

Abstract
Objective:
To evaluate urinary neurotrophin receptor p75 extracellular domain (p75ECD) levels as disease progression and prognostic biomarkers in amyotrophic lateral sclerosis (ALS).
Methods:
The population in this study comprised 45 healthy controls and 54 people with ALS, 31 of whom were sampled longitudinally. Urinary p75ECD was measured using an enzyme-linked immunoassay and validation included intra-assay and inter-assay coefficients of variation, effect of circadian rhythm, and stability over time at room temperature, 4°C, and repeated freeze-thaw cycles. Longitudinal changes in urinary p75ECD were examined by mixed model analysis, and the prognostic value of baseline p75ECD was explored by survival analysis.
Results:
Confirming our previous findings, p75ECD was higher in patients with ALS (5.6 ± 2.2 ng/mg creatinine) compared to controls (3.6 ± 1.4 ng/mg creatinine, p < 0.0001). Assay reproducibility was high, with p75ECD showing stability across repeated freeze-thaw cycles, at room temperature and 4°C for 2 days, and no diurnal variation. Urinary p75ECD correlated with the revised ALS Functional Rating Scale at first evaluation (r = −0.44, p = 0.008) and across all study visits (r = −0.36, p < 0.0001). p75ECD also increased as disease progressed at an average rate of 0.19 ng/mg creatinine per month (p < 0.0001). In multivariate prognostic analysis, bulbar onset (hazard ratio [HR] 3.0, p = 0.0035), rate of disease progression from onset to baseline (HR 4.4, p < 0.0001), and baseline p75ECD (HR 1.3, p = 0.0004) were predictors of survival.
Conclusions:
The assay for urinary p75ECD is analytically robust and shows promise as an ALS biomarker with prognostic, disease progression, and potential pharmacodynamic application. Baseline urinary p75ECD provides prognostic information and is currently the only biological fluid–based biomarker of disease progression.
Frustration over the continued failure of amyotrophic lateral sclerosis (ALS) clinical trials1,2 and the absence of therapeutic options for this fatal disease3 has fueled interest in the prospect that biomarkers may hold great promise for advancing therapy development efforts.4,5 Prognostic biomarkers, which aid in predicting the future course of disease, might be used to identify more homogeneous subsets of patients at the time of trial enrollment. Pharmacodynamic biomarkers, which have the potential to show that a biological response has occurred in a patient who has received an experimental therapeutic, may help in assessing the efficacy of drugs selected in phase II to advance to phase III clinical trials. Disease progression biomarkers (i.e., those that show a change over time as disease advances) may also serve as markers of pharmacodynamic effect.
Among the biological fluid–based biomarker candidates, the cytoskeletal proteins neurofilament light (NfL) and phosphorylated neurofilament heavy (pNfH) show great promise as prognostic markers and potential pharmacodynamic biomarkers. However, since neurofilament levels remain largely stable over time,6–8 they do not reflect disease progression. Therefore, we currently lack any biological fluid–based biomarkers of disease progression. This has led us to focus on the common neurotrophin receptor (p75) as a biomarker of motor neuron degeneration. Based on preliminary observations that the extracellular domain of p75 (p75ECD) is present at elevated levels in the urine of patients with ALS compared to healthy individuals9 and that urinary p75 increases in the SOD1G93A mouse as disease unfolds,10 we investigated the potential of urinary p75ECD as a potential disease progression and prognostic biomarker.
METHODS
Standard protocol approvals, registrations, and patient consents.
This was a prospective cohort study in which urine samples from patients with ALS and controls were collected from the South Australian MND Clinic (Adelaide, Australia) and the Kessenich Family ALS Center at the University of Miami (Miami, FL).
Written informed consent was obtained from all participants following ethics approval from the Flinders University of South Australia Southern Adelaide Clinical Human Research Ethics Committee and the University of Miami Human Subject Research Office. Patients with ALS (recruited between March 2011 and March 2015) were diagnosed according to the revised El Escorial criteria11 by experienced ALS neurologists (Australia: D.S.; United States: M.B.). Healthy controls (recruited between June 2008 and February 2015) were typically spouses and friends of patients and exclusion criteria included any neurologic condition or illness that affects kidney function. Sample size was driven by pragmatic considerations related to the availability of funding for this pilot project. Clinical information was collected by investigators blinded to urinary p75ECD results, including the revised ALS Functional Rating Scale (ALSFRS-R), and latencies from symptom onset and diagnosis to baseline assessment and sample collection. The estimated monthly decrease in ALSFRS-R by time of first assessment (baseline) was calculated as ΔFRS = (48 − ALSFRS-R at baseline)/number of months between symptom onset and baseline.12 Permanent assisted ventilation (PAV) was defined as the use of noninvasive ventilation for at least 23 h/d or tracheostomy with initiation of invasive ventilation. Urine samples were collected and stored in accordance with the Urine & Kidney Proteome Project Standards13 and coded to ensure anonymity. Samples collected in Miami were shipped on dry ice and all samples were stored at −80°C until analysis. Urinary creatinine and osmolarity measurements were performed using a Roche (Basel, Switzerland)/Hitachi (Tokyo, Japan) modular analyzer.
Urinary p75ECD measurement.
A sandwich ELISA was used to quantify p75ECD as previously described.9 Briefly, ELISA plates (96-well, Costar Corning [Manassas, VA]) were coated with mouse anti-human p75 MLR1 antibody14 for p75 capture and incubated for 18 hours at 4°C in bicarbonate buffer, pH 9.6. Wells were then blocked with sample buffer, phosphate-buffered saline (PBS) containing 2% bovine serum albumin, and 0.01% thimerosal, pH 7.4, for 1 hour at 37°C. Urine samples and recombinant human p75ECD standard (amino acids 29–250; R&D Systems, Minneapolis, MN) were diluted in sample buffer and incubated for 20 hours at room temperature. Goat anti-mouse p75ECD (Sigma-Aldrich, St. Louis, MO) and bovine anti-goat immunoglobulin G horseradish peroxidase (Jackson ImmunoResearch, West Grove, PA) antibodies were used for p75ECD detection for 1 hour each at room temperature, and the peroxidase reaction was developed using TMB (Bio-Rad, Hercules, CA) and stopped using 2M sulfuric acid. Plates were washed between steps (PBS, 0.05% Tween 20, 0.01% thimerosal, pH 7.4), and were read using a PerkinElmer (Waltham, MA) Victor-x4 plate reader.
The detection limit and reproducibility of p75ECD measurement was determined by testing recombinant human p75ECD standard (R&D Systems) and 3 urine samples across 8 plates. The lowest detection limit was calculated by plotting the p75ECD standard curve with a log10 x-axis. Intra-assay and inter-assay reproducibility was determined by testing 3 urine samples with low, medium, and high p75ECD/mL measurements in duplicate, across 8 plates, to determine the coefficient of variation (CV). In addition, the diurnal fluctuation of urinary p75ECD was determined in healthy individuals by comparing measurements made from a first void sample in the morning, a random spot urine in the afternoon/evening, and a sample collected over 24-hours. p75ECD stability was determined by repeatedly assaying samples stored at room temperature (∼23°C) or 4°C for up to 7 days. In addition, samples were subjected to 1–4 freeze-thaw cycles switching between −80°C and room temperature (∼23°C) for 15 minutes per cycle and p75ECD measured after each freeze/thaw.
Statistical analysis.
Intra-assay and inter-assay reproducibility of ELISA measurements are expressed as CVs. Linear regression was used to describe the linearity of a standard curve for human p75ECD. Urinary p75ECD levels were compared between patients with ALS and controls by 2-sample t test. The relationships among baseline age, urinary p75ECD, creatinine, urinary osmolarity, and ALSFRS-R scores were assessed by Pearson correlation. The correlation between urinary p75ECD and ALSFRS-R were further assessed by including data from baseline as well as longitudinal follow-ups, with and without adjustment for repeated measures.15 In the 31 patients with ALS who had longitudinal samples, the rate of urinary p75ECD increase over time—with time defined as months since diagnosis (primary analysis), symptom onset, or baseline (first urine collection)—was ascertained by mixed model analysis; quadratic and interaction terms were considered. In addition, the association between baseline urinary p75ECD levels and survival (time to death or PAV) was evaluated by Cox proportional hazards model, and graphically illustrated by dividing the patients into those with baseline p75ECD above vs below the median value and plotting their Kaplan-Meier survival curves. Summary statistics are presented as mean ± SD, median, range, or frequency and percentage. A p value of <0.05 (2-sided) was considered statistically significant. Longitudinal and survival analyses were performed using SAS 9.3 (SAS Institute, Cary, NC); all other analyses were performed, and figures generated, using GraphPad Prism 6 (GraphPad Software, La Jolla, CA).
RESULTS
Study population.
The study population includes 45 healthy controls and 54 patients with ALS (table 1). In the subset of 31 patients with ALS with longitudinal follow-up, 26 (84%) reached PAV or death. Median disease duration (from diagnosis) among all 54 patients was 18.4 months (25th–75th percentile: 11.1–32.4).
Table 1.
Participant characteristics
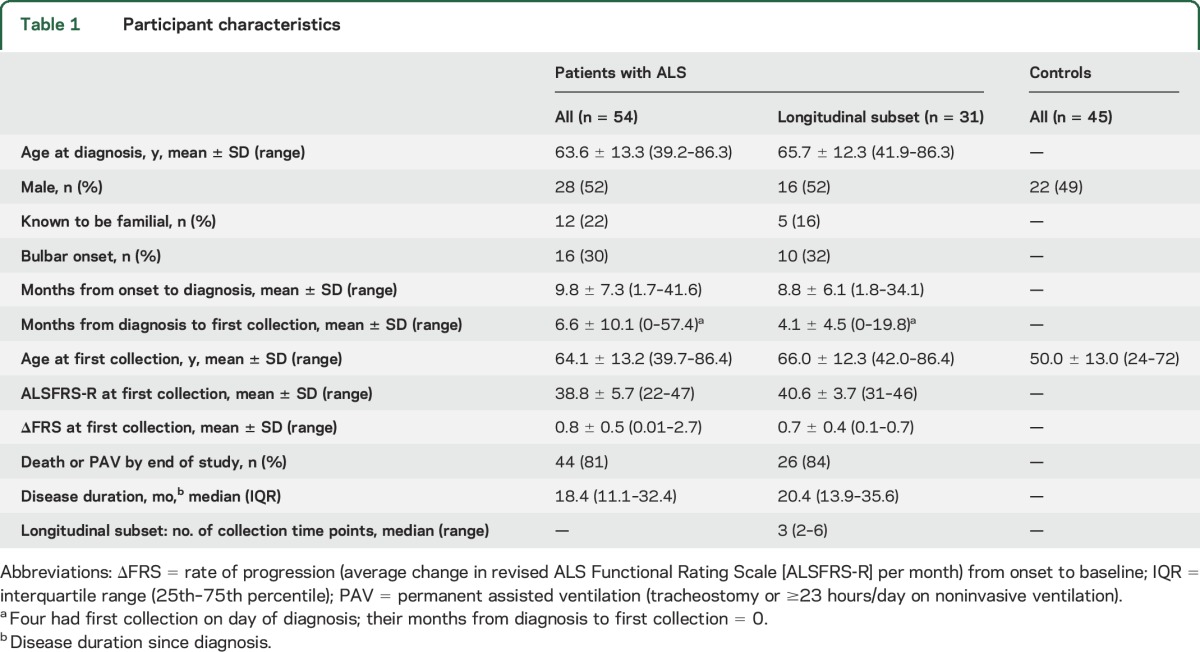
Urinary p75ECD in healthy controls and patients with ALS.
Among healthy controls, urinary p75ECD correlates with age (Pearson r = 0.31, p = 0.04) but increases only by 0.32 ng/mg creatinine for each advancing decade. Although the control group was younger than the ALS population, the potential utility of p75ECD as a disease progression or prognostic marker is independent of comparison to the control group and therefore not affected by this age difference. Sex is not a significant determinant of urinary p75ECD.
Confirming our previous findings,9 urinary p75ECD levels are higher in patients with ALS (5.6 ± 2.2 ng/mg creatinine) at first study visit (baseline) compared to controls (3.6 ± 1.4 ng/mg creatinine, p < 0.0001, figure 1). Baseline urinary p75ECD levels do not differ significantly between patients with limb vs bulbar onset disease, even after controlling for baseline disease severity.
Figure 1. Urinary extracellular domain of p75 (p75ECD) is elevated in amyotrophic lateral sclerosis (ALS).
Urinary p75ECD levels are higher in patients with ALS (n = 54) than in controls (n = 45) at first study visit (p < 0.0001, 2-sample t test).
Analytic validation of urinary p75ECD.
Confirming our previous observation of the linearity of the standard curve (absorbance and human urinary p75ECD concentration) for our in-house ELISA,9 here we show an assay sensitivity of ∼70 pg/mL and a linear standard curve up to 2.5 ng/mL (figure e-1, A–C, at Neurology.org). Our assay for human urinary p75ECD demonstrates reliability with an intra-assay CV of 6.6% and an inter-assay CV of 12.5% (table e-1). Urinary p75ECD levels remained steady after multiple freeze-thaw cycles (figure e-1D). The level of p75ECD detected was also stable in 3 samples after 2-day of storage both at room temperature and at 4°C, although from 2 to 7 days there was some variation from that measured in freshly collected urine (i.e., at time 0; figure e-1E). Diurnal variation in p75ECD was tested in controls (n = 2), between first void in the morning, a spot urine sample in the afternoon, and 24-hour samples. The variation between the spot urine and the first void in addition to the spot and the 24-hour samples was modest (below 17%). When urinary dilution was considered by correcting with creatinine (as we routinely do for reporting results), the difference between spot and first void and spot and 24-hour sample decreased to 7% (figure e-1F). In addition, creatinine was a reliable measure of urinary dilution, based on the strong correlation with urine osmolarity (figure e-1G, r = 0.88, p < 0.0001).
Urinary p75ECD as biomarker of disease progression.
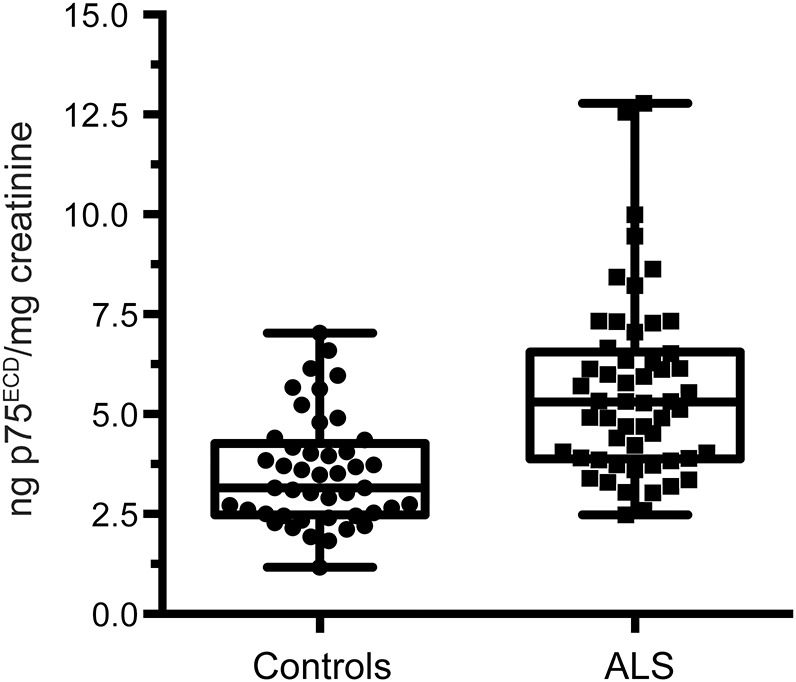
The potential utility of p75ECD as a biomarker of disease progression was explored in the 31 patients with ALS with longitudinal data and urine samples (median of 3 time points, range 2–6; table 1). To assess change in p75ECD over time, we considered 3 options for the time base—namely, time since symptom onset, time since diagnosis, and time since the initial evaluation and sample collection (i.e., baseline)—and the limitations of each. Symptom onset relies upon subjective report of symptoms and the decision of which symptoms represent the onset of disease. On the other hand, baseline date is somewhat arbitrary, as patients were enrolled at variable stages in the course of disease. While the delay from symptom onset to diagnosis can be highly variable, the date of diagnosis is reliably obtained from clinic notes, patient self-report, and medical record review. For these reasons, we elected a priori in our longitudinal analyses to use time from diagnosis for primary analysis. Results of our mixed model analysis show that urinary concentration of p75ECD increases linearly and on average by 0.19 ng/mg creatinine each month (p < 0.0001; figure 2A). Adjusting for baseline p75ECD does not significantly affect the slope, suggesting a fairly uniform rate of p75ECD change over time, irrespective of concentration at the earliest time point (data not shown). Adjusting for age similarly does not affect the slope of increase of p75ECD, likely because the effect of age is seen over decades rather than years, and median follow-up duration in this study was 10.8 months (range 0.4–43 months). Models using time from symptom onset and time from baseline show similar results (figure 2, B and C).
Figure 2. Longitudinal changes in urinary extracellular domain of p75 (p75ECD) in amyotrophic lateral sclerosis (ALS) as disease progresses.
(A) Individual patient urinary p75ECD trajectories increase since time of diagnosis at an average rate of 0.19 ng/mg creatinine per month (95% confidence interval [CI] 0.15–0.24). (B) p75ECD trajectories also increase since time from symptom onset (0.18 ng/mg, 95% CI 0.13–0.22), and (C) time since the initial evaluation and sample collection (i.e., baseline) (0.20 ng/mg, 95% CI 0.15–0.25). Total 31 patients with ALS, each sampled at least twice.
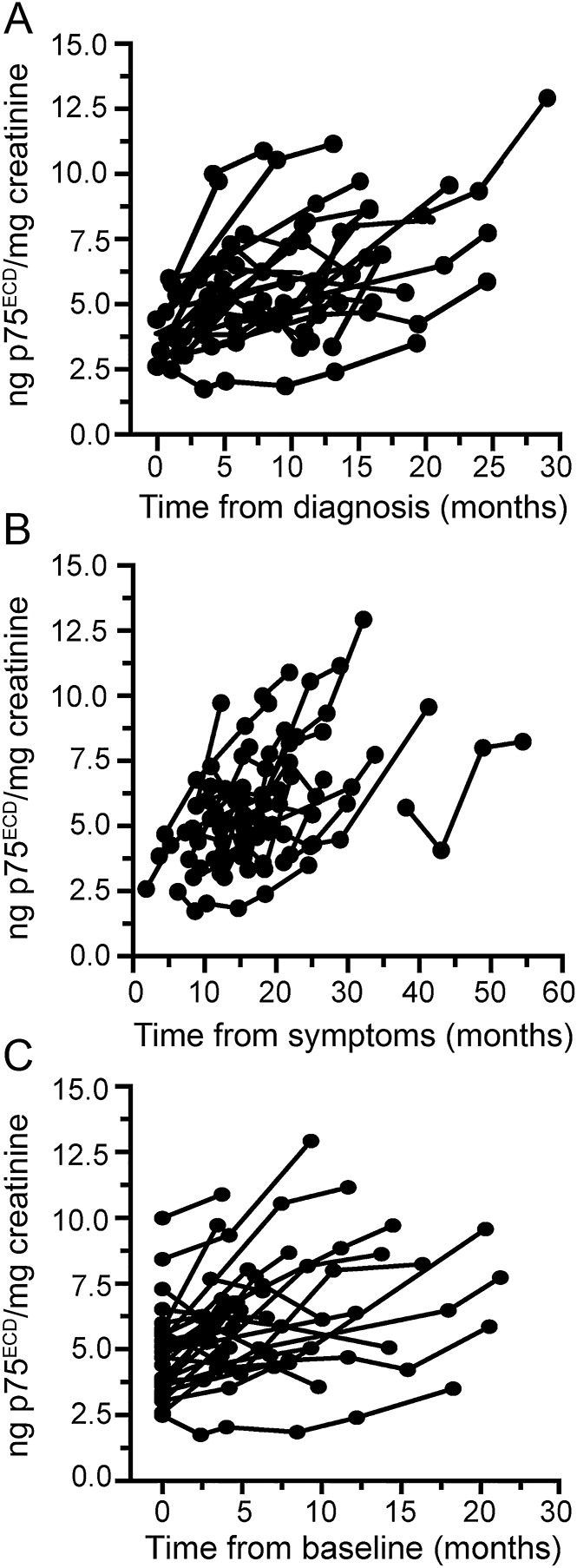
For comparison, we performed similar mixed model analyses with ALSFRS-R as the outcome measure. In the same 31 patients, average rate of ALSFRS-R decline was 0.82 points/month (95% CI 0.68–0.97, p < 0.0001), comparable to reported rate in other longitudinal observational studies and clinical trials.16 Similar to p75ECD, adjustment for baseline ALSFRS-R had little effect on the slope (data not shown). Moreover, using data from all 115 person-visits, there was a correlation between ALSFRS-R and p75ECD (r = −0.36, p < 0.0001), which was essentially unchanged after adjusting for the within-person correlation due to repeated measures (figure 3A). This correlation was also apparent when comparing ALSFRS-R and p75ECD at baseline for each of the 54 patients (figure 3B; r = −0.44, p = 0.0008).
Figure 3. Correlation between urinary extracellular domain of p75 (p75ECD) and revised ALS Functional Rating Scale (ALSFRS-R).
(A) Using p75ECD measures from all 115 person-visits, urinary p75ECD correlates with ALSFRS-R (r = −0.36, p < 0.0001). After adjusting for the within-person correlation that arises from repeated measures of the longitudinal subset, the correlation remains essentially unchanged (r = −0.37). (B) Urinary p75ECD also correlates with ALSFRS-R at first study visit for each of the 54 patients with amyotrophic lateral sclerosis in the study (r = −0.44, p = 0.008).
Urinary p75ECD as prognostic biomarker.
To explore the potential utility of baseline p75ECD as a predictor of prognosis, we performed a survival analysis using Cox proportional hazards model, including data from all 54 patients with ALS. In univariate analyses, neither age at diagnosis nor sex was associated with survival, but in agreement with prior studies,12,17 both bulbar onset (p = 0.0085) and ΔFRS (p < 0.0001) were predictors of survival. In multivariate models, adjusting for bulbar onset (hazard ratio [HR] 3.0, 95% confidence interval [CI] 1.4–6.3, p = 0.0035) and ΔFRS (HR 4.4, 95% CI 2.4–8.0, p < 0.0001), baseline p75ECD was also a predictor of survival (HR 1.3, 95% CI 1.1–1.5, p = 0.0004), indicating that baseline p75ECD provides additional prognostic value over and above what can be gleaned from routinely available clinical parameters. To visually illustrate the relationship between baseline p75ECD and survival, we divided the 54 patients into high vs low p75ECD groups (i.e., based on their baseline value being above or below the median), and present their Kaplan-Meier survival plot (figure 4).
Figure 4. Urinary extracellular domain of p75 (p75ECD) at baseline predicts future survival.
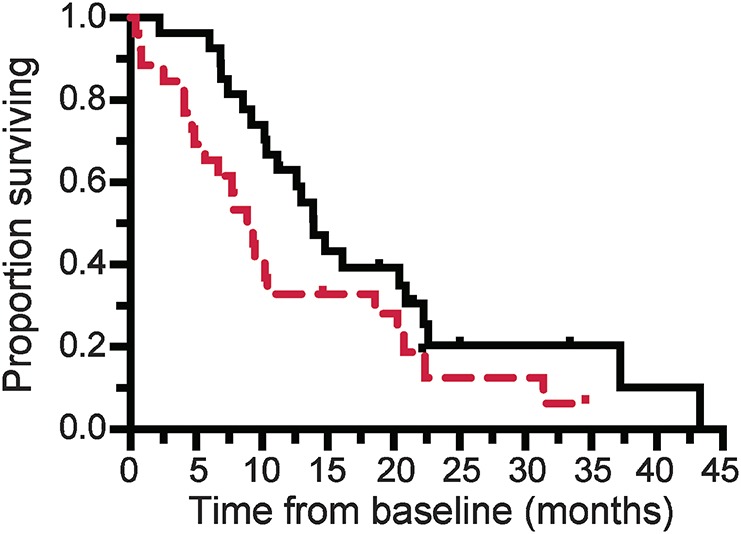
Kaplan-Meier survival estimates comparing 27 patients with amyotrophic lateral sclerosis who had baseline urinary p75ECD above (red dashed line) and 27 with values below (black line) the median (5.3 ng/mg creatinine), illustrates the longer median survival in those with lower compared to higher urinary p75ECD levels (13.9 vs 8.9 months; Wilcoxon test p = 0.024).
DISCUSSION
Our pursuit of urinary p75ECD as a potential ALS biomarker was informed by the biology of the neurotrophin receptor p75. Rodent motor neurons express p75 during development18 but this disappears soon after birth only to be re-expressed after injury,19,20 including apoptotic motor neurons of SOD1G93A mice.21 p75ECD is cleaved from cell membranes post injury,19 with elevated levels detected 40 days prior to onset of disease in the urine of SOD1G93A mice, progressively rising as symptoms unfold.9,10 p75 is also re-expressed on motor neurons22 and Schwann cells23 in postmortem tissue of patients with ALS. Hence, the presence of p75ECD in urine is indicative of underlying motor neuron degeneration. While ALS is heterogeneous in etiology, biology, and phenotypic manifestations,24 it is always characterized by degeneration and death of motor neurons. As a generic biomarker of motor neuron degeneration, therefore, urinary p75ECD is expected to be useful as biomarker in all forms of ALS, irrespective of etiology. This stands in contrast, for example, to C9RANT dipeptides, which may have utility as a pharmacodynamic biomarker only in patients with ALS due to a C9ORF72 repeat expansion.5
The most striking and important aspect of our findings is that urinary p75ECD changes over time, increasing as disease progresses and motor function declines. This is true even in patients with slowly progressive disease in which the absolute values of p75ECD remain relatively low (figure 2). In this respect, p75ECD is currently the only potential biochemical biomarker of ALS disease progression. In addition, p75ECD may have utility as a potential pharmacodynamic biomarker insofar as showing that an experimental therapeutic that blunts the increase, stabilizes, or reduces urinary p75ECD over time would provide evidence of an underlying biological effect of the treatment.25 Urinary p75ECD therefore joins CSF and blood NfL6,26–32 and CSF pNfH7,33–35 as the lead candidates for further development as pharmacodynamic biomarkers. The important difference is that neurofilament levels, although elevated in ALS compared to controls,6,7,26–34,36 are largely stable over time, with the possible exception of patients with rapidly progressive disease, in whom CSF NfL may increase,6 and levels of blood pNfH may decrease over time.7 These data suggest that NfL and pNfH may also have potential utility as pharmacodynamic biomarkers.
In addition to its value as a disease progression and pharmacodynamic biomarker, urinary p75ECD may have value as a prognostic biomarker. Essential to this claim is the observation that baseline p75ECD informs the probability of survival in a way that supplements the prognostic value of readily available clinical parameters such as site of disease onset and ΔFRS, even though the HR for the effect of p75ECD is modest. While other biochemical biomarkers including pNfH8,34,36 and NfL6,31 levels in blood and CSF have been reported to have prognostic utility, published data have not (yet) demonstrated that they add prognostic value over and beyond what can be learned from clinical parameters.
Moreover, p75ECD is currently the sole ALS biomarker that is measurable in urine, a biological fluid that is readily accessible and which most patients are willing to provide. While blood is also readily accessible, not all patients are willing or able to undergo lumbar puncture to obtain CSF, especially longitudinally. Since patient comfort and compliance are important pragmatic considerations, especially in clinical trials, obtaining urine for p75ECD quantification may be more practical than the more invasive and logistically complex option of obtaining CSF. In addition, the less complex nature of the urinary proteome than that of blood is an advantage in assaying proteins such as p75ECD. Notwithstanding these considerations, we are also exploring measurement of p75ECD in blood, which will enable validation of these findings using banked samples previously collected from patients.
This study is not without its limitations. Most notably, the study population represents a sample of convenience, with the attendant risk that the cohort is biased towards patients with more slowly progressive and perhaps more advanced disease. Moreover, the limited number of samples and assessments available from each patient precluded reliable estimation of the individual rates of ALSFRS-R decline,25 thereby limiting our ability to explore whether baseline p75ECD is useful in predicting prognosis in terms of future rate of ALSFRS-R decline, in addition to survival. These shortcomings, however, will be addressed through an ongoing validation study being performed as part of the Clinical Research in ALS and Related Disorders for Therapeutic Development (CReATe) consortium. In this study (NCT02327845), urine samples are collected every 3–6 months (over an 18- to 24-month period), along with rigorously and prospectively collected deep clinical phenotypic data.
Urinary p75ECD currently stands alone as a biofluid-based biomarker that adds prognostic value to readily available clinical parameters, and changes longitudinally in individual patients with ALS as disease progresses. Urinary p75ECD and blood and CSF quantification of pNfH and NfL represent the most promising potential pharmacodynamic biomarkers available today that are suitable for further investigation in the context of future clinical trials.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the research participants for their participation, Brad Rumbelow from SA Pathology for creatinine analysis, and the clinical research coordinators at the Miami site for assistance with patient recruitment.
GLOSSARY
- ALS
amyotrophic lateral sclerosis
- ALSFRS-R
revised ALS Functional Rating Scale
- CI
confidence interval
- CV
coefficient of variation
- HR
hazard ratio
- NfL
neurofilament light
- p75ECD
extracellular domain of p75
- PAV
permanent assisted ventilation
- PBS
phosphate-buffered saline
- pNfH
phosphorylated neurofilament heavy
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
S.R. Shepheard: experiments, manuscript preparation. J. Wuu: supervision of participant recruitment and sample collection (Miami site), statistical analysis, results interpretation, manuscript preparation. M. Cardoso: experiments. L. Wilkendt: data analysis. P. Dinning: consultant on data analysis. T. Chataway: design of experiments. D. Schultz: participant recruitment and sample collection (Flinders site). M. Benatar: supervision of participant recruitment and sample collection (Miami site), hypothesis generation, results interpretation, manuscript preparation. M.-L. Rogers: design of experiments, data analysis, manuscript preparation.
STUDY FUNDING
This study was supported by the NIH RDCRN CReATe Consortium (U54 NS092091), Motor Neurone Disease Research Institute of Australia (MNDRIA, Grant in Aid and Rosalind Nicholson Grant), Flinders University Centre for Neuroscience and FMC Foundation (Seeding Grants), ALS Association (grants 15-IIP-193 and 2015), Muscular Dystrophy Association (grants 4365 and 172123), ALS Recovery Fund, and Australian Rotary Health (Neville and Jeanne York PhD Family Scholarship for Motor Neuron Disease). The Clinical Research in ALS and Related Disorders for Therapeutic Development (CReATe) Consortium (U54 NS092091) is a part of the NIH Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), National Center for Advancing Translational Sciences (NCATS). CReATe is funded through collaboration between NCATS and the National Institute of Neurological Disorders and Stroke (NINDS).
DISCLOSURE
S. Shepheard received a PhD scholarship from Australian Rotary Health. J. Wuu received support from NIH RDCRN CReATe Consortium, ALS Association, Muscular Dystrophy Association, and ALS Recovery Fund. M. Cardoso, L. Wiklendt, P. Dinning, and T. Chataway report no disclosures relevant to the manuscript. D. Schultz received support from the ALS Association. M. Benatar received funding from the NIH RDCRN CReATe Consortium, ALS Association, Muscular Dystrophy Association, and ALS Recovery Fund. M. Rogers received support from the MNDRIA, FMC Foundation, ALS Association, and the NIH RDCRN CReATe Consortium. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Cudkowicz ME, Katz J, Moore DH, et al. Toward more efficient clinical trials for amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2010;11:259–265. [DOI] [PubMed] [Google Scholar]
- 2.Benatar M. ALS therapy development: challenges and opportunities, 14th International Congress on Neuromuscular Diseases, Canada. J Neuromuscul Dis 2016;3:S1–S10. [DOI] [PubMed] [Google Scholar]
- 3.Mitsumoto H, Brooks BR, Silani V. Clinical trials in amyotrophic lateral sclerosis: why so many negative trials and how can trials be improved? Lancet Neurol 2014;13:1127–1138. [DOI] [PubMed] [Google Scholar]
- 4.Otto M, Bowser R, Turner M, et al. Roadmap and standard operating procedures for biobanking and discovery of neurochemical markers in ALS. Amyotroph Lateral Scler 2012;13:1–10. [DOI] [PubMed] [Google Scholar]
- 5.Benatar M, Boylan K, Jeromin A, et al. ALS biomarkers for therapy development: state of the field and future directions. Muscle Nerve 2016;53:169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu CH, Macdonald-Wallis C, Gray E, et al. Neurofilament light chain: a prognostic biomarker in amyotrophic lateral sclerosis. Neurology 2015;84:2247–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu CH, Petzold A, Topping J, et al. Plasma neurofilament heavy chain levels and disease progression in amyotrophic lateral sclerosis: insights from a longitudinal study. J Neurol Neurosurg Psychiatry 2015;86:565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boylan K, Yang C, Crook J, et al. Immunoreactivity of the phosphorylated axonal neurofilament H subunit (pNF-H) in blood of ALS model rodents and ALS patients: evaluation of blood pNF-H as a potential ALS biomarker. J Neurochem 2009;111:1182–1191. [DOI] [PubMed] [Google Scholar]
- 9.Shepheard SR, Chataway T, Schultz DW, Rush RA, Rogers ML. The extracellular domain of neurotrophin receptor p75 as a candidate biomarker for amyotrophic lateral sclerosis. PLoS One 2014;9:e87398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matusica D, Alfonsi F, Turner BJ, et al. Inhibition of motor neuron death in vitro and in vivo by a p75 neurotrophin receptor intracellular domain fragment. J Cell Sci 2016;129:517–530. [DOI] [PubMed] [Google Scholar]
- 11.Brooks BR, Miller RG, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 12.Kimura F, Fujimura C, Ishida S, et al. Progression rate of ALSFRS-R at time of diagnosis predicts survival time in ALS. Neurology 2006;66:265–267. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto T, Langham RG, Ronco P, Knepper MA, Thongboonkerd V. Towards Standard Protocols and Guidelines for Urine Proteomics: A Report on the Human Kidney and Urine Proteome Project (HKUPP) Symposium and Workshop, October 6, 2007, Seoul, Korea and November 1, 2007, San Francisco, CA. Proteomics; 2008;2156–2159. [DOI] [PubMed] [Google Scholar]
- 14.Rogers M-L, Atmosukarto I, Berhanu DA, Matusica D, Macardle P, Rush RA. Functional monoclonal antibodies to p75 neurotrophin receptor raised in knockout mice. J Neurosci Methods 2006;158:109–120. [DOI] [PubMed] [Google Scholar]
- 15.Hamlett A, Ryan L, Wolfinger R. On the use of PROC MIXED to estimate correlation in the presence of repeated measures. Paper 198-29 of the Proceedings of the Twenty-ninth Annual SAS Users Group International Conference, 2004. Cary, NC: SAS Institute Inc. Available at: www2.sas.com/proceedings/sugi29/198-29.pdf. Accessed December 22, 2016. [Google Scholar]
- 16.Atassi N, Berry J, Shui A, et al. The PRO-ACT database: design, initial analyses, and predictive features. Neurology 2014;83:1719–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Georgoulopoulou E, Fini N, Vinceti M, et al. The impact of clinical factors, riluzole and therapeutic interventions on ALS survival: a population based study in Modena, Italy. Amyotroph Lateral Scler Frontotemporal Degener 2013;14:338–345. [DOI] [PubMed] [Google Scholar]
- 18.Ernfors P, Henschen A, Olson L, Persson H. Expression of nerve growth factor receptor mRNA is developmentally regulated and increased after axotomy in rat spinal cord motoneurons. Neuron 1989;2:1605–1613. [DOI] [PubMed] [Google Scholar]
- 19.DiStefano PS, Johnson EM. Identification of a truncated form of the nerve growth factor receptor. Proc Natl Acad Sci USA 1988;85:270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taniuchi M, Clark HB, Johnson EM. Induction of nerve growth factor receptor in Schwann cells after axotomy. Proc Natl Acad Sci USA 1986;83:4094–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith KS, Rush RA, Rogers ML. Characterization and changes in neurotrophin receptor p75-expressing motor neurons in SOD1(G93A) G1H mice [corrected]. J Comp Neurol 2015;523:1664–1682. [DOI] [PubMed] [Google Scholar]
- 22.Seeburger J, Tarras S, Natter H, Springer J. Spinal cord motoneurons express p75NGFR and p145trkB mRNA in amyotrophic lateral sclerosis. Brain Res 1993;621:111–115. [DOI] [PubMed] [Google Scholar]
- 23.Kerkhoff H, Jennekens FGI, Troost D, Veldman H. Nerve growth factor receptor immunostaining in the spinal cord and peripheral nerves in amyotrophic lateral sclerosis. Acta Neuropathol 1991;81:649–656. [DOI] [PubMed] [Google Scholar]
- 24.Fournier C, Glass JD. Modeling the course of amyotrophic lateral sclerosis. Nat Biotechnol 2015;33:45–47. [DOI] [PubMed] [Google Scholar]
- 25.Moore DH II, Miller RG. Improving efficiency of ALS clinical trials using lead-in designs. Amyotroph Lateral Scler Other Motor Neuron Disord 2004;5(suppl 1):57–60. [DOI] [PubMed] [Google Scholar]
- 26.Gaiottino J, Norgren N, Dobson R, et al. Increased neurofilament light chain blood levels in neurodegenerative neurological diseases. PLoS One 2013;8:e75091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Norgren N, Rosengren L, Stigbrand T. Elevated neurofilament levels in neurological diseases. Brain Res 2003;987:25–31. [DOI] [PubMed] [Google Scholar]
- 28.Reijn TS, Abdo WF, Schelhaas HJ, Verbeek MM. CSF neurofilament protein analysis in the differential diagnosis of ALS. J Neurol 2009;256:615–619. [DOI] [PubMed] [Google Scholar]
- 29.Rosengren LE, Karlsson JE, Karlsson JO, Persson LI, Wikkelso C. Patients with amyotrophic lateral sclerosis and other neurodegenerative diseases have increased levels of neurofilament protein in CSF. J Neurochem 1996;67:2013–2018. [DOI] [PubMed] [Google Scholar]
- 30.Tortelli R, Copetti M, Ruggieri M, et al. Cerebrospinal fluid neurofilament light chain levels: marker of progression to generalized amyotrophic lateral sclerosis. Eur J Neurol 2015;22:215–218. [DOI] [PubMed] [Google Scholar]
- 31.Tortelli R, Ruggieri M, Cortese R, et al. Elevated cerebrospinal fluid neurofilament light levels in patients with amyotrophic lateral sclerosis: a possible marker of disease severity and progression. Eur J Neurol 2012;19:1561–1567. [DOI] [PubMed] [Google Scholar]
- 32.Zetterberg H, Jacobsson J, Rosengren L, Blennow K, Andersen PM. Cerebrospinal fluid neurofilament light levels in amyotrophic lateral sclerosis: impact of SOD1 genotype. Eur J Neurol 2007;14:1329–1333. [DOI] [PubMed] [Google Scholar]
- 33.Boylan KB, Glass JD, Crook JE, et al. Phosphorylated neurofilament heavy subunit (pNF-H) in peripheral blood and CSF as a potential prognostic biomarker in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2013;84:467–472. [DOI] [PubMed] [Google Scholar]
- 34.Brettschneider J, Petzold A, Süssmuth SD, Ludolph AC, Tumani H. Axonal damage markers in cerebrospinal fluid are increased in ALS. Neurology 2006;66:852–856. [DOI] [PubMed] [Google Scholar]
- 35.Levine TD, Bowser R, Hank NC, et al. A pilot trial of pioglitazone HCl and tretinoin in ALS: cerebrospinal fluid biomarkers to monitor drug efficacy and predict rate of disease progression. Neurol Res Int 2012;2012:582075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ganesalingam J, An J, Bowser R, Andersen PM, Shaw CE. pNfH is a promising biomarker for ALS. Amyotroph Lateral Scler Frontotemporal Degener 2013;14:146–149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.