

Abstract
Objective:
Based on the observation that a subset of patients originally diagnosed with pure autonomic failure (PAF) eventually develops extrapyramidal or cerebellar involvement consistent with multiple system atrophy (MSA), Parkinson disease (PD), or dementia with Lewy bodies (DLB), we aimed to identify predictors of progression of PAF to more sinister synucleinopathies.
Methods:
In this retrospective cohort study, we reviewed patients seen at Mayo Clinic Rochester by autonomic specialists between 2001 and 2011 and during initial evaluation diagnosed with orthostatic hypotension consistent with PAF (possible PAF). In order to assess for the presence or absence of progression, we identified patients with 3 years or more of in-person follow-up (stable PAF) or documented progression to another synucleinopathy (converters). To identify predictors of conversion, we assessed odds of conversion based on clinical, autonomic, and laboratory variables.
Results:
Among 318 patients fulfilling criteria for possible PAF, we identified 41 with stable PAF and 37 (12%) converters. Of those who evolved, 22 developed MSA, 11 developed PD/DLB, and 4 remained indeterminate. Several variables were identified to predict conversion to MSA: (1) mild degree of cardiovagal impairment, (2) preganglionic pattern of sweat loss, (3) severe bladder dysfunction, (4) supine norepinephrine >100 pg/mL, and (5) subtle motor signs at first presentation. Separate variables were found to predict conversion to PD/DLB. Composite conversion scores were generated based on individual predictors.
Conclusions:
Over 10% of patients originally diagnosed with PAF eventually evolve to develop CNS involvement, most commonly MSA. A combination of variables allows for prediction of conversion.
Multiple system atrophy (MSA) is a sporadic, progressive neurodegenerative disorder characterized clinically by autonomic failure along with motor dysfunction that may primarily consist of parkinsonism or cerebellar dysfunction.1,2 Glial cytoplasmic α-synuclein inclusions and associated neuronal loss in selective regions represent the pathologic hallmark of this invariably fatal disease and characterize it as one of the synucleinopathies, which also include Parkinson disease (PD) and dementia with Lewy bodies (DLB).3–5
Pure autonomic failure (PAF), originally described by Bradbury and Eggleston,6 is a syndrome characterized by orthostatic hypotension, fixed pulse rate, anhidrosis, erectile dysfunction, and constipation without motor features. The pathology of PAF is characterized by degeneration of peripheral autonomic neurons along with α-synuclein–positive, Lewy body–like inclusions in sympathetic ganglia and widespread α-synuclein deposits in autonomic neurons, establishing PAF as a restricted, nonmotor form of the synucleinopathies.7,8 Nevertheless, involvement of central structures is suggested by the presence of REM sleep disorder and anosmia, as well as by the spinal fluid catechol profile suggesting involvement of central noradrenergic and dopaminergic neurons.9–11 It therefore comes as no surprise that progression from a PAF phenotype to MSA, PD, and DLB has been described, or in other words that patients with MSA, PD, and DLB can present with isolated autonomic failure earlier in the disease course.12,13 This presentation of synucleinopathies with delayed motor involvement can make a distinction from stable PAF difficult, yet presents a unique opportunity to diagnose patients at a very early, premotor stage, if certain characteristics could predict later motor involvement.
METHODS
Standard protocol approvals, registrations, and patient consents.
This study was reviewed and approved by Mayo Clinic's institutional review board.
Patients.
We retrospectively reviewed the records of all patients seen at Mayo Clinic Rochester by an autonomic specialist between July 1, 2001, and July 1, 2011, who were between 18 and 80 years old and demonstrated to have orthostatic hypotension (OH) on standardized autonomic reflex testing, defined as drop in blood pressure (BP) of ≥30 mm Hg systolic or ≥15 mm Hg diastolic during a 5-minute head-up tilt.14
Patients were excluded from further analysis if any of the following criteria were fulfilled:
Autonomic testing not consistent with neurogenic OH (normal BP recovery time on Valsalva maneuver).
Clinical evidence of peripheral neuropathy, Adie pupils, or prominent upper gastrointestinal symptoms.
Treatment response to immunomodulatory therapy.
Clinical evidence of central neurodegeneration (i.e., parkinsonism, cerebellar ataxia, or dementia).
Abnormal paraneoplastic antibody panel.
Antibody-positive Sjögren syndrome.
Medical conditions or medications that could interfere with results of autonomic testing, including heart disease, neck radiation, and chemotherapy.
Expert diagnosis not consistent with PAF.
Patients fulfilling inclusion and exclusion criteria were considered possible PAF. In order to assess for the presence of absence of progression to motor involvement, we identified patients who (1) had 3 years or more of neurologic in-person follow-up without evidence of parkinsonism or cerebellar ataxia (stable PAF) or (2) had documented progression to motor involvement (converters). Categorization of converters to MSA, PD, or DLB was based on established clinical consensus criteria (MSA) or clinical evaluation by a movement disorders specialist (PD, DLB).
Detailed clinical, autonomic, and laboratory data were extracted for all patients fulfilling the above described criteria.
Clinical variables.
Medical record data were reviewed, extracted, and coded for all patients. Those variables utilized in further analysis included the degree of bladder dysfunction (absent or limited to frequency/urgency vs severe dysfunction such as urinary incontinence, urinary retention, or need for bladder catheterization), the presence of REM sleep behavior disorder (RBD), obstructive sleep apnea, and subtle/questionable signs of parkinsonism or cerebellar dysfunction (too mild for the clinician to definitively discern from a normal variant or normal aging).
Autonomic variables.
Autonomic testing at Mayo Clinic Rochester is highly standardized and composed of (1) the autonomic reflex screen (ARS), a test battery to assess postganglionic sudomotor, cardiovagal, and cardiovascular adrenergic function; and (2) the thermoregulatory sweat test (TST), which quantitatively assesses preganglionic and postganglionic sudomotor pathways over the entire anterior body surface. Patients are routinely instructed to hold medications that may interfere with autonomic function at least 48 hours prior to autonomic function testing.
Autonomic reflex screen.
Postganglionic sympathetic sudomotor function was assessed using a quantitative sudomotor axon reflex test (QSART) utilizing acetylcholine iontophoresis at 4 standardized sites.15 Cardiovagal function was assessed using heart rate responses to deep breathing and the Valsalva maneuver.16 Cardiovascular adrenergic function was evaluated by assessing BP responses to the Valsalva maneuver and passive head-up tilt.16 In order to quantify and compare sudomotor, cardiovagal, and cardiovascular adrenergic function independent of age and sex, a Composite Autonomic Severity Score (CASS) was derived. The CASS score is a validated instrument to quantify the overall severity and distribution of autonomic failure utilizing findings on the ARS. It allocates 0–3 points for sudomotor, 0–3 points for cardiovagal, and 0–4 points for cardiovascular adrenergic impairment, resulting in a total score ranging from 0 (no impairment) to 10 (severe autonomic failure).14,16
Thermoregulatory sweat test.
The TST provides a quantitative evaluation of thermoregulatory sweat function.17 It was performed under standardized conditions in a cabinet with a hot and humid environment using indicator powder that changes color when exposed to sweat. Digital photographs of the sweat distribution were then used to quantitate the percentage of anhidrosis (0%–100%) of the anterior body surface.18 A comparison of sudomotor findings on TST with those on QSART allowed for determination of the presence of a preganglionic or central sudomotor lesion (thermoregulatory sweat loss at a site with preserved axon reflex sweating).
Variables were extracted from autonomic testing at the time of the original diagnosis of PAF. Those included total CASS, CASS subscores, percentage of TST anhidrosis, and the presence of a preganglionic pattern of sweat loss. All autonomic variables were derived by a single investigator blinded to the final diagnostic classification.
Laboratory and imaging variables.
Laboratory variables extracted from the record included supine and orthostatic fractionated catecholamines at the time of original PAF diagnosis. Supine and orthostatic norepinephrine (NE; pg/mL) was utilized for further analysis. If performed at the time of PAF diagnosis, MRI (brain) reports and stored MRI were carefully reviewed for signs consistent with or suggestive of CNS involvement. Those included characteristic T2 signal abnormalities such as posterior putaminal hypointensity, hyperintense lateral putaminal rim, or cruciform pontine hyperintensity, as well as atrophy of putamen, pons, cerebellar peduncles, or cerebellum.2,19
Statistical analysis.
Descriptive statistics were used to describe demographic and parameter characteristics, including mean, SD, median, interquartile range (IQR), frequencies, and percentages as appropriate. Categorical variables were compared using χ2 test or Fisher exact test as appropriate. Continuous variables were compared using Student t test or Wilcoxon rank-sum test as appropriate. To identify the strength of predictors of conversion, we assessed the odds of conversion (OR) for those variables with significant group differences (stable PAF vs MSA and stable PAF vs PD/DLB). Statistical analyses were performed using SAS version 9.3 with significance set at p < 0.05.
RESULTS
Of 1,427 patients fulfilling diagnostic criteria for OH and seen by an autonomic expert in the specified timeframe, 318 fulfilled criteria for possible PAF. Of those, we identified 41 patients with stable PAF and 37 (12%) converters (figure 1). The majority of converters developed MSA (n = 22, 59%), while 11 patients (30%) developed PD or DLB. Figure 2 shows pathologic confirmation in a case of MSA and DLB conversion, respectively. The diagnosis of 4 patients who developed clear motor involvement remained indeterminate at the time of last follow-up.
Figure 1. Study flowchart.

DLB = dementia with Lewy bodies; MSA = multiple system atrophy; PAF = pure autonomic failure; PD = Parkinson disease.
Figure 2. Pathologic confirmation of multiple system atrophy (MSA) and dementia with Lewy bodies (DLB) conversion.

(A–E) Central Lewy body pathology in an 81-year-old woman who converted from pure autonomic failure (PAF) to DLB (postmortem delay [PMD] 12 hours). (A) Hematoxylin & eosin (H&E) of midbrain (arrows; Lewy bodies). (B) Midbrain, α‐synuclein immunostain in the substantia nigra. (C) H&E of anterior cingulate. (D) Anterior cingulate, α‐synuclein immunostain. (E) Basal ganglia, α‐synuclein immunostain. (F) MSA pathology (glial cytoplasmic inclusions) in a 53-year-old man who converted to MSA (PMD 10 hours); basal ganglia, α‐synuclein immunostain. Bar = 50 μm.
The median time to conversion to MSA was 2.4 (IQR 1.9–3.3) years, demonstrating that the conversion of the majority of patients ultimately meeting consensus criteria for MSA occurs during the first 3 years after the diagnosis of possible PAF. On the other hand, the median time to conversion to PD or DLB was notably longer: 3.9 (IQR 1.9–8.4) years. Those with stable PAF had a median time of in-person follow-up of 6.6 (IQR 4.2–10.6) years.
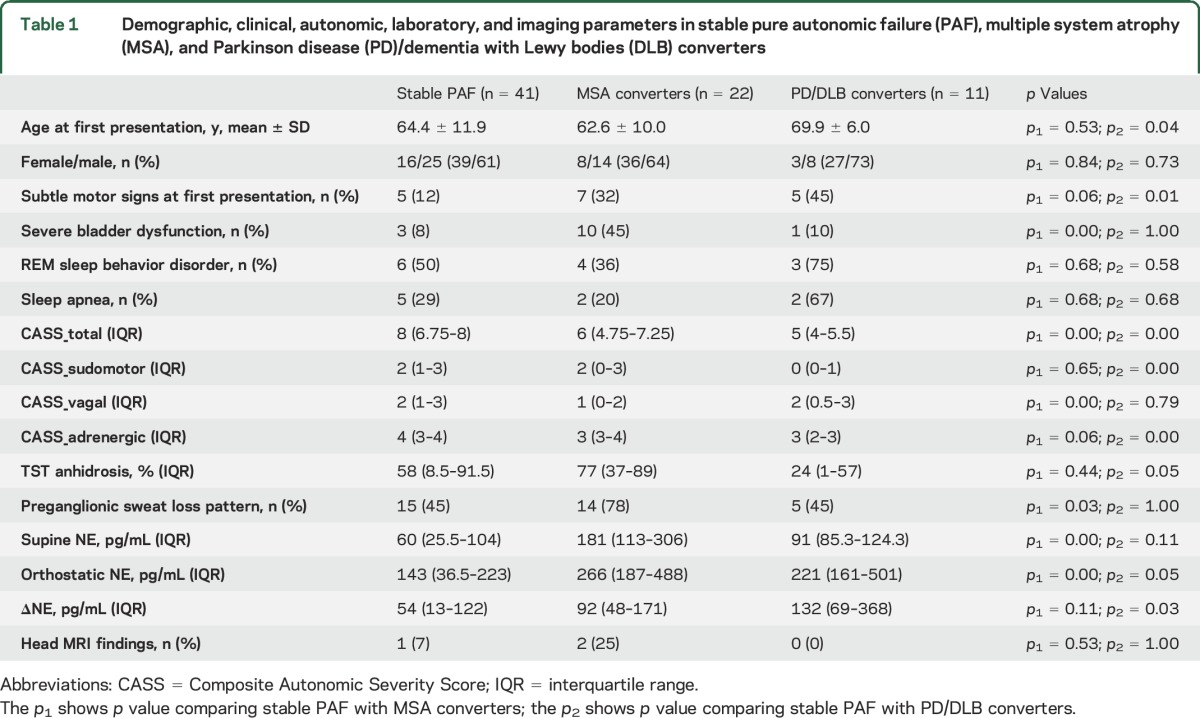
Demographic, clinical, autonomic, and laboratory parameters at the time of first diagnosis of PAF are summarized in table 1 and grouped by those with stable PAF vs MSA and PD/DLB converters. Highly significant differences between stable PAF and MSA converters were seen in the degree of bladder dysfunction, total CASS score, vagal CASS subscore, the presence of a preganglionic pattern of sudomotor deficits, and supine and orthostatic NE. Subtle motor signs at first presentation were more frequently seen in MSA converters (32%) vs stable PAF (12%, p = 0.06). The lower cardiovagal CASS subscore was the main contributor to the overall lower CASS score seen in MSA converters. Significant differences between stable PAF and PD/DLB converters were seen in subtle motor signs at first presentation, total CASS score, sudomotor and adrenergic CASS scores, as well as the orthostatic increase of NE. Also, the PD/DLB converters were slightly older, had a lower degree of sweat loss on TST, and had higher NE on standing.
Table 1.
Demographic, clinical, autonomic, laboratory, and imaging parameters in stable pure autonomic failure (PAF), multiple system atrophy (MSA), and Parkinson disease (PD)/dementia with Lewy bodies (DLB) converters
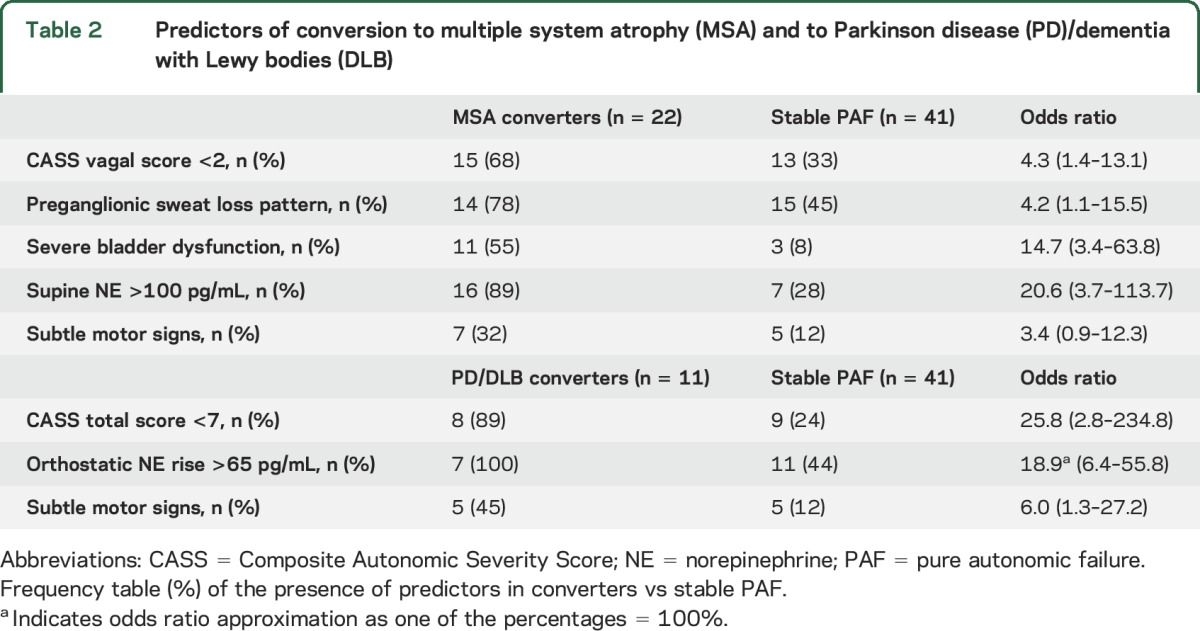
Based on these differences, 5 independent variables were identified as predictors for future conversion to MSA, and 3 variables for future conversion to PD/DLB. Table 2 shows ORs for developing CNS involvement based on the best cutoff values for these parameters. The highest OR for developing MSA was seen for severe bladder dysfunction and supine NE >100 pg/mL, while a total CASS score of less than 7 and an orthostatic rise in NE >65 pg/mL showed the highest OR for developing PD/DLB.
Table 2.
Predictors of conversion to multiple system atrophy (MSA) and to Parkinson disease (PD)/dementia with Lewy bodies (DLB)
MSA and PD/DLB conversion scores (0–5 and 0–3) were generated allotting 1 point for the presence of individual predictors (figure 3). Utilizing these conversion scores, the later development of MSA or PD/DLB could be predicted with high accuracy. A score of 0 or 1 on each score affirmed a diagnosis of stable PAF and was associated with a low risk of conversion to MSA or PD/DLB, respectively. A MSA conversion score greater than 2 and a PD/DLB conversion score greater than 1 strongly predicted future conversion to MSA or PD/DLB, respectively. An MSA conversion score greater than 2 predicted later conversion with a sensitivity of 68% (confidence interval [CI] 45%–86%) and specificity of 98% (CI 87%–100%), while a PD/DLB conversion score greater than 1 predicted subsequent conversion with a sensitivity of 73% (CI 39%–94%) and specificity of 90% (CI 77%–97%).
Figure 3. Multiple system atrophy (MSA) and Parkinson disease (PD)/dementia with Lewy bodies (DLB) conversion scores.

Conversion scores derived from identified risk factors for future (A, C) MSA and (B, D) PD/DLB conversion in stable pure autonomic failure (light columns) vs converters (dark columns). An MSA conversion score >2 is highly predictive of future conversion to MSA, while a PD/DLB conversion score >1 predicts future conversion to PD or DLB with high sensitivity and specificity.
DISCUSSION
PAF is generally considered a limited or restricted synucleinopathy affecting predominantly postganglionic autonomic fibers and ganglia. There have been reports of patients with a PAF phenotype eventually developing motor involvement and converting to MSA, PD, or DLB, but this phenomenon has not been formally investigated. Here, we studied a large group of carefully selected patients presenting with a PAF phenotype and applied further careful selection to define a group of patients with stable PAF, a group of converters to MSA, and a group of converters to PD or DLB.
Our findings include the following: (1) A considerable proportion of those presenting with PAF phenotype eventually develop motor involvement and convert to MSA, PD, or DLB. Assuming that those who converted had similar likelihood of long-term follow-up at our institution as those who remained PAF, the likelihood of conversion would be almost 50%. However, considering our role as a tertiary referral center, one has to assume that patients would be more likely to be re-referred if new symptoms develop that are inconsistent with the original diagnosis. A conservative estimation would therefore use as a denominator all 318 patients originally considered possible PAF, resulting in a conversion rate of 12%. The true conversion rate should therefore fall in the lower range between 12% and 48%, still a considerable and concerning percentage. (2) Almost twice as many patients convert to MSA than to PD or DLB, which may—considering the more rapidly progressive and disabling nature of MSA—in part relate to referral bias. (3) The vast majority of those who convert to MSA do so within the first 3 years of their PAF diagnosis, while patients converting to PD or DLB convert in a wider time range, though most convert within 5 years. (4) A set of clinical, autonomic, and laboratory parameters is associated with the conversion risk, and most of those parameters differ between MSA and PD/DLB, allowing for differential prognostication.
The presence of subtle motor signs at time of PAF diagnosis was—not surprisingly—confirmed as a risk factor for future conversion to both MSA and PD/DLB. Our clinical experience has long suggested that those patients in whom subtle motor findings such as slight gait imbalance or subtle tremor exist are more likely to eventually develop a motor synucleinopathy, even if those findings at the time are too subtle to make a diagnosis of either parkinsonism or cerebellar dysfunction.
One of the classic defining features of PAF is the finding of low circulating NE as would be expected in a disease primarily affecting postganglionic autonomic neurons.20–22 The finding that higher supine NE values (>100 pg/mL) are associated with higher risk of conversion to MSA is therefore intuitive. On the other hand, patients with MSA as well as PAF are known to lack a significant orthostatic rise in NE. We could demonstrate that this occurs to a lesser extent in those subsequently converting to PD or DLB, defining an orthostatic NE rise of more than 65 pg/dL as a predictive feature of that subgroup.
TST assesses the thermoregulatory system from the hypothalamus to the eccrine sweat glands and lesions anywhere in this pathway can lead to thermoregulatory sudomotor deficits. Sudomotor deficits on TST in areas of preserved sudomotor axon reflex testing are therefore indicative of central lesions and more consistent with MSA than PAF, 21 which is reflected in our finding that a preganglionic pattern of sweat loss is associated with higher odds of conversion to MSA.
The finding of less severe cardiovagal impairment being associated with higher odds of MSA conversion may be slightly surprising considering the severe and widespread autonomic failure often encountered in MSA and pathologic studies showing comparable involvement of the dorsal vagal nucleus in MSA and Lewy body disorders.23 However, there is solid evidence based on PET and SPECT imaging as well as pathologic data that there is vast postganglionic denervation of the heart in PAF affecting sympathetic, but also parasympathetic, vagal fibers, less commonly seen in MSA.24–26 PD and DLB converters were characterized by an overall lesser degree of autonomic failure. In fact, a total CASS score of 6 or less was highly predictive of future conversion to PD or DLB. Autonomic failure of DLB and PD has been well-described and while exceptions with profound autonomic failure occur, the majority of patients with these disorders are characterized by milder degrees of autonomic failure compared to MSA and PAF.27
Surprisingly, RBD was not found to be associated with a higher risk of conversion. Although this could be explained by the fact that its presence or absence was not consistently documented, RBD was seen in about half of patients with stable PAF and therefore is common even in those who do not convert. This conclusion underlines our clinical experience, although previous small case series on this association report conflicting evidence.28,29
The factors reported here associated with higher odds of conversion to one of the motor synucleinopathies largely reflect clinical experience and are supported by our current understanding of these disorders; our data provide confirmation, further insight, and quantification of these variables, and the provided scores predicting conversion should be particularly relevant and clinically helpful. Being able to predict if a patient presenting with a PAF phenotype will eventually evolve to develop a more sinister disorder affecting motor function is of importance in itself and for patient counseling, yet the ability to predict conversion with high sensitivity and specificity is a first step towards diagnosing motor synucleinopathies at a premotor stage. This is highly relevant in an era where disease-modifying therapies for these disorders are coming closer to reality.
Our study has limitations. The single-center design introduces the potential of referral bias. The retrospective design limits fully standardized data collection and also holds a potential of ascertainment and referral/re-referral bias. On the other hand, all data were extracted from the notes of autonomic experts at our institution with detail and high standardization of data collection and documentation; the autonomic test battery has been standardized and stable since 1983; conversion to a motor synucleinopathy was confirmed by expert review and application of diagnostic consensus criteria; and data points considered crucial such as autonomic testing were a prerequisite for inclusion. The requirement of at least 3 years of in-person neurologic follow-up was based on clinical experience, yet was somewhat arbitrary. While some patients who ultimately converted to PD or DLB did so after 3 years following initial presentation, the vast majority of patients in our stable PAF group was followed for 5 years or more, some over 15 years. Although the number of PAF patients included in this study may be considered large considering the rare nature of this disease, the precision of reported ORs, sensitivities, and specificities is limited by the sample size, as evident in the rather wide CIs. Another limitation of this study is that further validation of the described predictors in a confirmation cohort is lacking; an ongoing prospective study at our institution will eventually fill this gap. Finally, TST is limited to selected tertiary referral centers, yet required for the determination of a preganglionic sweat loss pattern. On the other hand, PAF is a rare disease commonly referred to specialized centers for diagnostic confirmation. In addition, the described conversion factors are still relevant in isolation or some combination without all variables necessarily needed to judge conversion risk.
Our study demonstrates that a considerable proportion of patients originally diagnosed with PAF eventually converts to motor synucleinopathies. This conversion of the clinical phenotype generally occurs within the first few years following diagnosis, and occurs earlier in those converting to MSA than those converting to PD or DLB. Specific risk factors for conversion were identified and when combined with conversion scores allow for prediction of conversion with high sensitivity and specificity.
GLOSSARY
- ARS
autonomic reflex screen
- BP
blood pressure
- CASS
Composite Autonomic Severity Score
- CI
confidence interval
- DLB
dementia with Lewy bodies
- IQR
interquartile range
- MSA
multiple system atrophy
- NE
norepinephrine
- OH
orthostatic hypotension
- PAF
pure autonomic failure
- PD
Parkinson disease
- QSART
quantitative sudomotor axon reflex test
- RBD
REM sleep behavior disorder
- TST
thermoregulatory sweat test
AUTHOR CONTRIBUTIONS
Wolfgang Singer, MD: concept, design, acquisition and analysis of the data, drafting/editing the manuscript. Sarah Berini, MD: acquisition and analysis of data, drafting/editing the manuscript. Paola Sandroni, MD: acquisition of data. Robert D. Fealey, MD: acquisition of data. Elizabeth A. Coon, MD: acquisition and analysis of data. Mariana Suarez: acquisition of data. Eduardo E. Benarroch, MD: acquisition of data. Phillip A. Low, MD: concept, acquisition of the data, analysis, grant support, editing the manuscript.
STUDY FUNDING
This publication was made possible by NIH (P01 NS44233, U54 NS065736, R01 NS092625, K23 NS075141, UL1 TR000135), FDA (R01 FD004789), Cure MSA Foundation, and Mayo Funds. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH or FDA.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Gilman S, Low PA, Quinn N, et al. Consensus statement on the diagnosis of multiple system atrophy. J Auton Nerv Syst 1998;74:189–192. [PubMed] [Google Scholar]
- 2.Gilman S, Wenning GK, Low PA, et al. Second consensus conference on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dickson DW, Liu W, Hardy J, et al. Widespread alterations of alpha-synuclein in multiple system atrophy. Am J Pathol 1999;155:1241–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wakabayashi K, Takahashi H. Cellular pathology in multiple system atrophy. Neuropathology 2006;26:338–345. [DOI] [PubMed] [Google Scholar]
- 5.Benarroch EE, Schmeichel AM, Parisi JE. Depletion of mesopontine cholinergic and sparing of raphe neurons in multiple system atrophy. Neurology 2002;59:944–946. [DOI] [PubMed] [Google Scholar]
- 6.Bradbury S, Eggleston C. Postural hypotension: an autopsy upon a case. Am Heart J 1925;1:73–86. [Google Scholar]
- 7.Hague K, Lento P, Morgello S, Caro S, Kaufmann H. The distribution of Lewy bodies in pure autonomic failure: autopsy findings and review of the literature. Acta Neuropathol 1997;94:192–196. [DOI] [PubMed] [Google Scholar]
- 8.Kaufmann H, Hague K, Perl D. Accumulation of alpha-synuclein in autonomic nerves in pure autonomic failure. Neurology 2001;56:980–981. [DOI] [PubMed] [Google Scholar]
- 9.Goldstein DS, Holmes C, Sato T, et al. Central dopamine deficiency in pure autonomic failure. Clin Auton Res 2008;18:58–65. [DOI] [PubMed] [Google Scholar]
- 10.Goldstein DS, Holmes C, Sharabi Y. Cerebrospinal fluid biomarkers of central catecholamine deficiency in Parkinson's disease and other synucleinopathies. Brain 2012;135:1900–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldstein DS, Sewell L. Olfactory dysfunction in pure autonomic failure: implications for the pathogenesis of Lewy body diseases. Parkinsonism Relat Disord 2009;15:516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coon EA, Sletten DM, Suarez MD, et al. Clinical features and autonomic testing predict survival in multiple system atrophy. Brain 2015;138:3623–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaufmann H, Nahm K, Purohit D, Wolfe D. Autonomic failure as the initial presentation of Parkinson disease and dementia with Lewy bodies. Neurology 2004;63:1093–1095. [DOI] [PubMed] [Google Scholar]
- 14.Low PA, Denq JC, Opfer-Gehrking TL, Dyck PJ, O'Brien PC, Slezak JM. Effect of age and gender on sudomotor and cardiovagal function and blood pressure response to tilt in normal subjects. Muscle Nerve 1997;20:1561–1568. [DOI] [PubMed] [Google Scholar]
- 15.Low PA, Caskey PE, Tuck RR, Fealey RD, Dyck PJ. Quantitative sudomotor axon reflex test in normal and neuropathic subjects. Ann Neurol 1983;14:573–580. [DOI] [PubMed] [Google Scholar]
- 16.Low PA. Autonomic nervous system function. J Clin Neurophysiol 1993;10:14–27. [DOI] [PubMed] [Google Scholar]
- 17.Low PA, Sletten DM. Laboratory evaluation of autonomic failure. In: Low PA, Benarroch EE, eds. Clinical Autonomic Disorders, 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 2008:130–163. [Google Scholar]
- 18.Fealey RD, Low PA, Thomas JE. Thermoregulatory sweating abnormalities in diabetes mellitus. Mayo Clinic Proc 1989;64:617–628. [DOI] [PubMed] [Google Scholar]
- 19.Matsusue E, Fujii S, Kanasaki Y, et al. Putaminal lesion in multiple system atrophy: postmortem MR-pathological correlations. Neuroradiology 2008;50:559–567. [DOI] [PubMed] [Google Scholar]
- 20.Bannister R, ed. Introduction and Classification. Oxford: Oxford Medicl Publications; 1988. [Google Scholar]
- 21.Low PA, Bannister R. Multiple system atrophy and pure autonomic failure. In: Low PA, ed. Clinical Autonomic Disorders: Evaluation and Management, 2nd ed. Philadelphia: Lippincott-Raven; 1997:555–575. [Google Scholar]
- 22.Goldstein DS, Polinsky RJ, Garty M, et al. Patterns of plasma levels of catechols in neurogenic orthostatic hypotension. Ann Neurol 1989;26:558–563. [DOI] [PubMed] [Google Scholar]
- 23.Benarroch EE, Schmeichel AM, Sandroni P, Low PA, Parisi JE. Involvement of vagal autonomic nuclei in multiple system atrophy and Lewy body disease. Neurology 2006;66:378–383. [DOI] [PubMed] [Google Scholar]
- 24.Amino T, Orimo S, Itoh Y, Takahashi A, Uchihara T, Mizusawa H. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Pathol 2005;15:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldstein DS, Holmes C, Cannon RO III, Eisenhofer G, Kopin IJ. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med 1997;336:696–702. [DOI] [PubMed] [Google Scholar]
- 26.Kimpinski K, Iodice V, Burton DD, et al. The role of autonomic testing in the differentiation of Parkinson's disease from multiple system atrophy. J Neurol Sci 2012;317:92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thaisetthawatkul P, Boeve BF, Benarroch EE, et al. Autonomic dysfunction in dementia with Lewy bodies. Neurology 2004;62:1804–1809. [DOI] [PubMed] [Google Scholar]
- 28.Plazzi G, Cortelli P, Montagna P, et al. REM sleep behaviour disorder differentiates pure autonomic failure from multiple system atrophy with autonomic failure. J Neurol Neurosurg Psychiatry 1998;64:683–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tijero B, Gomez-Esteban JC, Berganzo K, Zarranz JJ. REM sleep behaviour disorder and “pure” autonomic failure: presentation of two cases [in Spanish]. Neurologia 2012;27:55–57. [DOI] [PubMed] [Google Scholar]