

Abstract
Objective:
To identify clinical features that reliably differentiate individuals with cognitive impairment due to corticobasal degeneration (CBD) and Alzheimer disease (AD).
Methods:
Clinical features were compared between individuals with autopsy-proven CBD (n = 17) and AD (n = 16). All individuals presented with prominent cognitive complaints and were evaluated annually with semistructured interviews, detailed neurologic examinations, and neuropsychological testing.
Results:
Substantial overlap was observed between individuals with dementia due to CBD and AD concerning presenting complaints, median (range) duration of symptoms before assessment (CBD = 3.0 [0–5.0] years, AD = 2.5 [0–8.0] years; p = 0.96), and median (range) baseline dementia severity (Clinical Dementia Rating Sum of Boxes: CBD = 3.5 [0–12.0], AD = 4.25 [0.5–9.0], p = 0.49). Subsequent emergence of asymmetric motor/sensory signs, hyperreflexia, gait abnormalities, parkinsonism, falls, urinary incontinence, and extraocular movement abnormalities identified individuals with CBD, with ≥3 discriminating features detected in 80% of individuals within 3.1 years (95% confidence interval 2.9–3.3) of the initial assessment. Individuals with CBD exhibited accelerated worsening of illness severity and declines in episodic memory, executive functioning, and letter fluency. Semiquantitative pathologic assessment revealed prominent tau pathology within the frontal and parietal lobes of CBD cases. Comorbid AD neuropathologic change was present in 59% (10 of 17) of CBD cases but did not associate with the clinical phenotype, rate of dementia progression, or dementia duration.
Conclusions:
CBD may mimic AD dementia early in its disease course. Interval screening for discriminating clinical features may improve antemortem diagnosis in individuals with CBD and prominent cognitive symptoms.
The clinical presentation of corticobasal degeneration (CBD) is notoriously heterogeneous,1–5 contributing to diagnostic inaccuracy in 44% to 75% of clinically recognized cases.3,4,6–11 A recent review of 210 autopsy-confirmed cases of CBD recognized 5 distinct clinical phenotypes, including corticobasal syndrome (CBS; 37.1%), progressive supranuclear palsy syndrome (23.3%), frontal behavioral-spatial syndrome (13.8%), an Alzheimer disease (AD) dementia–like syndrome (8.1%), and a nonfluent/agrammatic variant of primary progressive aphasia syndrome (4.8%).9 Of these, only the AD dementia–like phenotype was excluded from proposed diagnostic criteria because of concerns that patients presenting with prominent cognitive impairment due to CBD or AD could not be reliably distinguished.9
Although early cognitive impairment was once thought to be rare in CBD,11,12 cognitive deficits are increasingly recognized in affected patients.1,3,5,8,9,13–15 This recognition has broadened the clinical phenotype of CBD but complicated the differentiation of individuals with cognitive complaints. Few studies have systematically considered the factors that distinguish individuals with memory complaints due to CBD from those with AD.4,8,13 The prevalence, time course, and sequence with which disease-defining symptoms and signs emerge remain unclear. To address this need, we considered whether individuals with neuropathologically confirmed CBD presenting with cognitive complaints could be distinguished from individuals with amnestic AD dementia by presenting symptoms, the emergence of abnormalities on neurologic examination, and performance on cognitive testing.
METHODS
Standard protocol approvals, registrations, and patient consents.
Seventeen participants with neuropathologically confirmed CBD were assessed within the Knight Alzheimer Disease Research Center (ADRC) longitudinal study of memory and aging (n = 12) or the affiliated outpatient Memory Diagnostic Center (MDC; Washington University in St. Louis, St. Louis, MO) from 1994 to 2012. All individuals presented with prominent cognitive (not motor) complaints. Sixteen Knight ADRC participants meeting clinical16,17 and neuropathologic criteria for AD,18 without infarction, infection, or other neurodegenerative pathology, were selected for comparison from an autopsy database including 529 potentially eligible controls enrolled from 2005 to 2012. Cases were selected on the basis of age and sex in an attempt to match the demographic features of individuals with CBD. When multiple brains were available from participants of similar age and sex, the most recently assessed participant was selected. All participants (or their delegate) consented to neuropathologic review and to the use of clinical information for research purposes. Study procedures and policies were approved by the Washington University School of Medicine Institutional Review Board.
Clinical and neuropsychological assessment.
Longitudinal symptoms and signs (appendix e-1 at Neurology.org) and results of neuropsychological testing and investigations were obtained from the prospectively assembled Knight ADRC research database or MDC records. Individuals were evaluated at each visit by experienced clinicians using a semistructured interview with a knowledgeable collateral source and the symptomatic individual, as well as a detailed neurologic examination. Two individuals with CBD and one with AD were evaluated only once. Knight ADRC participants underwent neuropsychological evaluations by experienced psychometrists within 2 weeks of the clinical assessment. Standard paper and pencil measures assessed global cognitive function, episodic memory, executive functioning, visuospatial ability, language, and semantic memory, normalized to z scores.19 MDC patients were assessed with a subset of the same measures (table e-1).
A clinical diagnosis of dementia was considered by study clinicians at the conclusion of each assessment, integrating results from the clinical assessment and bedside measures of cognitive function (Mini-Mental State Examination20 and the Short Blessed Test21 results were available in all participants; results from the brief neuropsychological battery were also available for MDC patients). Dementia was diagnosed according to National Institute of Neurological Disorders and Stroke criteria17 and National Institute on Aging–Alzheimer's Association Work Group criteria for participants assessed after 2011.16 Dementia was staged with the global Clinical Dementia Rating (CDR).22 Progression of cognitive impairment was indicated by annualized change in the CDR Sum of Boxes (CDR-SB).23
APOE and microtubule-associated protein tau genotyping.
DNA was extracted from fresh-frozen brain or antemortem blood samples from a subset of participants. APOE and microtubule-associated protein tau (MAPT) genotyping (limited to H1/H2 allele determination) was performed within the Knight ADRC Genetics Core with established techniques.24,25
Neuropathologic assessment.
Neuropathologic analysis was performed within the Knight ADRC Neuropathology Core by study authors (T.S.L. and N.J.C.). The brains were assessed macroscopically at the time of autopsy. Routinely, the right hemibrain was snap-frozen and preserved for biochemical studies with the use of established protocols.26 The left hemibrain was fixed in buffered saline for neuropathologic examination. Briefly, formalin-fixed tissue samples were embedded in paraffin wax, and 7-μm sections were cut. Sections were stained with hematoxylin & eosin and a modified Bielschowsky silver impregnation. Immunohistochemistry was performed with anti-phosphorylated tau (PHF1; a gift from Dr. P. Davies, The Feinstein Institute for Medical Research, Manhasset, NY), anti–phosphorylation-dependent α-synuclein (Wako Chemicals, Richmond, VA), anti–β-amyloid (10D5; Eli Lilly, Indianapolis, IN), and anti–phosphorylation-dependent TAR DNA-binding protein of 43 kDa (TDP-43; Cosmo Bio Co Ltd, Tokyo, Japan) antibodies on sections of the middle frontal gyrus, anterior cingulate gyrus, precentral gyrus, superior temporal gyrus, inferior parietal lobule, amygdala, hippocampus at the level of the lateral geniculate nucleus, caudate nucleus, putamen, basal forebrain nuclei, thalamus, midbrain, pons, medulla oblongata, and cerebellum. Neuropathologic diagnoses were assigned according to established criteria.18,27,28
Semiquantitative assessment of neuronal loss and gliosis, ballooned neurons, tau-positive neurons, astrocytic plaques, coiled bodies, and tau-positive threads was performed in CBD cases. Gray matter and white matter were sampled, and the density of pathologic features was assessed with templates18 and a predefined 4-point rating scale, where a score of 0 corresponds to no lesions; 1 corresponds to 1 to 5 inclusions or features per 1 mm2 (mild); 2 corresponds to ≥6 but <20 inclusions or features per 1 mm2 (moderate); and 3 corresponds to ≥20 inclusions or features per 1 mm2 (severe).
Statistical analyses.
Statistical analyses were conducted with R Statistical Computing Software (r-project.org). Group-wise comparisons were performed with the Mann-Whitney U test and Fisher exact tests for continuous and categorical measures, respectively. The Wilcoxon signed-rank test was used to compare neuronal loss and tau pathology between brain regions. A linear mixed-effects regression model examined the interactive effect of group (CBD vs AD) and time (years from symptom onset) on cognitive performance (with the lme4 package29). Covariates included fixed effects for age at symptomatic onset, sex, education, and CDR-SB. Random effects were specified for individuals and years from symptom onset. All mixed models were fit using restricted maximum likelihood estimation with the lmerTest package (version 2.0.3).30 Kaplan-Meier curves were generated for discriminating clinical findings with the survival package (version 3.0.2).31 Differences between curves were evaluated with the Mantel-Cox (log-rank) test. Statistical significance was established at p < 0.05.
RESULTS
Demographic features and clinically relevant symptoms and signs are summarized in table 1. Despite attempts at age matching, there was a trend toward younger median age at symptom onset in individuals with CBD (64 years, range 50–78 years) vs AD (74 years, range 58–91 years; p = 0.053). Symptoms were present for similar amounts of time (median [range]) before the initial assessment (CBD = 3.0 [0–5.0] years, AD = 2.5 [0–8.0] years; p = 0.96), with similar overall symptomatic disease duration (calculated from symptom onset to death: CBD = 8 [3–12] years, AD = 8 [6–14]; p = 0.23). There was a trend toward a higher frequency of APOE ε4 alleles in individuals with AD (10 of 16, 63%) vs CBD (3 of 12, 25%; p = 0.07). MAPT genotyping was performed in 9 individuals with CBD and 12 with AD. Seven with CBD (78%) and 8 with AD (67%) were homozygous for the MAPT H1 haplotype (p < 0.99); the remainder were MAPT H1/H2 heterozygotes. Hardy-Weinberg equilibrium was maintained. Clinical, genetic, and pathologic information for individuals with CBD is presented in table e-2.
Table 1.
Demographic features and prevalence of clinically relevant symptoms and signs divided by disease
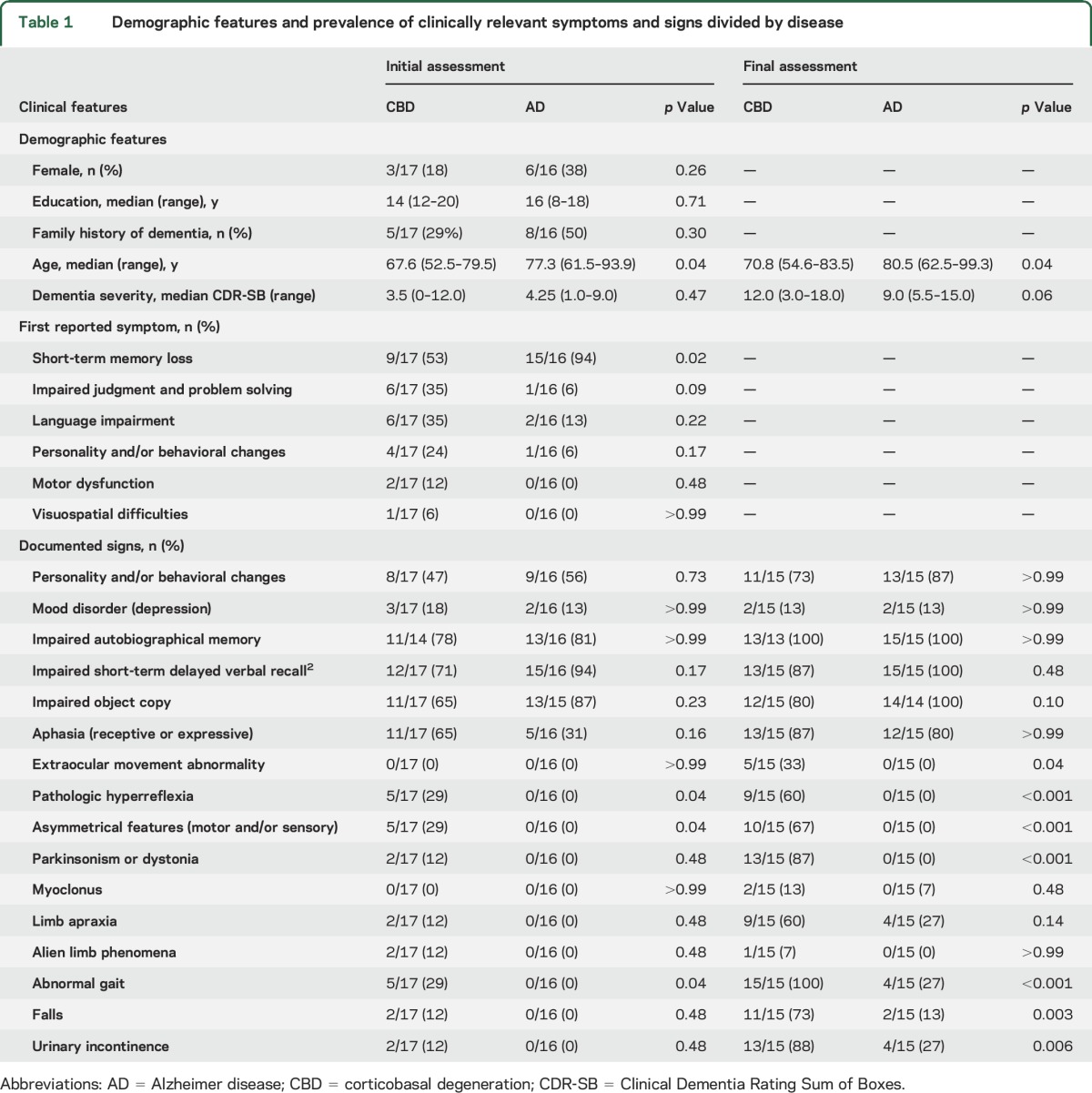
Clinical symptoms, signs, and diagnoses at initial assessment.
All individuals had cognitive complaints as the first clinical manifestation of disease. Short-term memory deficits were the most common initial complaint in both groups but were more frequently reported in individuals with AD (p = 0.02). Asymmetrical features, pathologic hyperreflexia, and abnormal gait patterns were detected in 5 individuals with CBD but in no individuals with AD (p = 0.04). Substantial overlap was observed in other neurologic findings, including features traditionally ascribed to CBS (i.e., parkinsonism or dystonia, myoclonus, limb apraxia, and alien limb phenomena).
The correct clinicopathologic diagnosis was established in 94% (15 of 16) of individuals with AD at presentation. The remaining individual presented with loss of emotional control, which was attributed to behavioral-variant frontotemporal dementia (bvFTD). In contrast, CBD was considered in only 12% (2 of 17) of individuals with corresponding pathology at first presentation (p < 0.001). Both individuals exhibited findings consistent with CBS, including parkinsonism, asymmetric motor and sensory findings, and alien limb phenomena. The remaining CBD cases were diagnosed with AD dementia (41%, 7 of 17), bvFTD (18%, 3 of 17), primary progressive aphasia (18%, 3 of 17), and posterior cortical atrophy (6%, 1 of 17). One individual with a history of optic neuritis presented with increasing falls, right-sided pyramidal signs, and multiple T2-hyperintense periventricular lesions on neuroimaging (determined neuropathologically to represent sequelae of small vessel disease) and was diagnosed with multiple sclerosis (6%, 1 of 17).
No differences were observed on measures estimating memory impairment (autobiographic or delayed verbal recall) or visuomotor skills (object copy; table 1). On more detailed neuropsychological testing, the AD group exhibited worse performance on story recall (Logical Memory; p = 0.004).
Longitudinal clinical symptoms, signs, and final diagnoses.
Clinical diagnoses were unchanged in participants with AD, with an accurate diagnosis established in 15 of 16 individuals (94%) with pathologically confirmed AD. Clinicopathologic concordance was established in 6 of 17 individuals (35%) with CBD (p < 0.001), with the majority of individuals given an antemortem diagnosis of AD dementia with atypical features (35%, 6 of 17), including extrapyramidal signs (n = 4), prominent aphasia (i.e., logopenic aphasia, n = 1), and visuospatial dysfunction (i.e., posterior cortical atrophy, n = 1). Others were presumed to have bvFTD (24%, 4 of 17) or multiple sclerosis (6%, 1 of 17). No clinical or pathologic differences were noted between individuals correctly diagnosed with CBD during life and those with cognitive impairment attributed to alternative neurodegenerative dementing illnesses (table e-3).
Discriminating CBD and AD on the basis of clinical features.
Asymmetric motor or sensory findings, pathologically brisk reflexes, and abnormal gait changes on neurologic examination were more common in individuals with CBD across follow-up (figure 1). By the final evaluation, parkinsonism or dystonia (p < 0.001), falls (p = 0.003), urinary incontinence (p = 0.006), and extraocular movement abnormalities (p = 0.04) emerged as additional discriminating clinical features (figure 1, A–G). The detection of any neurologic sign at the first assessment was associated with an increased probability of CBD (p = 0.02). Three or more discriminating features were present in the majority of individuals with CBD within 2.1 years (95% confidence interval [CI] 1.7–2.6) of initial assessment and 5.2 years (95% CI 3.5–7.0) from symptom onset and within 80% of individuals at 3.1 years (95% CI 2.9–3.3) from the initial assessment and 6.2 years (95% CI 6.0–6.4) from symptom onset (figure 1H). All individuals with CBD exhibited ≥3 features within 6.8 years from the initial assessment and 8.8 years from symptom onset. Two or fewer features were detected in 7 of 16 individuals (44%) with AD before death and were limited to gait abnormalities, falls, and urinary incontinence.
Figure 1. Onset of discriminating clinical features in individuals with CBD.

(A-G) Kaplan-Meier curves depicting the emergence of discriminating clinical features in individuals with corticobasal degeneration (CBD; red) and Alzheimer disease (blue). The time over which differentiating clinical features emerged (gray shading) was determined by generating Kaplan-Meier curves for sequential 2-year intervals (0–2, 0–4, 0–6 years from first clinical assessment). Differences between curves were evaluated with the Mantel-Cox (log-rank) test. p Values reflect differences between curves across the entire follow-up period. Dashed lines represent 95% confidence intervals. (H) Kaplan-Meier curve depicting the number of years from first clinical assessment to detection of ≥3 discriminating clinical features (black) or ≥1 CBD-specific clinical feature (gray). Short- and long-dashed lines represent time to detection in 50% (median time) and 80% of individuals with CBD, respectively.
Asymmetric motor/sensory features, pathologic hyperreflexia, parkinsonism/dystonia, or extraocular movement abnormalities were exclusively detected in individuals with CBD. Right-lateralizing signs were observed in 7 individuals with CBD and left-lateralizing signs in 5, with no clear effect on clinical manifestations (not shown). One or more CBD-specific features were detected in the majority of individuals with CBD within 2.0 years (95% CI 0.92–3.0) of initial assessment and 5.0 years (95% CI 2.8–7.1) from symptom onset and within 80% of individuals at 2.6 years (95% CI 2.4–2.8) from the initial assessment and 5.6 years (95% CI 5.4–5.8) from symptom onset. All individuals with CBD exhibited ≥1 CBD-specific feature within 6.8 years from the initial assessment and 8.8 years from symptom onset.
Across visits, individuals with CBD demonstrated greater rates of worsening in CDR-SB (p = 0.045), story recall (Logical Memory; p = 0.01), and letter fluency (p = 0.004; figure 2).
Figure 2. Estimated mean slopes and 95% confidence intervals overlaid on raw individual cognitive scores for participants with CBD (in red) and AD (in blue).

The x-axis represents time (in years) from symptom onset. The y-axis represents the change in cognitive performance (Clinical Dementia Rating [CDR] Sum of Boxes) from symptom onset. p Values represent the group-by-time interaction term. Patients with CBD had faster decline in clinical symptoms, episodic memory, and letter fluency but not in other areas of cognitive performance. AD = Alzheimer disease; CBD = corticobasal degeneration; WAIS = Wechsler Adult Intelligence Scale; WMS-R = Wechsler Memory Scale–Revised.
Neuropathologic findings.
Semiquantitative analysis of tau pathology was performed in individuals with CBD (table e-4). Neuronal loss and gliosis were greatest in the middle frontal gyrus, anterior cingulate gyrus, precentral gyrus, inferior parietal lobule, and substantia nigra. Ballooned neurons were observed in all 17 cases (figure e-1), most frequently in the anterior cingulate gyrus. The density of tau-immunoreactive tangles, coiled bodies, and tau-positive threads did not differ between the gray matter of different cortical regions. Tau-immunoreactive astrocytic plaques were frequently identified in the middle frontal gyrus, anterior cingulate gyrus, and inferior parietal lobule.
Comorbid AD neuropathologic change (ADNC) was identified in 10 of 17 cases (59%) with CBD (table 2), with 2 cases (12%) meeting National Institute on Aging–Alzheimer's Association criteria for high ADNC and 2 cases (12%) meeting criteria for intermediate ADNC.18 The remaining 6 cases (35%) had low ADNC. AD copathology had no effect on the median (range) symptomatic duration (no/low ADNC = 8 [3–12] years; intermediate/high ADNC = 7.5 [5–12] years; p = 0.39), dementia severity at the final clinical assessment (CDR-SB: no/low ADNC = 12.0 [3.5–18.0]; intermediate/high ADNC = 12.5 [3.0–18.0]; p = 0.14), or rate of change (∆CDR-SB per year: no/low ADNC = 2.0 [0.8–4.7]; intermediate/high ADNC = 2.5 [0.6–3.2]; p = 0.3). Furthermore, no individual incorrectly diagnosed with AD dementia exhibited greater than low ADNC at autopsy. Together, these findings suggest that the presence of ADNC had minimal effect on the clinical phenotype. Two cases (12%) met neuropathologic criteria for Lewy body disease.28 No cases met diagnostic criteria for frontotemporal lobar degeneration with TDP-43,27 although comorbid TDP-43 proteinopathy was present in the medial temporal lobes of 5 cases. Three cases had comorbid hippocampal sclerosis.
Table 2.
Other pathology in individuals with CBD and AD
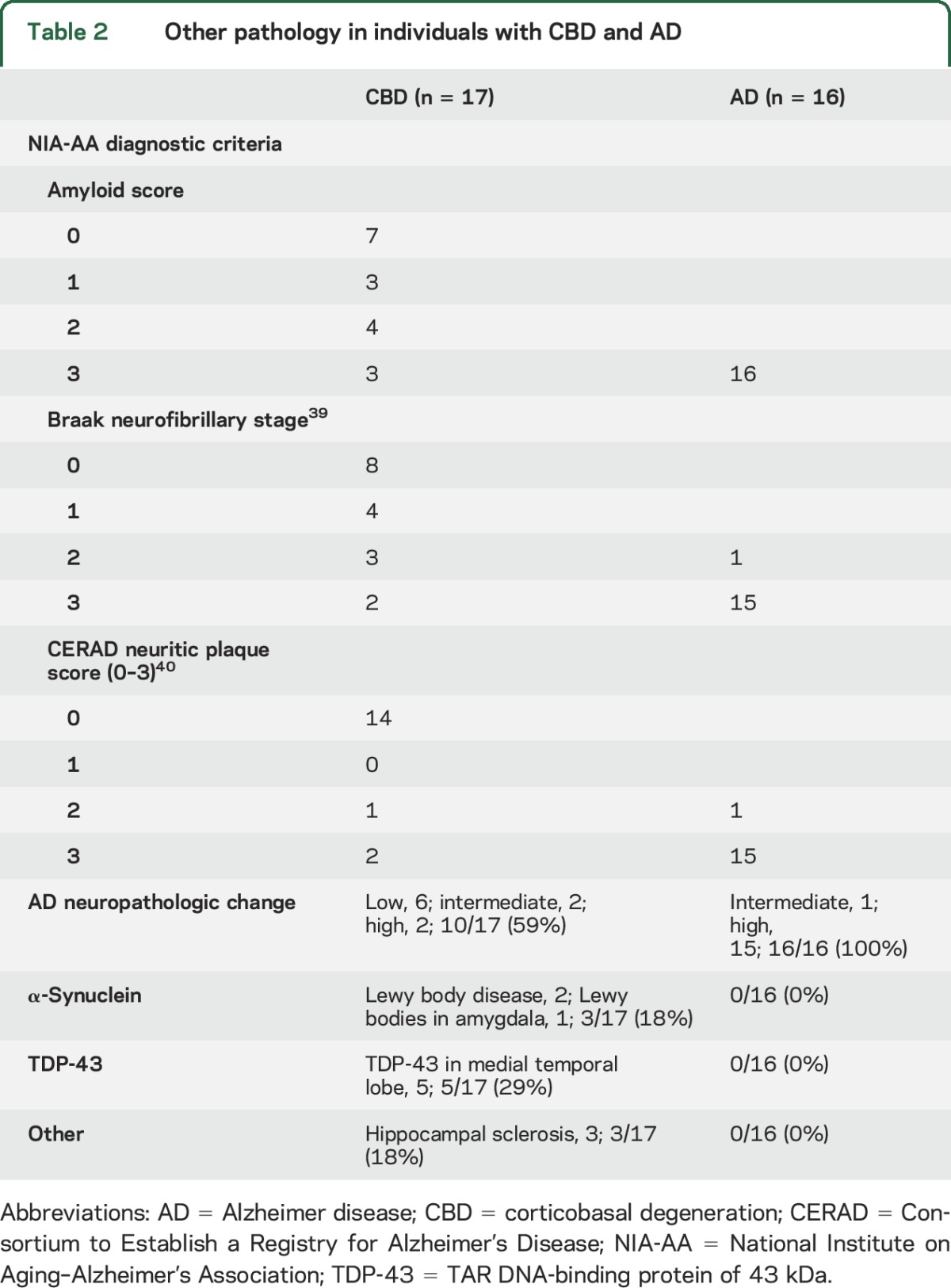
All individuals with AD dementia met National Institute on Aging–Alzheimer's Association criteria for ADNC.18
DISCUSSION
Individuals who presented with cognitive complaints later attributed to CBD were rarely suspected to have CBD at initial assessment. This observation highlights the challenges associated with differentiating individuals with cognitive impairment attributed to CBD from AD.3,4,7–10 In retrospect, the subsequent appearance of ≥3 discriminating clinical features on the neurologic examination (table 3), but not presenting complaints, reliably distinguished 80% of individuals with CBD from those with AD within 3.1 years of the initial assessment, with CBD-specific signs detected slightly earlier. Compared with individuals with AD, those with CBD had relative preservation of memory early in the disease course (despite subjective memory complaints) but an accelerated rate of decline on measures of story recall and letter fluency. Recognition of these features may help to distinguish patients with cognitive complaints due to CBD from those with complaints attributable to AD.
Table 3.
Clinical discriminating features between individuals with cognitive complaints due to CBD and AD divided by time of appearance

Memory loss was the most common presenting complaint in individuals with CBD in our sample. The prevalence of this symptom was surprising given the relatively minor impairments on tests of episodic memory and relative paucity of tauopathy in the mesial temporal lobes, a finding observed in other studies.4,32,33 The distribution of tau pathology was similar to that reported previously in patients with CBS,2,4,8,13,34–36 suggesting that the predominance of early memory complaints may reflect deficits in strategies for encoding and retrieval, owing to disruption of structures more frequently affected by CBD, including frontotemporal subcortical areas.15
Previous case series reported a low frequency of detection of classic CBS signs (i.e., asymmetric motor/sensory features, parkinsonism, and/or dystonia) at onset in patients with cognitive presentations of CBD.4,8,13 We report a similar frequency but note that extrapyramidal findings were detected in individuals with cognitive impairment due to CBD later in the disease, with a prevalence approaching that observed in CBS.9 The emergence of clinical findings ascribed to CBD in our population may reflect variability in the spatial distribution of pathology in patients presenting primarily with cognitive complaints, with early involvement of frontoparietal networks contributing to cognitive impairment3,4 and later involvement of surrounding cortical and subcortical areas preceding the emergence of pyramidal/extrapyramidal signs.
Individuals presenting with cognitive complaints due to CBD were commonly misdiagnosed before death (11 of 17, 65%), consistent with documented experience in movement disorder clinics6 and retrospective reviews of autopsy-confirmed CBD.3,4,8–10 Such findings have been interpreted to suggest that individuals with an AD dementia–like phenotype should be excluded from CBD diagnostic guidelines.9 As this study shows, it may be possible to detect individuals with cognitive complaints attributable to CBD by longitudinally screening for the emergence of discriminating clinical features. The detection of ≥1 abnormal features should prompt further diagnostic testing, including neuropsychological assessment, looking for patterns that may support the clinical diagnosis. Clinical suspicion of CBD should increase with the number of discriminating clinical features, with the highest probability of CBD assigned to individuals with ≥3 discriminating features and lower likelihood assigned to those in whom no discriminating features are detected within 3.1 years of the first clinical assessment.
Current CBD biomarkers are limited to detection of nonspecific genetic risk factors (i.e., the MAPT H1 haplotype). In contrast, AD biomarkers are increasingly used in research and clinical settings to determine individual risk of AD pathology and support clinical diagnoses.37 It may be challenging to use AD biomarkers to differentiate individuals with cognitive impairment due to CBD and AD because AD copathology was present in the majority of individuals with CBD (10 of 17, 59%) in this cohort (previously reported in 13%–27%2,9). Given the high prevalence of AD copathology, some individuals with CBD may test positive for AD-specific biomarkers, leading to misplaced confidence in the clinical diagnosis. This possibility has important implications for clinical trials that include AD biomarkers in the inclusion criteria: recognizing that a subset of biomarker-positive participants diagnosed with AD dementia may yet have an alternative cause of cognitive impairment. This realization emphasizes the value of routine surveillance for clinical features that discriminate between individuals with cognitive impairment due to CBD and AD and the need to define additional biomarkers that may distinguish CBD and AD.38
This study is subject to several limitations, including the limited sample size. With expanded recruitment of individuals with cognitive complaints and CBD, additional discriminating features may be detected, including clinical features approaching trend-level significance in our cohort. In addition, because recruitment was limited to individuals presenting with cognitive complaints, our findings may not be helpful in discriminating cognitive complaints attributable to CBD and other (non-AD) neurodegenerative dementing illnesses or differentiating between CBD and AD presenting as CBS (discussed elsewhere2,9). Finally, because sampling of hemibrains was completed before histopathologic review, no comment can be made concerning lateralization of pathology in our population. Previous studies have suggested relationships between laterality of pathology and cognitive vs motor phenotypes.15 Putative clinical-pathological correlations should be further evaluated through future well-designed prospective studies, recruiting individuals with cognitive and sensorimotor complaints in isolation and in combination.
Supplementary Material
ACKNOWLEDGMENT
The authors remain grateful for the commitment of participants, patients, and their families to advancing research and clinical care. The Knight ADRC Genetics Core provided information on APOE allele status.
GLOSSARY
- AD
Alzheimer disease
- ADNC
Alzheimer disease neuropathologic change
- ADRC
Alzheimer Disease Research Center
- bvFTD
behavioral-variant frontotemporal dementia
- CBD
corticobasal degeneration
- CBS
corticobasal syndrome
- CDR
Clinical Dementia Rating
- CI
confidence interval
- MAPT
microtubule-associated protein tau
- MDC
Memory Diagnostic Center
- SB
Sum of Boxes
- TDP-43
TAR DNA-binding protein of 43 kDa
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
G.S. Day participated in acquisition and interpretation of clinical data, statistical analysis, and drafting, revision, and finalization of the manuscript. T.S. Lim participated in study design, acquisition and interpretation of clinical and neuropathologic data, and drafting of the manuscript. J. Hassenstab participated in interpretation of clinical data, statistical analysis, and revision and finalization of the manuscript. A.M. Goate, E.A. Grant, and C.M. Roe participated in acquisition of clinical data and revision and finalization of the manuscript. N.J. Cairns participated in study design, acquisition and interpretation of neuropathologic data, and revision and finalization of the manuscript. Dr. Cairns had full access to all of the neuropathologic data and takes responsibility for the integrity of the data and the accuracy of the conclusions. J.C. Morris participated in study design, acquisition and interpretation of clinical data, and revision and finalization of the manuscript. Dr. Morris had full access to all of the clinical data and takes responsibility for the integrity of the data and the accuracy of the conclusions.
STUDY FUNDING
This project was supported by National Institute on Aging grants P50AG05681, P01AG03991, and P01AG26276 (J.C.M.). G.S.D. was supported by the Eugene M. Johnson, Jr. Weston Brain Institute Postdoctoral Fellowship, the Neiss-Gain Family Endowment for Alzheimer Disease Research, and an American Brain Foundation Clinical Research Training Fellowship.
DISCLOSURE
G. Day is involved in research supported by an in-kind gift of radiopharmaceuticals from Avid Radiopharmaceuticals and holds stocks (>$10,000) in ANI Pharmaceuticals (a generic pharmaceutical company). T. Lim and J. Hassenstab report no disclosures relevant to the manuscript. A. Goate is a member of the scientific advisory board of Denali Therapeutics and Cognition Therapeutics. She received research funding and honoraria from Genentech, Pfizer, and AstraZeneca during the course of this study. E. Grant, C. Roe, and N. Cairns report no disclosures relevant to the manuscript. J. Morris is currently participating in clinical trials of antidementia drugs (A4 Trial: the Anti-Amyloid Treatment in Asymptomatic Alzheimer's Disease), funded by the National Institute on Aging, Eli Lilly and Co, and several philanthropic organizations. Dr. Morris has served as a consultant for Lilly USA and Takeda Pharmaceuticals. He receives research support from Eli Lilly/Avid Radiopharmaceuticals and is funded by NIH grants P50AG005681, P01AG003991, P01AG026276, and UF01AG032438. Neither Dr. Morris nor his family owns stock or has equity interest (outside of mutual funds or other externally directed accounts) in any pharmaceutical or biotechnology company. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Kouri N, Murray ME, Hassan A, et al. Neuropathological features of corticobasal degeneration presenting as corticobasal syndrome or Richardson syndrome. Brain 2011;134:3264–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee SE, Rabinovici GD, Mayo MC, et al. Clinicopathological correlations in corticobasal degeneration. Ann Neurol 2011;70:327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ling H, O'Sullivan SS, Holton JL, et al. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain 2010;133:2045–2057. [DOI] [PubMed] [Google Scholar]
- 4.Murray R, Neumann M, Forman MS, et al. Cognitive and motor assessment in autopsy-proven corticobasal degeneration. Neurology 2007;68:1274–1283. [DOI] [PubMed] [Google Scholar]
- 5.Mathew R, Bak TH, Hodges JR. Diagnostic criteria for corticobasal syndrome: a comparative study. J Neurol Neurosurg Psychiatry 2012;83:405–410. [DOI] [PubMed] [Google Scholar]
- 6.Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain 2002;125:861–870. [DOI] [PubMed] [Google Scholar]
- 7.Boeve BF, Maraganore DM, Parisi JE, et al. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology 1999;53:795–800. [DOI] [PubMed] [Google Scholar]
- 8.Grimes DA, Lang AE, Bergeron CB. Dementia as the most common presentation of cortical-basal ganglionic degeneration. Neurology 1999;53:1969–1974. [DOI] [PubMed] [Google Scholar]
- 9.Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013;80:496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Litvan I, Agid Y, Goetz C, et al. Accuracy of the clinical diagnosis of corticobasal degeneration: a clinicopathologic study. Neurology 1997;48:119–125. [DOI] [PubMed] [Google Scholar]
- 11.Rinne JO, Lee MS, Thompson PD, Marsden CD. Corticobasal degeneration: a clinical study of 36 cases. Brain 1994;117:1183–1196. [DOI] [PubMed] [Google Scholar]
- 12.Riley DE, Lang AE, Lewis A, et al. Cortical-basal ganglionic degeneration. Neurology 1990;40:1203–1212. [DOI] [PubMed] [Google Scholar]
- 13.Bergeron C, Pollanen MS, Weyer L, Black SE, Lang AE. Unusual clinical presentations of cortical-basal ganglionic degeneration. Ann Neurol 1996;40:893–900. [DOI] [PubMed] [Google Scholar]
- 14.Graham NL, Bak TH, Hodges JR. Corticobasal degeneration as a cognitive disorder. Mov Disord 2003;18:1224–1232. [DOI] [PubMed] [Google Scholar]
- 15.Kertesz A, McMonagle P. Behavior and cognition in corticobasal degeneration and progressive supranuclear palsy. J Neurol Sci 2010;289:138–143. [DOI] [PubMed] [Google Scholar]
- 16.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 18.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012;123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson DK, Storandt M, Morris JC, Galvin JE. Longitudinal study of the transition from healthy aging to Alzheimer disease. Arch Neurol 2009;66:1254–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Folstein MF, Folstein SE, McHugh PR. “Mini-Mental State”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 21.Katzman R, Brown T, Fuld P, Peck A, Schechter R, Schimmel H. Validation of a short Orientation-Memory-Concentration Test of cognitive impairment. Am J Psychiatry 1983;140:734–739. [DOI] [PubMed] [Google Scholar]
- 22.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 23.Berg L, Miller JP, Storandt M, et al. Mild senile dementia of the Alzheimer type: 2: longitudinal assessment. Ann Neurol 1988;23:477–484. [DOI] [PubMed] [Google Scholar]
- 24.Kauwe JS, Cruchaga C, Mayo K, et al. Variation in MAPT is associated with cerebrospinal fluid tau levels in the presence of amyloid-beta deposition. Proc Natl Acad Sci USA 2008;105:8050–8054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pastor P, Roe CM, Villegas A, et al. Apolipoprotein Eepsilon4 modifies Alzheimer's disease onset in an E280A PS1 kindred. Ann Neurol 2003;54:163–169. [DOI] [PubMed] [Google Scholar]
- 26.Cairns NJ, Perrin RJ, Franklin EE, et al. Neuropathologic assessment of participants in two multi-center longitudinal observational studies: the Alzheimer Disease Neuroimaging Initiative (ADNI) and the Dominantly Inherited Alzheimer Network (DIAN). Neuropathology 2015;35:390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cairns NJ, Bigio EH, Mackenzie IR, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 2007;114:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005;65:1863–1872. [DOI] [PubMed] [Google Scholar]
- 29.Bates D, Maechler M, Walker S, et al. lme4: linear mixed-effects models using “Eigen” and S4. R Package Version 1.1–12; 2016. Available at: cran.r-project.org/web/packages/lme4/index.html. Accessed November 4, 2016. [Google Scholar]
- 30.Kuznetsova A, Christensen R, Brockhoff P. lmerTest: tests for random and fixed effects for linear mixed effect models (lmer objects of lme4 package). R Package Version 1.0–2; 2012. [Google Scholar]
- 31.Waring SC, Doody RS, Pavlik VN, Massman PJ, Chan W. Survival among patients with dementia from a large multi-ethnic population. Alzheimer Dis Assoc Disord 2005;19:178–183. [DOI] [PubMed] [Google Scholar]
- 32.Massman PJ, Kreiter KT, Jankovic J, Doody RS. Neuropsychological functioning in cortical-basal ganglionic degeneration: differentiation from Alzheimer's disease. Neurology 1996;46:720–726. [DOI] [PubMed] [Google Scholar]
- 33.Pillon B, Blin J, Vidailhet M, et al. The neuropsychological pattern of corticobasal degeneration: comparison with progressive supranuclear palsy and Alzheimer's disease. Neurology 1995;45:1477–1483. [DOI] [PubMed] [Google Scholar]
- 34.Forman MS, Zhukareva V, Bergeron C, et al. Signature tau neuropathology in gray and white matter of corticobasal degeneration. Am J Pathol 2002;160:2045–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Armstrong RA, Cairns NJ, Lantos PL. A quantitative study of the pathological lesions in the neocortex and hippocampus of twelve patients with corticobasal degeneration. Exp Neurol 2000;163:348–356. [DOI] [PubMed] [Google Scholar]
- 36.Armstrong RA, Cairns NJ. Clustering and spatial correlations of the neuronal cytoplasmic inclusions, astrocytic plaques and ballooned neurons in corticobasal degeneration. J Neural Transm 2009;116:1103–1110. [DOI] [PubMed] [Google Scholar]
- 37.Jack CR Jr, Bennett DA, Blennow K, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016;87:539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kikuchi A, Okamura N, Hasegawa T, et al. In vivo visualization of tau deposits in corticobasal syndrome by 18F-THK5351 PET. Neurology 2016;87:2309–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Braak H, Braak E. Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol Aging 1995;16:271–278; discussion 278–284. [DOI] [PubMed] [Google Scholar]
- 40.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD), part II: standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991;41:479–486. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.