, Crispin M, Scanlan CN, Zitzmann N, Dwek RA. Emerging principles for the therapeutic exploitation of glycosylation. Science. 2014;343:1235681. doi: 10.1126/science.1235681. [DOI] [PubMed] [Google Scholar]
- 8.Ibrahim NK, Murray JL, Zhou D, Mittendorf EA, Sample D, Tautchin M, et al. Survival Advantage in Patients with Metastatic Breast Cancer Receiving Endocrine Therapy plus Sialyl Tn-KLH Vaccine: Post Hoc Analysis of a Large Randomized Trial. Journal of Cancer. 2013;4:577–84. doi: 10.7150/jca.7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fremd C, Stefanovic S, Beckhove P, Pritsch M, Lim H, Wallwiener M, et al. Mucin 1-specific B cell immune responses and their impact on overall survival in breast cancer patients. Oncoimmunology. 2016;5:e1057387. doi: 10.1080/2162402X.2015.1057387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishimura H, Minato N, Nakano T, Honjo T. Immunological studies on PD-1 deficient mice: implication of PD-1 as a negative regulator for B cell responses. Int Immunol. 1998;10:1563–72. doi: 10.1093/intimm/10.10.1563. [DOI] [PubMed] [Google Scholar]
- 11.Haas KM. Programmed cell death 1 suppresses B-1b cell expansion and long-lived IgG production in response to T cell-independent type 2 antigens. J Immunol. 2011;187:5183–95. doi: 10.4049/jimmunol.1101990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haas KM. B-1 lymphocytes in mice and nonhuman primates. Ann N Y Acad Sci. 2015 doi: 10.1111/nyas.12760. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKay JT, Egan RP, Yammani RD, Chen L, Shin T, Yagita H, et al. PD-1 Suppresses Protective Immunity to Streptococcus pneumoniae through a B Cell-Intrinsic Mechanism. J Immunol. 2015;194:2289–99. doi: 10.4049/jimmunol.1401673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Good-Jacobson KL, Szumilas CG, Chen L, Sharpe AH, Tomayko MM, Shlomchik MJ. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat Immunol. 2010;11:535–42. doi: 10.1038/ni.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sage PT, Francisco LM, Carman CV, Sharpe AH. The receptor PD-1 controls follicular regulatory T cells in the lymph nodes and blood. Nat Immunol. 2013;14:152–61. doi: 10.1038/ni.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. 2013;14:1212–18. doi: 10.1038/ni.2762. [DOI] [PubMed] [Google Scholar]
- 17.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 18.Freeman-Keller M, Weber JS. Anti-programmed death receptor 1 immunotherapy in melanoma: rationale, evidence and clinical potential. Ther Adv Med Oncol. 2015;7:12–21. doi: 10.1177/1758834014551747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. PNAS. 2002;99:12293–7. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol. 2005;17:133–44. doi: 10.1093/intimm/dxh194. [DOI] [PubMed] [Google Scholar]
- 21.Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9:562–7. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- 22.Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64:1140–5. doi: 10.1158/0008-5472.can-03-3259. [DOI] [PubMed] [Google Scholar]
- 23.He YF, Zhang GM, Wang XH, Zhang H, Yuan Y, Li D, et al. Blocking programmed death-1 ligand-PD-1 interactions by local gene therapy results in enhancement of antitumor effect of secondary lymphoid tissue chemokine. J Immunol. 2004;173:4919–28. doi: 10.4049/jimmunol.173.8.4919. [DOI] [PubMed] [Google Scholar]
- 24.Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65:1089–96. [PubMed] [Google Scholar]
- 25.Tsuji T, Osawa T. Carbohydrate structures of bovine submaxillary mucin. Carbohydr Res. 1986;151:391–402. doi: 10.1016/s0008-6215(00)90358-6. [DOI] [PubMed] [Google Scholar]
- 26.Singhal A, Fohn M, Hakomori S. Induction of alpha-N-acetylgalactosamine-O-serine/threonine (Tn) antigen-mediated cellular immune response for active immunotherapy in mice. Cancer Res. 1991;51:1406–11. [PubMed] [Google Scholar]
- 27.Yin Z, Comellas-Aragones M, Chowdhury S, Bentley P, Kaczanowska K, Benmohamed L, et al. Boosting immunity to small tumor-associated carbohydrates with bacteriophage qbeta capsids. ACS chemical biology. 2013;8:1253–62. doi: 10.1021/cb400060x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haas KM, Blevins MW, High KP, Pang B, Swords WE, Yammani RD. Aging Promotes B-1b Cell Responses to Native, but Not Protein-Conjugated, Pneumococcal Polysaccharides: Implications for Vaccine Protection in Older Adults. J Infect Dis. 2014;209:87–97. doi: 10.1093/infdis/jit442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Boyle KP, Coatsworth S, Anthony G, Ramirez M, Greenwald E, Kaleya R, et al. Effects of desialylation of ovine submaxillary gland mucin (OSM) on humoral and cellular immune responses to Tn and sialylated Tn. Cancer Immun. 2006;6:5. [PubMed] [Google Scholar]
- 30.O’Boyle KP, Zamore R, Adluri S, Cohen A, Kemeny N, Welt S, et al. Immunization of colorectal cancer patients with modified ovine submaxillary gland mucin and adjuvants induces IgM and IgG antibodies to sialylated Tn. Cancer Res. 1992;52:5663–67. [PubMed] [Google Scholar]
- 31.Van den Eijnden DH, Evans NA, Codington JF, Reinhold V, Silber C, Jeanloz RW. Chemical structure of epiglycanin, the major glycoprotein of the TA3-Ha ascites cell. The carbohydrate chains. J Biol Chem. 1979;254:12153–9. [PubMed] [Google Scholar]
- 32.Hagmar B, Ryd W. Site dependency of TA3 Ha allotransplantability. Transplantation. 1977;23:93–7. doi: 10.1097/00007890-197701000-00016. [DOI] [PubMed] [Google Scholar]
- 33.Nagy JA, Herzberg KT, Dvorak JM, Dvorak HF. Pathogenesis of malignant ascites formation: initiating events that lead to fluid accumulation. Cancer Res. 1993;53:2631–43. [PubMed] [Google Scholar]
- 34.Nagy JA, Masse EM, Herzberg KT, Meyers MS, Yeo KT, Yeo TK, et al. Pathogenesis of ascites tumor growth: vascular permeability factor, vascular hyperpermeability, and ascites fluid accumulation. Cancer Res. 1995;55:360–8. [PubMed] [Google Scholar]
- 35.Lescar J, Sanchez JF, Audfray A, Coll JL, Breton C, Mitchell EP, et al. Structural basis for recognition of breast and colon cancer epitopes Tn antigen and Forssman disaccharide by Helix pomatia lectin. Glycobiology. 2007;17:1077–83. doi: 10.1093/glycob/cwm077. [DOI] [PubMed] [Google Scholar]
- 36.Holmberg LA, Sandmaier BM. Vaccination with Theratope (STn-KLH) as treatment for breast cancer. Expert Rev Vaccines. 2004;3:655–63. doi: 10.1586/14760584.3.6.655. [DOI] [PubMed] [Google Scholar]
- 37.Buonaguro L, Petrizzo A, Tornesello ML, Buonaguro FM. Translating tumor antigens into cancer vaccines. Clin Vaccine Immunol. 2011;18:23–34. doi: 10.1128/CVI.00286-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heimburg-Molinaro J, Lum M, Vijay G, Jain M, Almogren A, Rittenhouse-Olson K. Cancer vaccines and carbohydrate epitopes. Vaccine. 2011;29:8802–26. doi: 10.1016/j.vaccine.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Danussi C, Coslovi A, Campa C, Mucignat MT, Spessotto P, Uggeri F, et al. A newly generated functional antibody identifies Tn antigen as a novel determinant in the cancer cell-lymphatic endothelium interaction. Glycobiology. 2009;19:1056–67. doi: 10.1093/glycob/cwp085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DiLillo DJ, Yanaba K, Tedder TF. B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: therapeutic B cell depletion enhances B16 melanoma growth in mice. J Immunol. 2010;184:4006–16. doi: 10.4049/jimmunol.0903009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nelson BH. CD20+ B cells: the other tumor-infiltrating lymphocytes. J Immunol. 2010;185:4977–82. doi: 10.4049/jimmunol.1001323. [DOI] [PubMed] [Google Scholar]
- 42.Germain C, Gnjatic S, Dieu-Nosjean MC. Tertiary Lymphoid Structure-Associated B Cells are Key Players in Anti-Tumor Immunity. Front Immunol. 2015;6:67. doi: 10.3389/fimmu.2015.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yammani RD, Haas KM. Primate B-1 cells generate antigen-specific B cell responses to T cell-independent type 2 antigens. J Immunol. 2013;190:3100–08. doi: 10.4049/jimmunol.1203058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70. J Exp Med. 2011;208:67–80. doi: 10.1084/jem.20101499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baumgarth N. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol. 2011;11:34–46. doi: 10.1038/nri2901. [DOI] [PubMed] [Google Scholar]
- 46.Allavena P, Chieppa M, Bianchi G, Solinas G, Fabbri M, Laskarin G, et al. Engagement of the mannose receptor by tumoral mucins activates an immune suppressive phenotype in human tumor-associated macrophages. Clin Dev Immunol. 2010;2010:547179. doi: 10.1155/2010/547179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Monti P, Leone BE, Zerbi A, Balzano G, Cainarca S, Sordi V, et al. Tumor-derived MUC1 mucins interact with differentiating monocytes and induce IL-10highIL-12low regulatory dendritic cell. J Immunol. 2004;172:7341–9. doi: 10.4049/jimmunol.172.12.7341. [DOI] [PubMed] [Google Scholar]
- 48.Fung PY, Longenecker BM. Specific immunosuppressive activity of epiglycanin, a mucin-like glycoprotein secreted by a murine mammary adenocarcinoma (TA3-HA) Cancer Res. 1991;51:1170–6. [PubMed] [Google Scholar]
- 49.Hiltbold EM, Vlad AM, Ciborowski P, Watkins SC, Finn OJ. The mechanism of unresponsiveness to circulating tumor antigen MUC1 is a block in intracellular sorting and processing by dendritic cells. J Immunol. 2000;165:3730–41. doi: 10.4049/jimmunol.165.7.3730. [DOI] [PubMed] [Google Scholar]
- 50.Wu ZQ, Shen Y, Khan AQ, Chu CL, Riese R, Chapman HA, et al. The mechanism underlying T cell help for induction of an antigen-specific in vivo humoral immune response to intact Streptococcus pneumoniae is dependent on the type of antigen. J Immunol. 2002;168:5551–7. doi: 10.4049/jimmunol.168.11.5551. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.