

Abstract
One of the most common analysis tasks in genomic research is to identify genes that are differentially expressed (DE) between experimental conditions. Empirical Bayes (EB) statistical tests using moderated genewise variances have been very effective for this purpose, especially when the number of biological replicate samples is small. The EB procedures can however be heavily influenced by a small number of genes with very large or very small variances. This article improves the differential expression tests by robustifying the hyperparameter estimation procedure. The robust procedure has the effect of decreasing the informativeness of the prior distribution for outlier genes while increasing its informativeness for other genes. This effect has the double benefit of reducing the chance that hypervariable genes will be spuriously identified as DE while increasing statistical power for the main body of genes. The robust EB algorithm is fast and numerically stable. The procedure allows exact small-sample null distributions for the test statistics and reduces exactly to the original EB procedure when no outlier genes are present. Simulations show that the robustified tests have similar performance to the original tests in the absence of outlier genes but have greater power and robustness when outliers are present. The article includes case studies for which the robust method correctly identifies and downweights genes associated with hidden covariates and detects more genes likely to be scientifically relevant to the experimental conditions. The new procedure is implemented in the limma software package freely available from the Bioconductor repository.
Keywords and phrases: Empirical Bayes, outliers, robustness, gene expression, microarrays, RNA-seq
1. Introduction
Modern genomic technologies such as microarrays and RNA sequencing have made it routine for biological researchers to measure gene expression on a genome-wide scale. Researchers are able to measure the expression level of every gene in the genome in any set of cells chosen for study under specified treatment conditions. This article focuses on one of the most common analysis tasks, which is to identify genes that are differentially expressed (DE) between experimental conditions.
Gene expression experiments pose statistical challenges because the data are of extremely high dimension while the number of independent replicates of each treatment condition is often very small. Simply applying univariate statistical methods to each gene in succession can produce imprecise results because of the small sample sizes. Substantial gains in performance can be achieved by leveraging information from the entire dataset when making inference about each individual gene.
Empirical Bayes (EB) is a statistical technique that is able to borrow information in this way (Efron and Morris, 1973; Morris, 1983; Casella, 1985). EB has been applied very successfully in gene expression analyses to moderate the genewise variance estimators (Baldi and Long, 2001; Wright and Simon, 2003; Smyth, 2004). These articles assume a conjugate gamma prior for the genewise variances and produce posterior variance estimates that are a compromise between a global variance estimate and individual genewise variance estimates. The posterior variance estimators can be substituted in place of the classical estimators into linear model t-statistics and F-statistics. Wright and Simon (2003) and Smyth (2004) derived exact small sample distributions for the resulting moderated test statistics. They showed that the EB statistics follow classical t and F distributions under the null hypothesis but with augmented degrees of freedom. The additional degrees of freedom of the EB statistics relative to classical statistics represent the information that is indirectly borrowed from other genes when making inference about each individual gene.
EB assumes a Bayesian hierarchical model for the genewise variances but, instead of basing the prior distribution on prior knowledge as a Bayesian procedure would do, the prior distribution is estimated from the marginal distribution of the observed data. Smyth (2004) developed closed-form estimators for the parameters of the prior distribution from the marginal distribution of the residual sample variances. This procedure is implemented in the limma software package (Ritchie et al., 2015) and the resulting EB tests have been shown to offer improved statistical power and false discovery rate (FDR) control relative to the ordinary genewise t-tests, especially when the sample sizes are small (Kooperberg et al., 2005; Murie et al., 2009; Ji and Liu, 2010; Jeanmougin et al., 2010). The limma software has been used successfully in thousands of published biological studies using data from a variety of genomic technologies, especially studies using expression microarrays and RNA-seq.
This article improves the limma EB differential expression tests by robustifying the hyperparameter estimation procedure. As in the original method, we fit genewise linear models to the log-expression values and extract residual variances, but now we give special attention to residual variances that are exceptionally large or exceptionally small. Genes corresponding to extreme variances will be considered ‘outliers’. Following terminology used in the genomics literature, we refer to outlier genes with large variances as ‘hypervariable genes’. We show that, for certain genomic datasets, a small number of outlier genes can have an undesirable influence on the hyperparameter estimators, decreasing the effectiveness of the EB differential expression procedures. (Figure 1 plots residual standard deviations for three datasets. In each study there are special biological factors that cause a subset of the genes to have larger than expected residual variances.) We show that in such cases the effectiveness of EB can be restored by a robust approach that isolates the outlier genes. We develop a robust estimation scheme with a positive breakdown point for the hyperparameters and incorporate this into the differential expression procedure. The robust EB procedure has the effect of decreasing the informativeness of the prior distribution for hypervariable genes while increasing its informativeness for other genes. This effect has the double benefit of reducing the chance that hypervariable genes will be spuriously identified as DE while increasing statistical power for the main body of genes.
Fig 1.
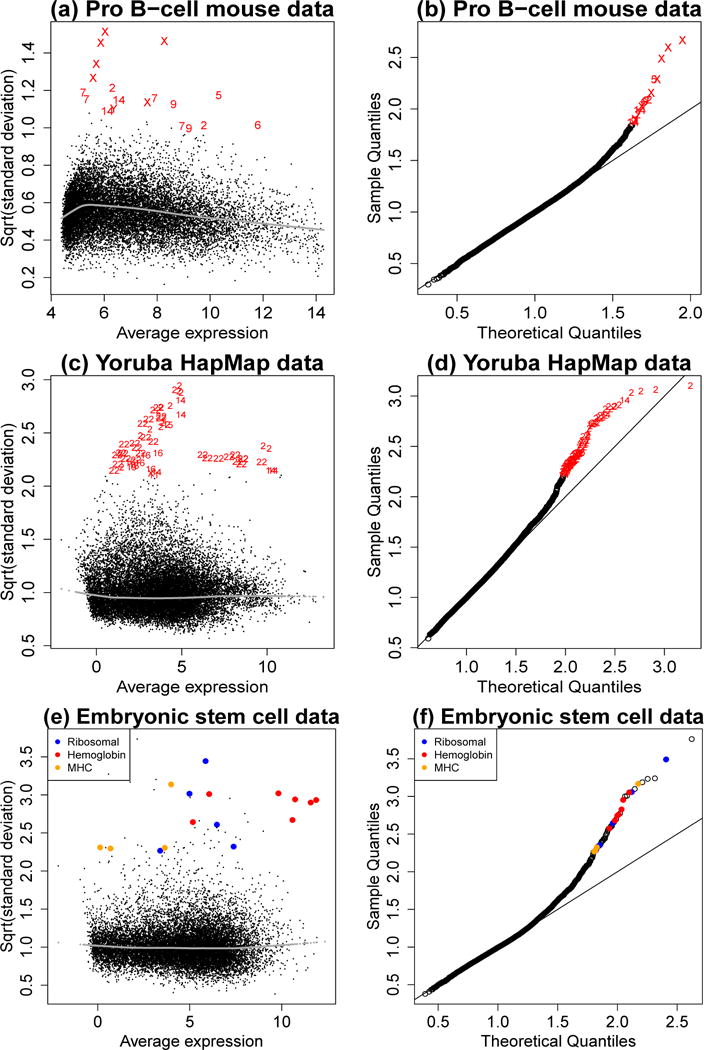
Hypervariable genes for the three case study datasets. Left panels plot genewise residual standard deviations vs average log2-expression. Right panels show probability plots of the standard deviations after transforming to equivalent normal deviates. Hypervariable genes are marked by chromosome number in panels a–d and by gene function in panels e–f. For each dataset the hypervariable genes have clear biological interpretations.
Our robust EB approach uses more diffuse prior distributions for the variances of hypervariable genes. A conjugate prior is still used for each gene and this allows us to preserve a key feature of the original EB differential expression procedures, which is the ability to derive exact small-sample null distributions for the test statistics. Our robust EB approach is fast and numerically stable without difficult convergence issues. It reduces exactly to the original EB procedure when there are no hypervariable genes. Simulation studies show that the robustified tests for differential expression have similar performance to the original tests in the absence of outlier genes but have greater power and robustness when they are present.
To the best of our knowledge there has been no previous work on robust EB for variances. Most robust EB schemes in other contexts have been based on heavy-tailed prior distributions. We have avoided such an approach because crucial advantages of the original DE procedures would thereby be lost, in particular the posterior mean variance estimators would no longer be available in closed form and the test statistics would no longer yield exact p-values. Efron and Morris (1972) proposed limited translation rules when estimating means of standard normal distributions. This proposal originated the idea of limited learning for extreme cases, but with the aim of limiting the bias in extreme cases rather than improving estimation of the hyperparameters. Gaver and O’Muircheartaigh (1987) analyzed a Poisson process using a heavy-tailed (log-Student) prior distribution for the Poisson mean. This approach achieves insensitivity to outliers but loses efficiency as well as being less mathematically tractable. Liao, McMurry and Berg (2014) estimated log-fold expression changes and achieved robustness with respect to misspecified working priors by conditioning on the rank of each estimated log-fold-change rather than on the actual observation. Again this is less mathematically tractable than our approach.
Our robustified EB procedure has been implemented in the limma software package and can be invoked by the option robust=TRUE in calls to the eBayes or treat (McCarthy and Smyth, 2009) functions. Invoking the option requires no other changes to the analysis pipelines from a user point of view. All downstream functions recognize and work with robust EB results as appropriate.
The following sections review the EB approach to differential expression, then derive the robust estimators and modified differential expression scheme. We evaluate the performance of the robustified procedure using simulations, then present three case studies in which the EB approach identifies genetic instabilities specific to each study. We give a detailed analysis of a microarray study of PRC2 function in pro-B cells for which a gender effect produces hypervariable genes. The EB procedure is effective at downweighting sex-linked genes associated with the unwanted covariate in favor of genes of more scientific interest. Finally we discuss possible generalizations of our robust EB approach to other contexts.
2. Linear models and moderated t-statistics
Consider a genomic experiment in which the expression levels of G genes are measured for n RNA samples. We follow the notation and linear model formulation introduced by Smyth (2004). Write ygi for the log-expression level of gene g in sample i. The log-expression values satisfy genewise linear models
where yg is the column vector (yg1,…, ygn)T, X is an n × p design matrix of full column rank representing the experimental design and βg is an unknown coefficient vector that parametrizes the average expression levels in each experimental condition. For each gene, the ygi are assumed independent with
where is an unknown variance and the wgi are known weights. The least squares coefficient estimator is
where Wg is the diagonal matrix with elements wg1,…, wgn. The residual sample variances are
where and dg is the residual degrees of freedom. Usually dg = n − p, but genes with missing y values or zero weights may have smaller values for dg. Conditional on , is assumed to follow a chisquare distribution with dg degrees of freedom, an assumption we write as
We assume a conjugate prior distribution for in order to stabilize the genewise estimators. The are assumed to be sampled from a scaled inverse chi-square prior distribution with degrees of freedom d0 and location ,
It follows that the posterior distribution of given is scaled inverse chi-square,
and the posterior expectation of given is with
The are the EB moderated variance estimators. The moderated t-statistic for a given coefficient βig is
where vi is the ith diagonal element of (XTWgX)−1. If the null hypothesis βgj = 0 is true, then follows a t-distribution on dg +d0 degrees of freedom (Smyth, 2004). In general, any ordinary genewise t or F-statistic derived from the linear model can be converted into an EB moderated statistic by substituting for , in which case the denominator degrees of freedom for the null distribution increase from dg to d0 + dg.
3. Robust hyperparameter estimation
Under the above hierarchical model, follows a scaled F-distribution on dg and d0 degrees of freedom,
The log-variances log follow Fisher’s z-distribution, which is roughly symmetric and has finite moments of all orders. The limma package estimates the hyperparameters and d0 by matching the theoretical mean and variance of the z-distribution to the observed sample mean and variance of the zg. The empirical estimates and d0 are then substituted into the above formulas to obtain and to conduct genewise statistical tests for differential expression.
As the observed variance of the log increases, the estimated value of d0 decreases, meaning that less information is borrowed from the prior to form the moderated t-statistics. In the examples shown in Figure 1, the variance of log would be much reduced if a small number of the most variable genes were excluded. We therefore seek to replace limma’s moment estimation scheme for the hyperparameters with a robust version.
Our approach is to apply moment estimation to the Winsorized sample variances. The idea of Winsorizing is to reset a specified proportion of the most extreme sample variances to less extreme values (Tukey, 1962). Let pu and pl be the maximum proportion of outliers allowed in the upper and lower tails of the respectively. Typical values are pl = 0.05 and pu = 0.1, although any values strictly between 0 and 0.5 are permissable. Let ql and qu be the corresponding quantiles of the empirical distribution of , so that pl of the variances are less than or equal to ql and pu are greater than or equal to qu. The empirical Winsorizing transformation is defined by
Write zg = log win for log-transformed Winsorized variances, and let and be the mean and variance of the observed values of zg.
Define the Winsorized F-distribution as follows. If then the Winsorized random variable is
where now ql and qu are the lower tail pl and upper tail pu quantiles of the distribution.
Write ν(dg, d0) and ϕ (dg, d0) for the expected value and variance of log win(f). An efficient and accurate algorithm for computing ν and ϕ using Gaussian quadrature is described in Section 10.
Assuming that the dg are all equal, the hyperparameter d0 is estimated by equating and solving for d0 using a modified Newton algorithm (Brent, 1973). Having estimated d0, the logarithm of the parameter is estimated by .
If the dg are not all equal, then the are transformed to equivalent random variables with equal dg before applying the above algorithm. Details of this transformation are given in Section 10.
4. Gene-specific prior degrees of freedom
Having estimated d0 and s0 robustly, we can identify genes that are outliers in that their variances are too large to have reasonably arisen from the estimated prior. The question naturally arises as to how to handle such outliers. One reasonable approach would be to ignore the prior information for such genes, on the basis that the prior appears to be inappropriate. This approach would assign d0 = 0 for such genes, meaning that ordinary t-tests would be used for these genes instead of EB moderated statistics. On the other hand, the prior should in principle still have some limited relevance even for the outliers. Our approach is to assign gene-specific prior degrees of freedom, d0g, whereby d0g = d0 for non-outlier genes but outlier genes are assigned smaller values depending on how extreme the outlier is. In effect we assume that each hypervariable gene g has a true variance sampled from with 0 < d0g < d0.
We start by identifying a lower bound for the d0g from the largest observed value. Specifically we find doutlier such that the maximum is equal to the median of the distribution. A fast stable numerical algorithm for finding doutlier is given in Section 10.
Next we evaluate the posterior probability that each gene is a hypervariable outlier. Let pg be the p-value for testing whether gene g is a outlier, defined by where . Let π0 be the prior probability that gene g is not an outlier and let rg be the marginal probability of observing a residual variance more extreme than . The posterior probability given that case g is not an outlier is πg = pgπ0/rg. Assuming that most genes are not outliers, we conservatively set π0 = 1. The marginal probability rq can be estimated empirically from the rank of amongst all the observed values of , i.e., by rg = (r − 0.5)/G where r is the rank of Substituting these values in the above formula yields a conservative estimate πg = pg/rg.
The initial estimate of πg is not necessarily monotonic in or pg. We ensure that πg is a non-decreasing function of pg in the following manner. First the cases are ordered in increasing order of pg. Then the cumulative mean is computed for each g. Let gm be the first value of g for which achieves its minimum. All πg for g = 1,…, gm are set to the minimum value of , to allow for the possibility that πg might be small for a group of cases but not for the most extreme case. Finally, a cumulative maximum filter is applied to the πg, after which the πg are non-decreasing. In practice, this process is nearly identical to using isotonic regression (Barlow et al., 1972) to enforce monotonicity.
Finally the genewise prior degrees of freedom are defined by
This process ensures that d0g = d0 for most genes, but any gene that is a clear outlier with a very small pg value will be assigned a much lower value.
5. Covariate dependent priors
In gene expression experiments, the variance of the log-expression values often depends partly on the magnitude of the expression level (Sartor et al., 2006; Law et al., 2014). It is therefore helpful to extend the EB principle to permit the prior variance to depend on the average log-expression Ag of each gene. This extension generalizes the prior distribution for to be gene-specific:
where varies smoothly with Ag. In other words, the prior distribution depends on the covariate Ag. Such a strategy is implemented in the limma package (Ritchie et al., 2015).
Our strategy for robust EB with a variance trend is as follows. First we fit a robust lowess trend (Cleveland, 1979) to log as a function of Ag We detrend the log by subtracting the fitted lowess curve, then apply the robust EB algorithm described above to the detrended variances. The final genewise prior values are the product of the unlogged lowess trend and the estimated from the detrended variances.
6. Software implementation
The robust hyperparameter estimation strategy described above is implemented in the limma function fitFDist-Robustly. The tail proportions for Winsorizing are user settable with defaults pl = 0.05 and pu = 0.1. This function is called by squeezeVar, which computes EB moderated variances. squeezeVar in turn is called by user-level functions including eBayes and treat in the limma package and estimateDisp and glmQLFit in the edgeR package (Robinson, McCarthy and Smyth, 2010). These functions integrate the robust EB strategy into analysis pipelines for gene expression microarrays and RNA-seq. glmQLFit is also called in analysis pipelines of the csaw and diffHic packages for the analysis of ChIP-seq and Hi-C sequencing data (Lun and Smyth, 2016, 2015a).
7. Evaluation using simulated data
Simulations were used to evaluate the performance of the robust hyperparameter estimators. Expression values were generated for 10,000 genes and 6 RNA samples. The RNA samples were assumed to belong to two groups, with three in each group, leading to a linear model with dg = 4 residual degrees of freedom. Genewise variances and expression values were generated according to the hierarchical model of Section 2 with s0 = 0.2 and with d0 = 2, 4 or 10. Both the standard and robust hyperparameter estimators were found to be accurate in the absence of outliers (Figure 2a, c). When 250 hypervariable genes were included, however, the robust estimators were considerably more accurate than the standard (Figure 2b, d). The hypervariable genes were simulated to have d0g = 0.5. Next we checked type I error rates for the EB t-tests in the absence of outliers. Both standard and robust tests were found to control the error rate correctly (Table 1). In all simulations, the tail proportions pl and pu were at their default values.
Fig 2.
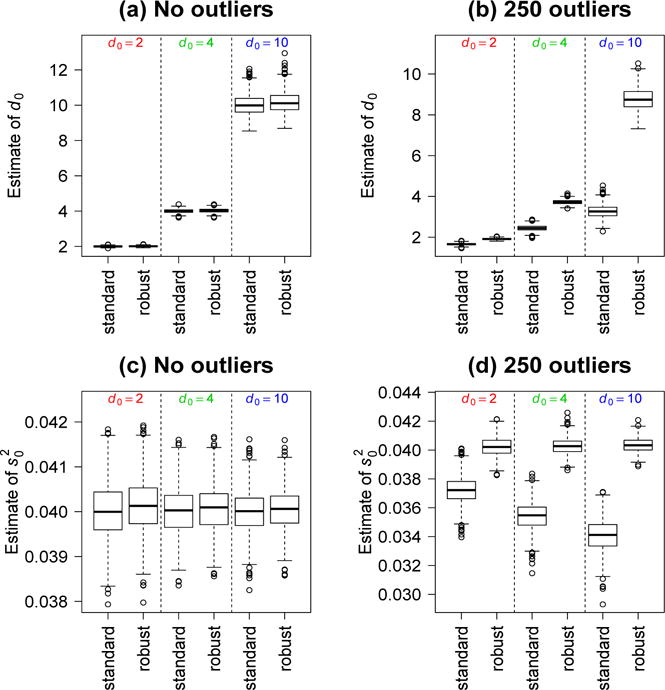
Boxplots of standard and robust hyperparameter estimates from 1000 simulated datasets. True values are 0.4 for and either 2, 4 or 10 for d0. Panels a, c show estimates when no outliers are present. Panels b, d show estimates when the data includes 250 hypervariable genes.
Table 1.
Type I error rates for standard and robust EB t-tests. Datasets were simulated with different d0 values but with no DE genes and or outliers. The table gives the mean error rate over all genes in 1,000 simulated datasets for various p-value cutoffs.
| d0 | Method | Nominal error rate
|
|||
|---|---|---|---|---|---|
| 0.001 | 0.01 | 0.05 | 0.1 | ||
| 2 | Standard | 0.000996 | 0.00998 | 0.05001 | 0.09994 |
| Robust | 0.000996 | 0.00998 | 0.04996 | 0.09983 | |
| 4 | Standard | 0.001008 | 0.01002 | 0.05008 | 0.10012 |
| Robust | 0.001013 | 0.01004 | 0.05006 | 0.10005 | |
| 10 | Standard | 0.001017 | 0.01005 | 0.05018 | 0.10008 |
| Robust | 0.001033 | 0.01010 | 0.05021 | 0.10005 | |
Finally we evaluated statistical power and FDR control. Each simulated dataset now included 500 DE genes, with log fold changes generated from a N(0, 4) distribution. In the absence of hypervariable genes, the standard and robust EB tests were indistinguishable in terms of false discoveries or power (Figure 3a,c). In the presence of 250 outliers, the robust tests consistently gave fewer false discoveries (Figure 3b) and higher power (Figure 3d). As expected, the improvement achieved by the robustified tests was greater for larger values of d0. For simplicity of interpretation, no genes were both DE and hypervariable in these simulations.
Fig 3.
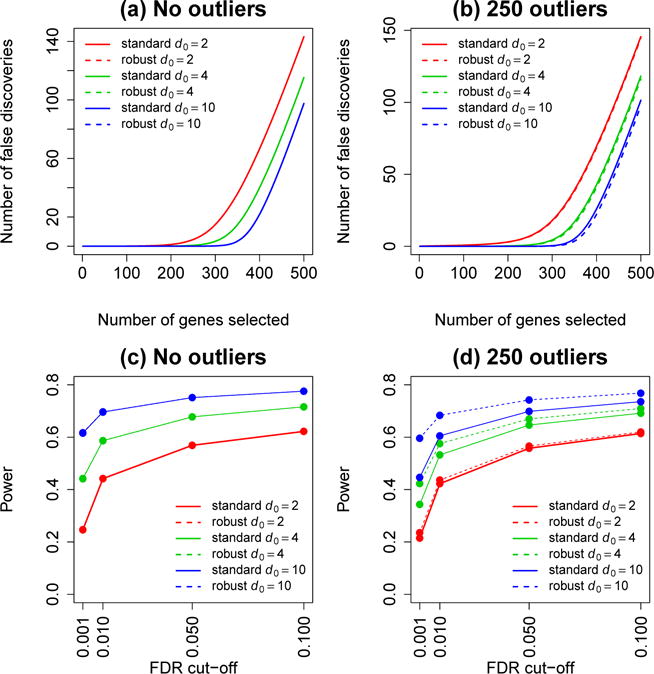
Detection of differential expression. Panels a–b show the number of false discoveries amongt the 500 top-ranked genes. Panels c–d show power, the proportion of truly DE genes selected as significant at various FDR cutoffs. Results are averaged over 1000 simulations. Simulations b and d include 250 hypervariable genes. Results are shown for both standard and robust EB tests and for three values of the prior degrees of freedom.
8. Case studies
8.1. Loss of polycomb repressor complex 2 function in pro B cells
Polycomb group proteins are transcriptional repressors that play a central role in the establishment and maintenance of gene expression patterns during development. Suz12 is a core component of Polycomb Repressive Complex 2 (PRC2). Majewski et al. (2008, 2010) studied mice with a mutation in the Suz12 gene that results in loss of function of the Suz12 protein and hence PRC2. They profiled gene expression in hematopoietic stem cells from these mice. Here we describe a gene expression study of a different hematopoietic cell type from the same Suz12 mutant mice strain. This study profiles gene expression in pro-B cells, an early progenitor immune cell intermediate in a series of development stages between hematopoietic stem cells and mature B-cells.
Our interest is to study development, so cells were isolated from 16-day embryonic mice. For this study, RNA was extracted from foetal pro B cells that were isolated from the liver of four wild-type mice and four Suz12 mutant mice. RNA was hybridized at the Australian Genome Research Facility to Illumina Mouse Whole-Genome-6 version 2 BeadChips, a microarray platform containing about 48,000 60-mer DNA sequences probing most genes in the genome. Intensities were background corrected, quantile normalized and transformed to the log2-scale using the neqc function (Shi, Oshlack and Smyth, 2010). One of the Suz12 mutant samples was discarded because it clustered with the wildtype instead of the Suz12 samples, leaving four wild-type and three Suz12 mutant samples. Probes were filtered from further analysis if they failed to achieve a detection p-value of less than 0.01 in at least two of the remaining samples, leaving 14,084 probes for analysis.
Linear modeling was applied to the normalized log-expression values, resulting in a residual sample variance on 5 degrees of freedom for each probe. Figure 1a shows the residual standard deviation plotted against the average log intensity for each probe. The gray curve shows the estimated trend for the prior variance. The robust algorithm identified a number of outlier variances (Figure 1b). The non-robust estimate of the prior degrees of freedom was 11.9. The robust algorithm estimated prior degrees of freedom 14.1 for most genes, but with prior degrees of freedom as low as 0.5 for the outlier variances (Figure 4a).
Fig 4.
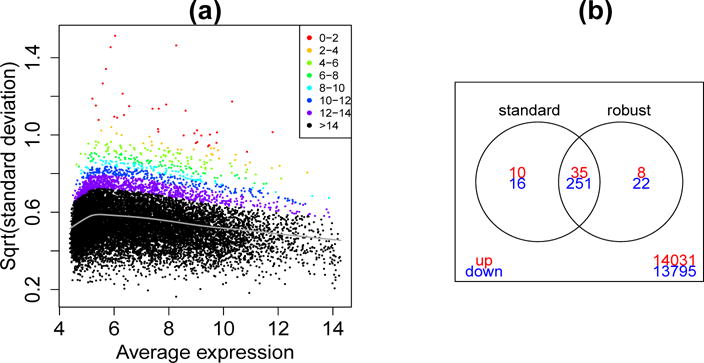
(a) Square-root standard deviation plotted against average log intensity for each probe. The gray line shows the trended estimate of the prior variance. Points are colored by the estimated prior degrees of freedom: probes with larger sample variances have smaller df. (b) Venn diagram showing overlap the numbers of significant genes for Suz12 versus wildtype using the standard and robust methods.
Further examination showed that many of the probes identified as outliers corresponded to genes known to have sex-linked expression, including many on the X or Y chromosomes (Figure 1ab). The most outlying variances corresponded to Y chromosome genes Erdr1 and Eif2s3y up-regulated in males, and X chromosome gene Xist, known to be up-regulated in females. Other outliers gene were ribosomal genes Rn18s and Rpl7a, suggesting ribosomal RNA retention in one or more samples, and hemoglobin genes Hbb-y and Hbb-b1 suggesting red blood or bone marrow content in some tissue samples. None of these genes should be related to the Suz12 mutation.
Differential expression between the Suz12 mutants and the wildtype mice was assessed using EB moderated t-statistics. P-values were adjusted to control the false discovery rate at less than 5% (Benjamini and Hochberg, 1995). The standard and robust procedures found 251 down-regulated and 35 up-regulated probes in common (Figure 4b). However 22 and 16 down-regulated genes were found only by the robust or standard procedures respectively. The non-robust unique genes tended to be sex-linked or hemoglobin related (Xist, Apoa2, Hbb-b1 etc) whereas the robust unique genes were related to programmed cell death (Bcl2l1), cell cycle (Ccne2) or chromatin remodeling (Myst2). For up-regulated genes, 8 and 10 unique probes were found by the robust and non-robust procedures respectively. The non-robust unique genes tended to be Y chromosome sex-linked genes (Ddx2y, Erdr1 etc) whereas the robust unique genes appeared related to the PRC2 process of interest.
Investigation after the analysis confirmed that two of the Suz12 mutant embryos were in fact female, whereas all the other mice were male. This sex imbalance was an unwanted complication in the experiment, difficult to avoid without sex-typing of the embryo mice at the time of tissue collection. The results show that the robust EB method was successful in identifying and downweighting genes that are associated with the hidden covariate. The robust procedure results in more statistical power to detect other genes that are more likely to be of scientific significance.
8.2. RNA-seq profiles of Yoruba HapMap individuals
As part of the International HapMap project, RNA-Seq profiles were made of cell lines derived from B lymphocytes from 69 different Yoruba individuals from Ibadan, Nigeria (Pickrell et al., 2010a,b). Genewise read counts were obtained from the tweeDEseqCountData package version 1.8.0 from Bioconductor and were transformed to log2-counts per million with precision weights using voom (Law et al., 2014). The analysis compares males to females. Figure 1c shows the genewise standard deviations and Figure 1d gives a probability plot of the standard deviations against the fitted distribution. The hypervariable genes were identified as B cell receptor segments on chromosomes 2 and 22. B cells contain a random selection of these segments in order to generate a repertoire of antigen binding sites. The robust analysis reveals that the cell lines were clonal, each cell line apparently derived from a single B cell or from a very small number of original cells. It would be appropriate to remove the receptor segments from the RNA-seq analysis, and the robust EB procedure effectively achieves that.
8.3. Embryonic stem cells
Sheikh et al. (2015) used RNA-seq to profile embryonic stem cells. Genewise read counts were transformed using voom. Figure 1e shows the genewise standard deviations and Figure 1f gives a probability plot of the standard deviations against the fitted distribution. The hyper-variable genes are predominately associated with the ribosome or with hemoglobin, suggesting inconsistenciees in cell purification and RNA processing. Other hyper-variable genes are located in the major histocompatibility complex, known to be one of the most variable parts of the genome.
9. Discussion
In recent years we have routinely checked for hyper-variable genes in gene expression studies. We have found that many studies harbor a subset of outlier genes. In many cases, the identities of the hyper-variable genes suggest a mechanism for their variability. We have analyzed studies, for example, where the hypervariable genes are enriched for sex-linked genes, for ribosomal genes, for mitochondrial genes, or for B cell receptor segments. In other cases, hypervariable genes may be associated with a particular cell type suggesting inconsistent cell population proportions in the different biological samples. In some cases the reasons why some genes are highly variable between individuals are unknown. The phenomenon is common enough to be viewed as an unavoidable part of cutting edge genomic research rather than a result of flawed experimental procedures. In most cases the studies are overall high quality.
Hypovariable genes can also arise, although usually for technical rather than biological reasons. Quantile normalization (Bolstad et al., 2003) of expression data can occasionally produce expression values that are numerically identical for all samples for a given gene when the number of samples is small. Sequencing data can also give rise to variances that are zero or very small because of the discreteness of read counts.
This article describes a robustified version of EB differential expression analysis. This procedure protects against hyper and hypovariable genes in the sense that it allows non-outlier genes to share information amongst themselves as if the outlier genes were not present. In many cases, this results in a gain in statistical power for the non-outlier genes. Hypervariable genes are not removed from the analysis but instead borrow less information from the ensemble and are assigned test statistics closer to ordinary t-statistics. Hypovariable genes on the other hand are moderated as for non-outlier genes—this increases their posterior variances closer to typical values.
A key feature of our procedure is that a conjugate Bayesian model is used for each gene, enabling closed-form posterior estimators and exact small-sample p-values. Robustness is achieved by fitting the prior distribution to non-outlier genes and by assigning lower degrees of freedom to the hyper-variable outliers. We have proposed a practical algorithm for assigning prior degrees of freedom to outlier genes. In practice, the list of DE genes is not sensitive to the exact values of the prior degrees of freedom, provided that d0g = d0 for non-outliers and d0g is substantially smaller for clear outliers. For many datasets the list of DE genes is unchanged for a range of reasonable values for doutlier.
The default values for the Winsorizing tail proportions pl and pu work well in our practical experience. Users however may choose to increase the default values for datasets where high proportions of outlier genes are expected.
Simulations show that the robustified EB procedure estimates the hyperparameters equally as accurately as the original method in the absence of outliers. When outliers are present, however, the robustified EB procedure was able to simultaneously increase power and decrease the false discovery rate when assessing differential expression.
The robust EB method developed here has been applied not only to microarray data, but also to data from RNA-seq (Good-Jacobson et al., 2014), ChIP-seq (Lun and Smyth, 2016, 2015b) and Hi-C (Lun and Smyth, 2015a) technologies. With microarrays, the linear models are fitted to normalized log-intensities. With RNA-seq, the number of sequence reads overlapping each gene can be counted and the EB models can be applied to the log-counts-per million (Law et al., 2014). Alternatively, the robust EB method can be applied to count data by way of quasi-generalized linear models. Lun, Chen and Smyth (2016) analyzed RNA-seq read counts used quasi-negative-binomial generalized linear models. The limma robust EB variance estimation method was applied to the genewise residual deviances leading to the construction of EB quasi-F-tests. The robust EB method has also been used to estimate the prior degrees of freedom for the weighted likelihood approach used in the edgeR package, again using residual deviances (Chen, Lun and Smyth, 2014).
In this article we view genes as outliers rather than individual expression values as outliers. An alternative robustifying approach would be to replace least squares estimation of the genewise linear models with robust regression (Gottardo et al., 2006; Zhou, Lindsay and Robinson, 2014), and the limma package has included an M-estimation option for this purpose for over a decade. In principle, the two approaches are complementary and both can be used simultaneously, i.e., one could apply the robust EB procedure of this article to variances estimated by robust regression. The robust regression approach assumes that the expression values for a gene contain one or two outliers that are “errors” whereas the other values are “correct”. Our experience suggests that this scenario is relatively rare for gene expression data. The outlier-gene approach of this article allows a more general context in which a hypervariable gene may produce an arbitrary number of inconsistent expression values that cannot be meaningfully categorized into correct and incorrect. The robust regression is only applicable to experimental designs with at least three expression values per experimental group, whereas the outlier-gene approach can be usefully applied to any experimental design, even down to studies with a single residual degree of freedom.
The robust EB strategy of this article could in principle be applied in other EB contexts. The basic idea is to estimate hyperparameters robustly, then to test for outlier cases, and finally to assign a more diffuse prior to outlier cases. This approach may be particularly attractive for use with conjugate Bayesian models and seems different from previous robust EB strategies.
It is interesting to contrast our approach with the large literature on robust Bayesian analysis (Berger, 1984, 1990; Insua and Ruggeri, 2000). Robust Bayesian analysis considers a class of possible prior distributions and tries to limit or at least quantify the range of posterior conclusions as the prior ranges over the class. The issues that concern us in this article are different in a number of important ways. The issues that we address are specific to empirical Bayes and do not arise in true Bayesian frameworks for which hyperparameters do not need to be estimated. Our aim is not to limit the influence of the prior but to increase it for the majority of genes. Our method applies only to large scale data with many cases (probes, genes or genomic regions) whereas robust bayesian analysis is typically concerned with individual cases.
Acknowledgments
This work was supported by The University of Melbourne (PhD scholarship to BP), by the National Health and Medical Research Council (Fellowship 1058892, Program Grant 1054618, Independent Research Institutes Infrastructure Support Scheme), and by a Victorian State Government OIS grant.
10. Appendix: Details of computational algorithms
10.1. Standard (non-robust) moment estimation of the hyperparameters
In order to calculate the EB moderated t-statistics, the hyperparameters d0 and need to be estimated. Here we summarize the original (non-robust) methods-of-moments strategy for estimating the hyperparameters proposed by Smyth (2004). The marginal distribution of the gene-wise residual variances is . It is convenient to work with the logged variances, defined by , as these have finite moments and a roughly normal distribution for which the methods of moments should be efficient. The theoretical mean and variance of zg are available as
and
where ψ and ψ′ represent the digamma and trigamma functions respectively. Write and for the observed mean and variance of the zg. These have theoretical expections
and
The equation for can be solved for d0, then that for can be solved for .
Alternatively a abundance-dependent trend can be put on s0. In that case, a regression spline with 4 df is used to model zg − ψ(dg/2) + log(dg/2) a function of average log2-expression, , and the fitted values from the regression are used in place of .
10.2. Tranforming samples variances to equal residual degrees of freedom
The ordinary non-robust moment estimators for the hyperparemeters do not require the residual degrees of freedom to be equal for all genes, but the robust estimation scheme does require this. In practice, the dg are usually equal, but occasionally some genes may have reduced dg because of missing expression values for some samples. If this is the case, we transform the sample variances so they can be treated as being on the same degrees of freedom. Let d be the maximum dg. First the hyperparameters d0 and are estimated by the non-robust algorithm. Then the are transformed to where denotes here the cumulative distribution function of the F-distribution on k1 and k2 degrees of freedom.
10.3. Computing Winsorized moments
Here we describe the computation of the mean ν(dg, d0) and variance ϕ(dg, d0) of the log Winsorized F-distribution. Let Z = log win(f) denote the log-Winsorized F random variable defined in Section 3 of the main paper. The expected value is
with plu = (1 − pl − pu). After transforming Z to the unit interval, the conditional expectation can be re-interpretted as
where U is uniformly distributed on the interval from a = ql/(1 + ql) to b = qu/(1 + qu) and
where pdf is the probability density function of the F-distribution on dg and d0 degrees of freedom. The expectation in terms of the uniform random variable can be evaluated efficiently by Gauss-Legendre quadrature, as described in the next section.
Having computed the mean, the variance of Z can be obtained as
where ν = ν(dg, d0). The second term containing the conditional expectation can be re-interpretted as (b − a) E{h(U)} with
to permit evaluation by Gauss-Legendre quadrature.
10.4. Evaluating an integral using Gaussian quadrature
To compute the Winzorized moments, we need E{h(U)} where U is uniformly distributed on interval [a, b]. We compute this expectation using Gauss-Legendre quadrature. Specifically, we use the function gauss.quad.prob from the statmod package (Smyth, 2015). This function computes Gauss quadrature weights and nodes using an adaption of the algorithm and Fortran code published by Golub and Welsch (1969). For any desired order k, nodes ui and weights wi can be computed such that
The approximation is exact if h(u) can be expressed as a polynomial of order 2k − 1 or less on [a, b]. The accuracy of Gauss-Legendre quadration is excellent if h(u) is a reasonably smooth function taking finite values on the interval.
All results reported in the article use k = 128 nodes. This is sufficient for close to double-precision accuracy provided a is bounded above zero and b is bounded below one.
10.5. Solving for doutlier
Let be the maximum of over all genes g. We wish to find doutlier such that , where is the right tail probability of the F distribution with dg and d0 degrees of freedom. Initializing doutlier = d0 and then repeating
converges monotonically to the required value. Sufficient accuracy is achieved after two or three iterations.
References
- Baldi P, Long AD. A Bayesian framework for the analysis of microarray expression data: regularized t-test and statistical inferences of gene changes. Bioinformatics. 2001;17:509–519. doi: 10.1093/bioinformatics/17.6.509. [DOI] [PubMed] [Google Scholar]
- Barlow RE, Bartholomew DJ, Bremner JM, Brunk HD. Statistical Inference under Order Restrictions: Theory and Application of Isotonic Regression. Wiley; London: 1972. [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B. 1995;57:289–300. [Google Scholar]
- Berger JO. The robust Bayesian viewpoint. In: Kadane J, editor. Robustness of Bayesian Analysis. Amsterdam: North-Holland; 1984. pp. 63–124. [Google Scholar]
- Berger JO. Robust Bayesian analysis: sensitivity to the prior. Journal of Statistical Planning and Inference. 1990;25:303–328. [Google Scholar]
- Bolstad BM, Irizarry RA, Åstrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Brent RP. Algorithms for minimization without derivatives. Prentice-Hall; Englewood Cliffs, N.J: 1973. [Google Scholar]
- Casella G. An introduction to empirical Bayes data analysis. American Statistician. 1985;39:83–87. [Google Scholar]
- Chen Y, Lun ATL, Smyth GK. Differential expression analysis of complex RNA-seq experiments using edgeR. In: Datta S, Nettleton DS, editors. Statistical Analysis of Next Generation Sequence Data. Springer; New York: 2014. pp. 51–74. [Google Scholar]
- Cleveland WS. Robust locally weighted regression and smoothing scatterplots. Journal of the American statistical association. 1979;74:829–836. [Google Scholar]
- Efron B, Morris C. Limiting the risk of Bayes and empirical Bayes estimators—Part II: the empirical Bayes case. Journal of the American Statistical Association. 1972;67:130–139. [Google Scholar]
- Efron B, Morris C. Stein’s estimation rule and its competitors—an empirical Bayes approach. Journal of the American Statistical Association. 1973;68:117–130. [Google Scholar]
- Gaver DP, O’;Muircheartaigh IG. Robust empirical Bayes analyses of event rates. Technometrics. 1987;29:1–15. [Google Scholar]
- Golub GH, Welsch JH. Calculation of Gauss quadrature rules. Mathematics of Computation. 1969;23:221–230. [Google Scholar]
- Good-Jacobson KL, Chen Y, Voss AK, Smyth GK, Thomas T, Tarlinton D. Regulation of germinal center responses and B-cell memory by the chromatin modifier MOZ. Proceedings of the National Academy of Sciences. 2014;111:9585–9590. doi: 10.1073/pnas.1402485111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottardo R, Raftery AE, Yee Yeung K, Bumgarner RE. Bayesian robust inference for differential gene expression in microarrays with multiple samples. Biometrics. 2006;62:10–18. doi: 10.1111/j.1541-0420.2005.00397.x. [DOI] [PubMed] [Google Scholar]
- Insua DR, Ruggeri F. Robust Bayesian Analysis. Lecture Notes in Statistics. Vol. 152. Springer; New York: 2000. [Google Scholar]
- Jeanmougin M, De Reynies A, Marisa L, Paccard C, Nuel G, Guedj M. Should we abandon the t-test in the analysis of gene expression microarray data: a comparison of variance modeling strategies. PloS ONE. 2010;5:e12336. doi: 10.1371/journal.pone.0012336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Liu XS. Analyzing ’omics data using hierarchical models. Nature Biotechnology. 2010;28:337–340. doi: 10.1038/nbt.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooperberg C, Aragaki A, Strand AD, Olson JM. Significance testing for small microarray experiments. Statistics in Medicine. 2005;24:2281–2298. doi: 10.1002/sim.2109. [DOI] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biology. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao JG, McMurry T, Berg A. Prior robust empirical Bayes inference for large-scale data by conditioning on rank with application to microarray data. Biostatistics. 2014;15:60–73. doi: 10.1093/biostatistics/kxt026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun ATL, Chen Y, Smyth GK. It’s DE-licious: a recipe for differential expression analyses of RNA-seq experiments using quasi-likelihood methods in edgeR. Methods in Molecular Biology. 2016;1418:391–416. doi: 10.1007/978-1-4939-3578-9_19. [DOI] [PubMed] [Google Scholar]
- Lun ATL, Smyth GK. diffHic: a Bioconductor package to detect differential genomic interactions in Hi-C data. BMC Bioinformatics. 2015a;16:258. doi: 10.1186/s12859-015-0683-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun ATL, Smyth GK. From reads to regions: a Bioconductor workflow to detect differential binding in ChIP-seq data. F1000Research. 2015b;4:1080. doi: 10.12688/f1000research.7016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun ATL, Smyth GK. csaw: a Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Research. 2016;44:e45. doi: 10.1093/nar/gkv1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski IJ, Blewitt ME, De Graaf CA, McManus EJ, Bahlo M, Hilton AA, Hyland CD, Smyth GK, Corbin JE, Metcalf D, et al. Polycomb repressive complex 2 (PRC2) restricts hematopoietic stem cell activity. PLOS Biology. 2008;6:e93. doi: 10.1371/journal.pbio.0060093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski IJ, Ritchie ME, Phipson B, Corbin J, Pakusch M, Ebert A, Busslinger M, Koseki H, Hu Y, Smyth GK, et al. Opposing roles of polycomb repressive complexes in hematopoietic stem and progenitor cells. Blood. 2010;116:731–739. doi: 10.1182/blood-2009-12-260760. [DOI] [PubMed] [Google Scholar]
- McCarthy DJ, Smyth GK. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics. 2009;25:765–771. doi: 10.1093/bioinformatics/btp053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris CN. Parametric empirical Bayes inference: theory and applications. Journal of the American Statistical Association. 1983;78:47–55. [Google Scholar]
- Murie C, Woody O, Lee AY, Nadon R. Comparison of small n statistical tests of differential expression applied to microarrays. BMC Bioinformatics. 2009;10:45. doi: 10.1186/1471-2105-10-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras JB, Stephens M, Gilad Y, Pritchard JK. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010a;464:768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Pai AA, Gilad Y, Pritchard JK. Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genetics. 2010b;6:e1001236. doi: 10.1371/journal.pgen.1001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartor MA, Tomlinson CR, Wesselkamper SC, Sivaganesan S, Leikauf GD, Medvedovic M. Intensity-based hierarchical Bayes method improves testing for differentially expressed genes in microarray experiments. BMC bioinformatics. 2006;7:538. doi: 10.1186/1471-2105-7-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh BN, Downer NL, Phipson B, Vanyai HK, Kueh AJ, McCarthy DJ, Smyth GK, Thomas T, Voss AK. MOZ and BMI1 play opposing roles during Hox gene activation in ES cells and in body segment identity specification in vivo. Proceedings of the National Academy of Sciences. 2015;112:5437–5442. doi: 10.1073/pnas.1422872112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Oshlack A, Smyth GK. Optimizing the noise versus bias trade-off for Illumina Whole Genome Expression BeadChips. Nucleic Acids Research. 2010;38:e204. doi: 10.1093/nar/gkq871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology. 2004;3 doi: 10.2202/1544-6115.1027. Article 3. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Statistical Modeling: The statmod package, 1.4.22. 2015 http://www.r-project.org.
- Tukey JW. The future of data analysis. Annals of Mathematical Statistics. 1962;33:1–67. [Google Scholar]
- Wright GW, Simon RM. A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics. 2003;19:2448–2455. doi: 10.1093/bioinformatics/btg345. [DOI] [PubMed] [Google Scholar]
- Zhou X, Lindsay H, Robinson MD. Robustly detecting differential expression in RNA sequencing data using observation weights. Nucleic Acids Research. 2014;42:e91. doi: 10.1093/nar/gku310. [DOI] [PMC free article] [PubMed] [Google Scholar]