

Abstract
Objective:
To investigate the association between a genetic risk score (GRS) and familial late-onset Alzheimer disease (LOAD) and its predictive value in families multiply affected by the disease.
Methods:
Using data from the National Institute on Aging Genetics Initiative for Late-Onset Alzheimer Disease (National Institute on Aging–Late-Onset Alzheimer's Disease Family Study), mixed regression models tested the association of familial LOAD with a GRS based on single nucleotide polymorphisms (SNPs) previously associated with LOAD. We modeled associations using unweighted and weighted scores with estimates derived from the literature. In secondary models, we adjusted subsequent models for presence of the APOE ε4 allele and further tested the interaction between APOE ε4 and the GRS. We constructed a similar GRS in a cohort of Caribbean Hispanic families multiply affected by LOAD by selecting the SNP with the strongest p value within the same regions.
Results:
In the NIA-LOAD families, the GRS was significantly associated with LOAD (odds ratio [OR] 1.29; 95% confidence interval 1.21–1.37). The results did not change after adjusting for APOE ε4. In Caribbean Hispanic families, the GRS also significantly predicted LOAD (OR 1.73; 1.57–1.93). Higher scores were associated with lower age at onset in both cohorts.
Conclusions:
High GRS increases the risk of familial LOAD and lowers the age at onset, regardless of ethnic group.
Late-onset Alzheimer disease (LOAD) is the leading cause of dementia in the elderly.1 The most important genetic risk factor is the APOE ε4 allele (attributable risk 20%2) but more than half of affected individuals do not carry this high-risk allele. Between 2009 and 2011, 9 risk loci were identified (CLU, PICALM, CR1, and BIN1, MS4A4A/MS4A4E/MS4A6E cluster, ABCA7, CD2AP, CD33, and EPHA1),3–7 and in 2013, 12 additional novel risk loci were found in a large genome-wide association study (GWAS) performed by the International Genomics of Alzheimer's Project (IGAP).8 Of note, CD33 and DSG2 were not replicated in stage II analysis. However, the effect sizes associated with these variants were small (odds ratios [ORs] ranged between 1.1 and 1.3), suggesting that a large part of the genetic component of LOAD remains unexplained.9
Polygenic risk scores (genetic risk scores [GRS]) represent an alternative strategy to summarize sparse genetic information and identify genetic risk profiles. An international case-control study with unrelated individuals10 confirmed that LOAD is enriched with a polygenic component, although sensitivity–specificity assessment showed limited disease prediction accuracy. The best area under the receiver operating characteristic (ROC) curve (AUC of 78%) was reached only when the model included APOE, sex, and age as predictors in addition to various GRS constructs. Similar results were found in the Flanders-Belgian Alzheimer disease (AD) study,11 which showed that GRS increased the risk of AD (OR 2.3) and was inversely associated with CSF Aβ1-42 and age at onset. Nevertheless, this study also confirmed the limited prediction accuracy, reporting a similar moderate AUC (i.e., 70%). An identical approach was used to study incident LOAD in a large sample of non-Hispanic white participants.12 The GRS was associated with a higher LOAD risk (particularly in participants with the APOE ε4 allele) and risk prediction after 7 years of follow-up showed a small improvement when the GRS was added to common risk factors such as age, sex, APOE ε4, and education. The value of GRS was also tested in 4 samples of participants diagnosed with mild cognitive impairment (MCI)13: the GRS was found not associated with higher conversion from MCI to LOAD, and did not interact with any of the known risk factors (age, sex, APOE ε4 allele).
These investigations imply that sporadic LOAD is characterized by a significant polygenic component. The GRS has never been investigated in familial LOAD or in other ethnic groups with familial LOAD. Thus, we will also test the significance of a GRS in a sample of Caribbean Hispanic families by employing the same set of loci used in the non-Hispanic white participants.
METHODS
Study samples.
In 2003, the National Institute on Aging–Late-Onset Alzheimer's Disease Family Study (NIA-LOAD) began to recruit families containing multiple individuals with LOAD at participating Alzheimer Disease Centers (ADCs) in the United States. Selection criteria included (1) a proband who received a diagnosis of definite or probable LOAD with age at onset of at least 60 years; (2) a full sibling with definite, probable, or possible LOAD with age at onset after 60 years; (3) a related family member (first-, second-, or third-degree relative) of the affected sibling pair and 60 years or older if unaffected, or 50 years or older if diagnosed with LOAD or MCI. Unaffected elderly individuals were included if documented cognitive testing and clinical examination confirmed the clinical designation. For participants who could not complete a detailed in-person evaluation, either because they had advanced disease or were living in a remote location, the site investigator conducted a detailed review of medical records to document the presence or absence of LOAD. The sample also included unrelated individuals; however, all cases had a family history of AD according to the study protocol inclusion criteria.
The Estudio Familiar de Influencia Genetica en Alzheimer (EFIGA) study was employed as a replication study. Similar to NIA-LOAD, this study recruited patients from families multiply affected by LOAD, but of Caribbean Hispanic ancestry from the Dominican Republic and New York. Families were recruited after confirming diagnoses in the probands. Family members with dementia were also interviewed and neurologically evaluated. Clinical diagnoses were made in a consensus diagnostic conference by a panel of neurologists, neuropsychologists, and psychiatrists. Detailed description is available elsewhere.14
For these family-based studies, we included data from families for which their members (1) were 60 years or older at the time of enrollment; (2) had a diagnosis of probable or possible LOAD according to National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association (NINDS-ADRDA) criteria; (3) had available pedigree information and covariates.
Standard protocol approvals, registrations, and patient consents.
Written informed consent was obtained from all participants (or caregivers in case the individual was deemed incapable of consent) using procedures approved by institutional review boards at each of the clinical research centers. Recruitment for each study was approved by the institutional review board of the Columbia University Medical Center and the Bioethics National Committee for Research in the Dominican Republic (for EFIGA participants).
Outcomes.
Primary outcomes of the study were clinically diagnosed probable or possible LOAD based on NINDS-ADRDA criteria.15 As a secondary outcome, we explored age at onset (AAO) of LOAD, restricting the analyses to affected individuals only.
Statistical methods.
NIA-LOAD families.
For non-Hispanic white participants, we computed the GRS for LOAD based on the genome-wide significant single nucleotide polymorphisms (SNPs) other than APOE, reported from the IGAP meta-analysis.8 Participants from the NIA-LOAD study were part of the IGAP meta-analysis, although the contribution consisted of a single individual from each family. For full description of genotype array, quality control, and imputation methods, see Wijsman et al.16 For each individual, we used 2 established approaches to calculate the GRS.17 First, the imputed probabilities of susceptibility risk alleles (ranging from 0 to 2) were summed (unweighted GRS). We then constructed a second GRS (weighted GRS) by multiplying each imputed probability by the β coefficient derived from the IGAP meta-analysis.8 In this way, we weighted each SNP by the expected effects on LOAD. The GRS was also recomputed in a conservative approach excluding the CD33 and DSG2 variants, because the relevant SNPs were not replicated in the second-stage analyses. We applied a rank invariant normal transformation to all GRS. A full description of SNPs and ORs used to derive the GRS is given in table e-1 at Neurology.org.
EFIGA families.
For Caribbean Hispanic families, we modified the approach for selecting the SNPs to be included in the GRS. This was justified based on the expected differences in minor allele frequency, unavailability of some SNPs due to imputation limits, and the inability to apply the IGAP coefficients previously estimated in a white, non-Hispanic meta-analysis. A univariate mixed model was employed to test the association between each SNP lying within the 20 IGAP genes and LOAD, adjusting for sex and age as fixed effects, and the kinship matrix as random effect. Only SNPs with an observed minor allele frequency >1% were included in the analyses. In addition, we selected only high-quality imputed SNPs (R2 ≥ 0.8) and added a 100 kb flanking region at either end of each locus, in order to cover potential regulatory regions and to account for different linkage disequilibrium patterns expected across different ethnic groups. Analyses were conducted employing the GEMMA software18 for the mixed model estimations; because of the admixed nature of the Caribbean Hispanic population, the kinship matrix was estimated through REAP software,19 which leverages the global admixture measures for each individual. A full description of the cohort can be found elsewhere14; detailed procedures are shown in appendix e-1 and figure e-1.
A single SNP within each locus was then selected, based on the strongest p value obtained by the above described mixed model, and its imputed probability was summed across the 20 loci as described previously. Therefore, the GRS for Caribbean Hispanics was constructed through an additive method, contrary to what has been done for non-Hispanic white participants, which received both additive and weighted GRS constructs.
We then applied a rank invariant normal transformation20 as implemented in the GenAbel R package to all GRS scores; the resulting distribution was then used in all subsequent analyses.
The list of SNPs used to derive the GRS is given in table e-2.
The association between the GRS and LOAD was studied in separate age- and sex-adjusted generalized mixed logistic regression models that included the ascertainment site (ADC center) as well as the family as random effects to adjust for possible center variability and intrafamilial correlations (model 1). Secondary models included the presence of at least 1 APOE ε4 allele as an additional covariate (model 2). A third statistical model explored the interaction between the GRS and the APOE ε4 allele (model 3). For AAO analyses, we employed a linear mixed model with an identical covariate-adjustment strategy, restricting the analyses to cases only.
ORs were computed for each model, and represent the increased risk of LOAD per SD increase of GRS. We also calculated sensitivity, specificity, and AUC by comparing the observed LOAD affected-unaffected status and the predicted probability estimated by employing a nonparametric analysis of clustered ROC curves21 in in R 3.0 (cran.org), with each cluster identified as a family pedigree. The AUC reflects the ability of the score to classify correctly those with and without the disease. A cutoff of 80% was considered a good to excellent grade for the selected test.
RESULTS
A full description of NIA-LOAD and EFIGA individuals is reported in table 1. A total of 4,792 NIA-LOAD individuals were included, 44% of those affected (73.4 cases' age at onset vs 74.7 age at last evaluation for the unaffected individuals). A total of 3,324 participants were included from the EFIGA study, 65% affected; the age distribution in the EFIGA family significantly differed between cases and controls (78.2 vs 69.8, respectively, p < 0.01). Distribution of the GRS for the NIA-LOAD and EFIGA study can be found in figure 1.
Table 1.
Clinical characteristics of National Institute on Aging–Late-Onset Alzheimer's Disease Family Study (NIA-LOAD) families
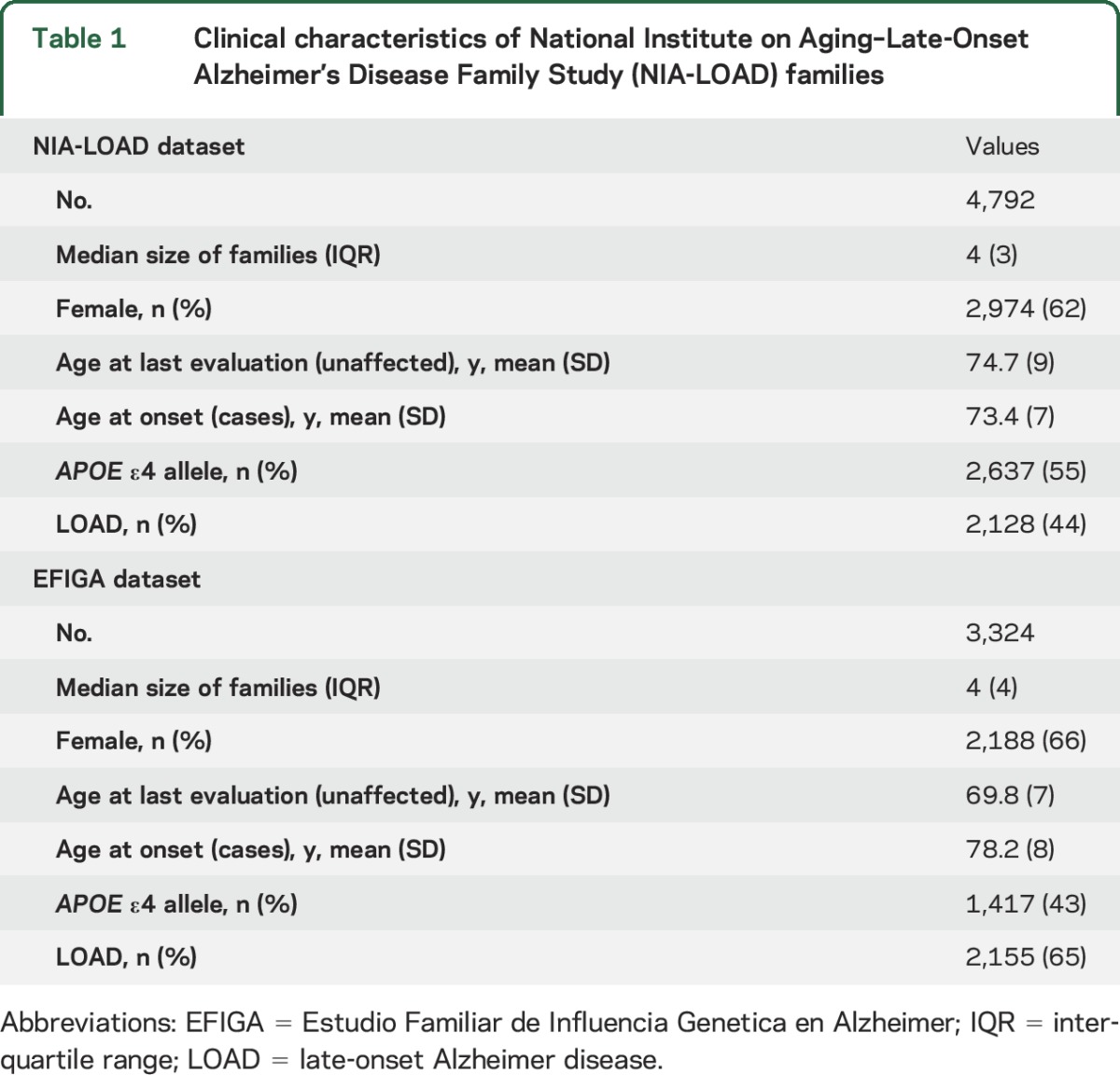
Figure 1. Polygenic risk scores (GRS) in the National Institute on Aging–Late-Onset Alzheimer's Disease Family Study (NIA-LOAD) and Estudio Familiar de Influencia Genetica en Alzheimer (EFIGA).

Distribution of the GRS in the NIA-LOAD (A) and EFIGA (B) datasets.
Generalized mixed models.
For NIA-LOAD families, the unweighted GRS conferred greater risk of LOAD (OR 1.29; 95% confidence interval [CI] 1.21–1.37). The weighted GRS also conferred increased risk of LOAD (OR 1.29; 95% CI 1.21–1.37) (model 1; table 2). Figure 2 graphically represents the effect sizes (and 95% CI) obtained from the mixed model for each SNP and the corresponding weighting log OR derived from the IGAP meta-analysis. The resulting effect size for the GRS is also depicted as a solid red line (plot created using the gtx R package (cran.r-project.org/web/packages/gtx). As expected, presence of an APOE ε4 allele was significantly associated with LOAD risk (OR 4.87, 95% CI 4.22–5.63). However, the risk of LOAD was similar after adjusting for the presence of an APOE ε4 allele (table 2, model 2), and there was no evidence of statistical interaction between the GRS and APOE ε4 allele (table 2, model 3). In both APOE ε4 allele carriers and noncarriers, GRS showed similar association with LOAD.
Table 2.
Results from the generalized mixed models for National Institute on Aging–Late-Onset Alzheimer's Disease Family Study and Estudio Familiar de Influencia Genetica en Alzheimer families
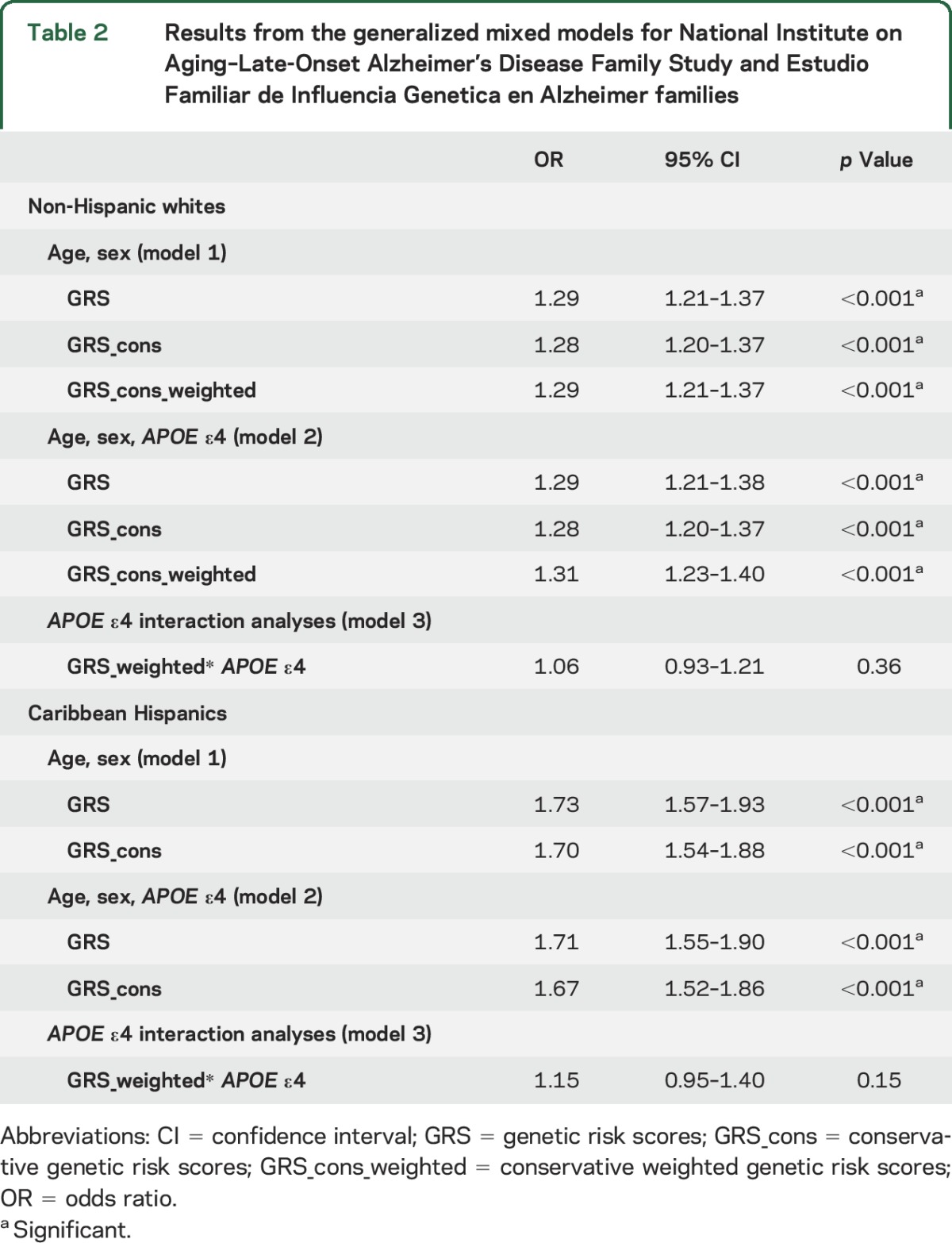
Figure 2. National Institute on Aging–Late-Onset Alzheimer's Disease Family Study (NIA-LOAD) polygenic risk score (GRS) and its weighting coefficients.

Construction of the NIA-LOAD polygenic risk score. Each single nucleotide polymorphism is plotted by the weighting coefficient extracted from the International Genomics of Alzheimer's Project analysis (x axis) vs the estimated effect size (and 95% confidence interval [CI] depicted by vertical gray lines) obtained by the mixed model (y axis). The solid red line indicates the effect size estimate for the GRS on late-onset Alzheimer disease (dotted lines represent the 95% CI). SLC24A4, ZCWPW1, and PTK2B share the same position on the graph, and for graphic clarity are not labeled.
GRS was significantly associated with AAO in cases: for each SD increase, AAO was ∼8 months lower (β −0.70, SE 0.15, p < 0.001).
In the Caribbean Hispanic families, the GRS increased the risk of LOAD, before and after APOE ε4 adjustment (OR 1.73, 95% CI 1.57–1.93; OR 1.71, 95% CI 1.55–1.90, respectively) (table 2, models 1 and 2). The presence of an APOE ε4 allele increased the risk of LOAD (OR 2.0, 95% CI 1.7–2.49). Again, there was no evidence of statistical interaction between the GRS and APOE ε4 allele (table 2, model 3). Results from the logistic mixed model for each SNP included in the EFIGA GRS can be found in table e-2 and appendix e-2. Among all the selected SNPs, the best association was observed for rs1742703, which lies within the SLC24A4-RIN3 region (p = 1 × 10−6). An example of a family included in the sample, with corresponding GRS scores across the pedigree, can be found in figure e-2.
In EFIGA participants, each SD of the GRS score again lowered AAO ∼10 months (β −0.86 SE 0.22, p < 0.001).
Clustered ROC curve.
None of the GRS constructs in NIA-LOAD showed a good or excellent AUC result (figure e-3). The greatest AUC was observed with the weighted GRS (AUC 57%, asymptotic 95% CI 55%–59%); similar results were obtained in the EFIGA study (AUC 59%, 95% CI 57%–61%).
DISCUSSION
The current investigation confirmed a polygenic component in familial LOAD by demonstrating statistically significant effects in families multiply affected by LOAD. In the NIA-LOAD families, non-Hispanic whites of European and North American ancestry, the GRS score was found to increase the risk of LOAD independently of the risk derived from having at least one copy of the APOE ε4 allele. The ORs in our models were similar to those previously reported in case-control studies.10,11 No interaction was found between the GRS and the APOE ε4 allele. We also demonstrated an effect of the polygenic component on age at onset, finding that individuals with higher genetic burden (represented by a higher GRS) had a lower age at onset of the disease. Despite the differences between the 2 GRSs, which prevent the possibility of comparing the effect sizes between 2 ethnic groups (mostly due to the different set of variants employed), we noticed their relative importance as compared to the APOE ε4 effect: in the non-Hispanic whites, the latter showed a ∼3-fold times the GRS OR; in the Caribbean Hispanics, the disparity was attenuated.
We previously confirmed the association between single variants lying within the IGAP genes and LOAD in Caribbean Hispanic families.14 In fact, for the CELF1, FERMT2, RIN3, and CD33 genes, the same SNP reported in the IGAP meta-analysis was also nominally significant in the Caribbean Hispanic GWAS.14 For the GRS construction, we chose the SNP most strongly associated with LOAD from the IGAP genes. Due to the limitations of imputation, several IGAP variants did not pass quality control. In addition, there was the expected variation in allele frequency across ethnic groups. Nevertheless, this modified GRS further confirmed an association with LOAD and AAO, ultimately indicating that a common multigenic profile is present in LOAD across ethnic groups.
Finally, the GRS showed little predictive ability as demonstrated by the low AUC reported. This finding is consistent with the previous investigations using unrelated cases and controls10,11 where, albeit with higher scores, GRSs achieved AUCs <80%, i.e., poor or fair grades.
Further investigations will be required when high-throughput sequence data become available for these large cohorts. In fact, several studies have suggested that signals identified by GWAS signals tag or show synthetic association22 with other variants, i.e., genes containing common variants with modest effects on complex traits (as is the case of LOAD) may also contain rare variants (i.e., those that occur in less than 1% of the world's population) with larger effects.23 We previously reported a ∼3-fold enrichment of nonsynonymous mutations in non-Hispanic whites with LOAD as compared to healthy controls and mutation significantly associated with LOAD in ABCA7, CD2AP, EPHA1, and BIN1, all loci identified by previous GWAS.24 Our and other groups also identified rare variants associated with the disease in loci indicated by large GWAS: SORL1,25 ABCA7.26 This overlap between GWAS and sequencing studies' findings has been validated in conditions other than LOAD.27 On the other side, it has to be noted that for several loci prioritized by GWAS in LOAD and other diseases,28–30 lukewarm evidence of disease-associated rare variants has been provided so far by sequencing studies. This could be due to methodologic issues (i.e., sample size) or mechanisms other than single-variant causative association, such as epistasis or polygenic contribution to the conditions. GWAS findings are based on statistical significance, which is very different from good prediction (as further proved by our ROC curves). In fact, higher significance does not imply stronger predictivity and highly predictive variables do not necessarily appear as highly significant.31 One of the possible explanations relies on the fact that variables may be significantly associated with the outcome simply for a small group of individuals in the population, thereby leading to poor prediction on the population. This is even more true for rare variants and ultimately might explain the discussed disconnection between lack of significant rare variants within GWAS signals. Rare genetic variants may be disproportionately important (although not significant genome-wide) and might improve GRS construction and its prediction accuracy.
This study has limitations. The GRS for the non-Hispanic whites was constructed with loci derived from the IGAP meta-analysis, which included the NIA-LOAD study. Nevertheless, the IGAP study only included one individual per family, whereas we included all individuals per family passing the inclusion criteria. In addition, we added families not originally included in the IGAP study. Second, the SNPs selected for the EFIGA study were based on analyses performed on the same cohort. Nevertheless, the selection of the loci came from a completely independent study (i.e., IGAP meta-analysis) and thus the positive results within this diverse ethnic group serve as an additional validation of the existence of a polygenic component across populations. Third, the studies included in this study differ greatly. Compared to the NIA-LOAD study, the age distribution between affected and unaffected individuals in the EFIGA study was greater. However, these differences did not affect the outcome.
Identifying individuals with high risk of developing the disease based on their genetic profile would benefit clinical trial enrollment as well as precision medicine for personalized drug development.
Supplementary Material
ACKNOWLEDGMENT
The article is submitted on behalf of the NIA-LOAD/NCRAD Family Study Group.
GLOSSARY
- AAO
age at onset
- AD
Alzheimer disease
- ADC
Alzheimer Disease Center
- AUC
area under the receiver operating characteristic curve
- CI
confidence interval
- EFIGA
Estudio Familiar de Influencia Genetica en Alzheimer
- IGAP
International Genomics of Alzheimer's Project
- LOAD
late-onset Alzheimer disease
- GRS
genetic risk score
- GWAS
genome-wide association study
- MCI
mild cognitive impairment
- NIA-LOAD
National Institute on Aging–Late-Onset Alzheimer's Disease Family Study
- NINDS-ADRDA
Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association
- OR
odds ratio
- ROC
receiver operating characteristic
- SNP
single nucleotide polymorphism
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Dr. Mayeux had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: Drs. Tosto, Schupf, Ottman, Sweet, Mayeux. Acquisition of data: Dr. Tosto, K. Faber, Drs. Bird, Bennett, Rosenberg, Boeve, Graff-Radford, Goate, Farlow, Sweet, Lantigua, Manly, Schupf, Stern, Foroud, Mayeux. Analysis and interpretation of data: Drs. Tosto, Rosenberg, Boeve, Graff-Radford, Sweet, Ottman, Schaid, Foroud, Mayeux. Drafting of the manuscript: Drs. Tosto, Mayeux. Critical revision of the manuscript for important intellectual content: all authors. Statistical analysis: Drs. Tosto, Mayeux. Obtained funding: Drs. Bennett, Sweet, Ottman, Foroud, Mayeux. Administrative, technical, or material support: K. Faber, Drs. Foroud, Bennett, Graff-Radford, Goate, Farlow, Lantigua, Mayeux. Study supervision: Drs. Tosto, Mayeux.
STUDY FUNDING
Study participants were enrolled under federal grants R01AG041797, U24AG026395, RO1AG037212, PO1AG007232, and U24AG21886 from the NIA. Research reported in this article was supported by the Office of Research Infrastructure of the NIH under award S10OD018522.
DISCLOSURE
G. Tosto, T. Bird, D. Tsuang, and D. Bennet report no disclosures relevant to the manuscript. B. Boeve was an investigator for clinical trials sponsored by Cephalon, Inc., Allon Pharmaceuticals, and GE Healthcare; receives royalties from the publication of Behavioral Neurology of Dementia (Cambridge Medicine, 2009); has received honoraria from the American Academy of Neurology; serves on the Scientific Advisory Board of the Tau Consortium; and receives research support from the National Institute on Aging (P50 AG016574, U01 AG006786, RO1 AG032306, RO1 AG041797) and the Mangurian Foundation. C. Cruchaga, K. Faber, and T. Faroud report no disclosures relevant to the manuscript. M. Farlow has received grant and research support from Accera, Biogen, Eisai Med Res, Eli Lilly & Company, Genentech, MedAvante/AstraZeneca, and Navidea; serves on the speaker's bureau at Eisai Med Res, Pfizer Inc., Forest, Novartis, and Eli Lilly & Company; and serves as consultant/on advisory boards at Accera, Alltech, Avanir, Eisai Med Res, Inc., Helicon, Medavante, Medivation, Inc., Merck and Co., Inc., Novartis, Pfizer Inc., Prana Biotech, QR Pharma, Roche, Sanofi-Aventis, Schering-Plough, Toyama Pharm, Eli Lilly & Company, UCB Pharma, and Elan. A. Goate, S. Bertelsen, N. Graff-Radford, M. Medrano, R. Lantigua, J. Manly, and R. Ottman report no disclosures relevant to the manuscript. R. Rosenberg holds a US patent for Amyloid β Gene Vaccines and serves on the editorial board of the Journal of the Neurologic Sciences. D. Schaid, N. Schupf, Y. Stern, R. Sweet, and R. Mayeux report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Alzheimer's Association. 2013 Alzheimer’s disease facts and figures. Alzheimers Dement 2013;9:208–245. [DOI] [PubMed] [Google Scholar]
- 2.Slooter AJ, Cruts M, Kalmijn S, et al. Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: the Rotterdam Study. Arch Neurol 1998;55:964–968. [DOI] [PubMed] [Google Scholar]
- 3.Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet 2009;41:1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet 2011;43:429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lambert J, Heath S, Even G, et al. ; European Alzheimer's Disease Initiative Investigators. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 2009;41:1094–1099. [DOI] [PubMed] [Google Scholar]
- 6.Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet 2011;43:436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seshadri S, Fitzpatrick AL, Ikram MA, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010;303:1832–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013;45:1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tosto G, Reitz C. Genome-wide association studies in Alzheimer's disease: a review. Current Neurol Neurosci Rep 2013;13:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Escott-Price V, Sims R, Bannister C, et al. Common polygenic variation enhances risk prediction for Alzheimer’s disease. Brain 2015;138:3673–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sleegers K, Bettens K, De Roeck A, et al. A 22-single nucleotide polymorphism Alzheimer's disease risk score correlates with family history, onset age, and cerebrospinal fluid Abeta 42. Alzheimers Dement 2015;11:1452–1460. [DOI] [PubMed] [Google Scholar]
- 12.Chouraki V, Reitz C, Maury F, et al. Evaluation of a genetic risk score to improve risk prediction for Alzheimer’s disease. J Alzheimers Dis 2016;53:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lacour A, Espinosa A, Louwersheimer E, et al. Genome-wide significant risk factors for Alzheimer's disease: role in progression to dementia due to Alzheimer's disease among subjects with mild cognitive impairment. Mol Psychiatry 2017;22:153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tosto G, Fu H, Vardarajan BN, et al. F-box/LRR-repeat protein 7 is genetically associated with Alzheimer's disease. Ann Clin Transl Neurol 2015;2:810–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease report of the NINCDS-ADRDA work group under the auspices of Department of Health and Human Services Task Force on Alzheimer's disease. Neurology 1984;34:939. [DOI] [PubMed] [Google Scholar]
- 16.Wijsman EM, Pankratz ND, Choi Y, et al. Genome-wide association of familial late-onset Alzheimer's disease replicates BIN1 and CLU and nominates CUGBP2 in interaction with APOE. PLoS Genet 2011;7:e1001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Che R, Motsinger-Reif AA. A new explained-variance based genetic risk score for predictive modeling of disease risk. Stat Appl Genet Mol Biol 2012;11:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet 2012;44:821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thornton T, Tang H, Hoffmann TJ, Ochs-Balcom HM, Caan BJ, Risch N. Estimating kinship in admixed populations. Am J Hum Genet 2012;91:122–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aulchenko YS, Ripke S, Isaacs A, Van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics 2007;23:1294–1296. [DOI] [PubMed] [Google Scholar]
- 21.Obuchowski NA. Nonparametric analysis of clustered ROC curve data. Biometrics 1997;53:567–578. [PubMed] [Google Scholar]
- 22.Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB. Rare variants create synthetic genome-wide associations. PLoS Biol 2010;8:e1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature 2009;461:747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vardarajan BN, Ghani M, Kahn A, et al. Rare coding mutations identified by sequencing of Alzheimer disease genome-wide association studies loci. Ann Neurol 2015;78:487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vardarajan BN, Zhang Y, Lee JH, et al. Coding mutations in SORL1 and Alzheimer disease. Ann Neurol 2015;77:215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steinberg S, Stefansson H, Jonsson T, et al. Loss-of-function variants in ABCA7 confer risk of Alzheimer's disease. Nat Genet 2015;47:445–447. [DOI] [PubMed] [Google Scholar]
- 27.Gloyn AL, McCarthy MI. Variation across the allele frequency spectrum. Nat Genet 2010;42:648–650. [DOI] [PubMed] [Google Scholar]
- 28.Hunt KA, Mistry V, Bockett NA, et al. Negligible impact of rare autoimmune-locus coding-region variants on missing heritability. Nature 2013;498:232–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang H, Jin X, Li Y, et al. A large-scale screen for coding variants predisposing to psoriasis. Nat Genet 2014;46:45–50. [DOI] [PubMed] [Google Scholar]
- 30.Fuchsberger C, Flannick J, Teslovich TM, et al. The genetic architecture of type 2 diabetes. Nature 2016;536:41–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lo A, Chernoff H, Zheng T, Lo SH. Why significant variables aren't automatically good predictors. Proc Natl Acad Sci USA 2015;112:13892–13897. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.