

Abstract
Aims
Determine the subacute time course of mitochondria disruption, cell death, and inflammation in a rat model of unilateral motor cortical ischemic stroke.
Main Methods
Rats received unilateral ischemia of the motor cortex and were tested on behavioral tasks to determine impairments. Animals were euthanized at 24 h, 72 h and 144 h and mRNA expression of key mitochondria proteins and indicators of inflammation, apoptosis and potential regenerative processes in ipsilesion cortex and striatum, using RT-qPCR. Mitochondrial proteins were examined at 144 h using immunoblot analysis.
Key Findings
Rats with stroke induced-behavioral deficits had sustained, 144 h post-lesion, decreases in mitochondrial-encoded electron transport chain proteins NADH dehydrogenase subunit-1 and cytochrome c oxidase subunit-1 (mRNA and protein) and mitochondrial DNA content in perilesion motor and sensory cortex. Uncoupling-protein-2 gene expression, but not superoxide dismutase-2, remained elevated in ipsilateral cortex and striatum at this time. Cortical inflammatory cytokine, interleukin-6, was increased early and was followed by increased macrophage marker F4/80 after stroke. Cleaved caspase-3 activation was elevated in cortex and growth associated protein-43 was elevated in the cortex and striatum six days post-lesion.
Significance
We identified a relationship between three disrupted pathways, (1) sustained loss of mitochondrial proteins and mitochondrial DNA copy number in the cortex linked to decreased mitochondrial gene transcription; (2) early inflammatory response mediated by interleukin- 6 followed by macrophages; (3) apoptosis in conjunction with the activation of regenerative pathways. The stroke-induced spatial and temporal profiles lay the foundation to target pharmacological therapeutics to these three pathways.
Keywords: cerebral ischemia, respiratory complexes, oxidative stress, sensorimotor cortex, ET-1
INTRODUCTION
Stroke is the fourth leading cause of death and the leading cause of long-term disability in the US [1]. Annually, stroke is responsible for 130,000 deaths with an estimated cost of 34 billion dollars for medications, health care, and disability services [2, 3]. Ischemic strokes make up 87% of all strokes [4] and treatment for ischemic stroke patients is limited. Many studies have focused on neuroprotective drugs that are administered prior to or within minutes to hours after stroke. While tissue plasminogen activator (TPA) has been found to be effective at reducing stroke-induced tissue loss, TPA has a small window of effectiveness (<4hr post-stroke) and can have severe consequences in hemorrhagic patients. Finally, only 25–46% of patients arrive at the hospital within 3 hours after stroke warning signs [5–7]; therefore, there remains a need for treatment options beyond the first few hours after stroke.
Within minutes to hours after injury, apoptosis starts to occur, normally due to calcium influx and mitochondria dysfunction [8]. Degeneration of distal axons, also known as Wallerian degeneration occurs days to weeks following injury due to onset of deleterious metabolic pathways which leads to expansion of infarct size and worsening of clinical outcome. The area undergoing secondary injury that surrounds the core of the ischemic lesion is termed the penumbra and this peri-infarct tissue is clinically attractive due to the delayed onset of pathogenic mechanisms, which may be amenable to therapeutic interventions [9].
In addition to synthesizing ATP, the mitochondrion is also important in cell metabolism, calcium homeostasis, free radical production, and apoptosis [10–12]. During the secondary phase of ischemic injury, these mitochondria-dependent pathways are disrupted leading to increased reactive oxygen species, intracellular calcium and induction of pro-apoptotic cascades [8, 13, 14]. Thus, the development of pharmacological agents to promote recovery of mitochondria and ATP-dependent cellular functions may limit secondary neuronal damage in peri-infarct tissue.
Mitochondria abundance and the integrity of mitochondrial DNA (mtDNA) is disrupted following subacute brain insult [15] and is crucial for recovery of cellular function following ischemic injury [16, 17]. Therefore, the aim of this study was to examine the subacute time course of mitochondria dysregulation by examining genes that encode for respiratory chain subunits and link these mitochondria changes to common pathological pathways such as neuroinflammation and cell death, following ET-1 induced stroke in the caudal forelimb area of the sensorimotor cortex (SMC) [18]. Because previous studies have shown that rescued function of SMC is dependent on preservation of the peri-infarct motor and sensory cortex and dorsal lateral striatum, we hypothesized that disruptions in cortical and striatal mitochondrial homeostasis in concert with neuroinflammation and cell death will result in impaired behavior outcomes. There are limited studies that explore the effect of mitochondrial dysregulation in the peri-infarct cortex and less is known about mitochondria function in brain regions interconnected via neural pathways, such as the striatum following a motor cortical stroke. Thus, this study will also elucidate the pattern of damage in the striatum to better understand secondary injury following ischemic stroke.
MATERIALS AND METHODS
Animals
Long Evans male rats (n=86, 3–4 months old) received food and water ad libitum and were kept on a 12:12hr light:dark cycle. Rats were randomly assigned to one of six groups that received either a sham or stroke procedure and were euthanized at one of three time points: 24 h (sham=10; stroke=10), 72 h (sham=14; stroke=22), or 144 h (sham=10; stroke=20). All animal protocols followed the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals, and were approved by the Medical University of South Carolina Animal Care and Use Committee.
Surgical Procedures
Rats were anesthetized with ketamine (1.1mg/kg I.P.) and Xylazine (0.7mg/kg I.P.). Unilateral ischemic lesions were induced via ET-1 (American Peptide, Inc) applied to the cortical surface of the forelimb area of the SMC (Fl-SMC). Briefly, a craniotomy was performed at 1.0 mm posterior and 2.0 mm anterior to bregma and 3.0–5.0 mm lateral to midline and dura was gently retracted. ET-1 was applied on the brain surface at approximately 1ul/min, with a 2 min wait between applications using a total of 4ul. After the final 1ul of ET-1, the brain was left undisturbed for 5 min and then the craniotomy was covered with gel film (Invotec International) and dental acrylic. The stroke hemisphere was randomized. Sham animals had all procedures up to craniotomy. All animals received buprenorphine (0.5mg/kg S.C.) prior to incision for pain.
Ladder Task
To assess ischemia-induced impairments of forelimb function and compare these to mitochondria homeostasis markers, all animals were tested on the ladder task on days 0, 1, 3, and 6. The ladder task was used to assess coordinated forelimb use, stepping accuracy, and limb placement and is sensitive to motor cortex damage [19].The ladder apparatus is made of two plexiglass walls, with 3mm diameter pegs spaced 1cm apart from each other. The ladder is raised ~20cm off the ground with a neutral start cage and the animal’s home cage at the end. Through slow-motion video replay, all forelimb placements were qualitatively scored on a 0–6 rating scale over three trials (three traverses across the ladder). A perfectly placed limb received a score of 6. Errors were scored as follows: 0 = limb missed the ladder rung and the limb fell through the rungs; 1 = the limb was placed the limb but when weight bearing either fell (score of 1) or slipped (score of 2) [19]. Percent errors was calculated as: sum of errors (0+1+2)/(total steps) per test day.
Tissue Collection
Animals were deeply anesthetized with Euthasol (0.1mg/kg) and brains were removed to obtain fresh tissue punches from the ipsilesional sensory and motor cortex and the striatum. Samples were taken medial and anterior to the injury based on specific lesions and anatomical observation, no tissue was sampled that contained the lesion core. Tissue was placed on dry ice to preserve mRNA and protein levels. Samples remained in a −80 freezer until RNA isolation or western blot analysis was performed. We choose to investigate the motor and sensory cortex that did not include the lesion core because these areas are highly connected to the primary area of injury, undergo secondary degeneration, and are important for recovery of sensorimotor function following caudal forelimb injuries and thus are targets for future intervention [20–22]. Additionally, we examined the entire striatum because it is known to undergo functional plasticity following stroke and is thought to be critical to intervention [23]. Investigation of the dorsal-lateral striatum would have provided more targeted information.
RNA Isolation and Real-Time PCR
Total RNA was extracted from cortex and striatum using TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. Reverse transcription was performed using the RevertAid First Strand cDNA kit (Thermo Fisher Scientific) with 0.5–1 ug of RNA. 5 uL of cDNA template was used to amplify PCR products using 2× Maxima SYBR green qPCR master mix (Thermo Fisher Scientific). The primer sequences used in the qPCR protocol are listed in Table 1. Fold changes in mRNA expression were normalized to tubulin were calculated using the ΔΔ- Ct analysis method detailed previously by Wills et. al. [24]. Tubulin has been established to be a relevant control used to determine changes in mitochondria genes [15, 24].
Table 1. Primer pairs used for qRT-PCR.
List of rat primer pairs used in qRT-PCR.
| Gene | Primer Setuence | |
|---|---|---|
| PGC-lα | Sense Antisense |
5′-AGGAAATCCGAGCGGAGCTGA-3′ 5′-GCAAGAAGGCGACACATCGAA-3′ |
| NDUFS1 | Sense Antisense |
5′-AGATGATTTGGGAACAACGG-3′ 5′-TAAGGCTTAGAGGTTAGGGC-3′ |
| COX1 | Sense Antisense |
5′-CCTGAGCAGGAATAGTAGGG-3′ 5′-AGTGGTACAAGTCAGTTCCC-3′ |
| ND1 | Sense Antisense |
5′-TGAATCCGAGCATCCTACC-3′ 5′-ATTCCTGCTAGGAAAATTGG-3′ |
| SOD2 | Sense Antisense |
5′-CAAGGGAGATGTTACAACTCAGG-3′ 5′-CTTAGGGCTCAGGTTTGTCCA-3′ |
| UCP2 | Sense Antisense |
5′-GAGATACCAGAGCACTGTCG-3′ 5′-GCTCAGTACAGTTGACAATGG-3′ |
| F4/80 | Sense Antisense |
5′-TCCTCTTCTGGGGCTTCAGT-3′ 5′-CCATTGCTGGGCAGAAAACC-3′ |
| IL-6 | Sense Antisense |
5′-TTCAGAGCAATACTGAAACCC-3′ 5′-GATGGTCTTGGTCCTTAGCC-3′ |
| Tubulin | Sense Antisense |
5′-CTCTCTGTCGACTACGAAAG-3′ 5′-TGGTGAGGATGGAATTGTAGG-3′ |
| Actin | Sense Antisense |
5′-TAAGGAACAACCCAGCATCC-3′ 5′-CAGTGAGGCCAGGATAGAGC-3′ |
Mitochondrial DNA Content
Relative mtDNA content in rat cortex and striatum samples was measured using real-time qPCR. DNA was isolated from tissues using the DNEasy Blood and Tissue Kit (Qiagen) and 5 ng of cellular DNA was used to perform qPCR. Relative quantity of mtDNA was assessed by expression of NADH dehydrogenase 1 (ND1), a mitochondrial gene, and normalized to nuclear-encoded β-actin. Primer sequences for ND1 and β-actin were ND1 sense: 5′-TGAATCCGAGCATCCTACC-3′; ND1 antisense: 5′-ATTCCTGCTAGGAAAATTGG-3′; β-actin sense: 5′-TAAGGAACAACCCAGCATCC-3′; and β-actin antisense: 5′-CAGTGAGGCCAGGATAGAGC-3′. The ΔΔ-Ct analysis method was used to calculate fold changes in expression [24]. β-actin is used as the housekeeping gene in this study because it does not change in response to ischemic injury. Because replication and expression of mtDNA are primarily controlled by nuclear genes, β-actin has proven to be an appropriate housekeeping gene [15, 25].
Immunoblot Analysis
Rat cortex and striatum tissue was homogenized in 150 μL of protein lysis buffer and protease inhibitors (1% Triton X-100, 150 mM NaCl, 10 mM Tris-HCl, pH 7.4; 1 mM EDTA; 1 mM EGTA; 2 mM sodium orthovanadate; 0.2 mM phenylmethylsulfonyl fluoride; 1 mM HEPES, pH 7.6; 1 μg/ml leupeptin; and 1 μg/ml aprotinin) using a Polytron homogenizer. Then the samples were sonicated and centrifuged at 14,000 g for 15 min at 4°C. The supernatant was collected and protein quantified using a bicinchoninic acid kit (Sigma). Proteins (30 μg) were separated on 4 to 20% gradient SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Membranes were blocked in 5% BSA or milk in TBST (0.1% Tween 20 in 1× Tris-buffered saline) and incubated with primary antibodies overnight at 4°C. Primary antibodies used in this study included: COX1 (1:2000 Abcam); ND1 (1:2000, Abcam); NDUFS1 (1:2000, Abcam); cleaved caspase 3 (1:1000, Cell Signaling, Danvers); Caspase 3 (1:1000, Santa Cruz); GAP-43 (1:1000, Cell Signaling) and GAPDH (1:10000, Fitzgerald). After incubation for 1hr at room temperature with secondary rabbit (1:2000, Abcam) or mouse (1:20000, Abcam) antibodies conjugated with horseradish peroxidase, membranes were detected by chemiluminescence. Densitometric analysis was performed using ImageJ [26].
Statistical Analysis
All data are reported as group means with ±S.E.M. Repeated analysis of variance (rANOVA) was used to test for behavioral differences with post-hoc comparisons for each post-operative day. Single comparison of molecular data was performed using the Student t-test, whereas data found to not have a normal distribution were subjected to a Mann-Whitney U-test. Sample size was determine based on a power analysis for AVOVA with effect size equal to 0.25, α= 0.05, β= 0.8 and previous behavioral studies. Data were considered statistically significantly different at p ≤ 0.05.
RESULTS
Stroke Induced Motor Impairment
As demonstrated previously, unilateral ET-1 lesions to the (Fl-SMC) result in lasting impairments in the forelimb opposite the lesion [19]. Animals exhibited more limb placement errors while walking across a horizontal ladder with their impaired limb at 24 h, 72 h, and 144 h post-injury compared to sham animals. (Fig. 1A) There were no differences seen with the non-impaired forelimb compared to sham animals or pre-stroke number of errors. (Fig. 1B)
Fig. 1. Stroke Induced Motor Impairment.
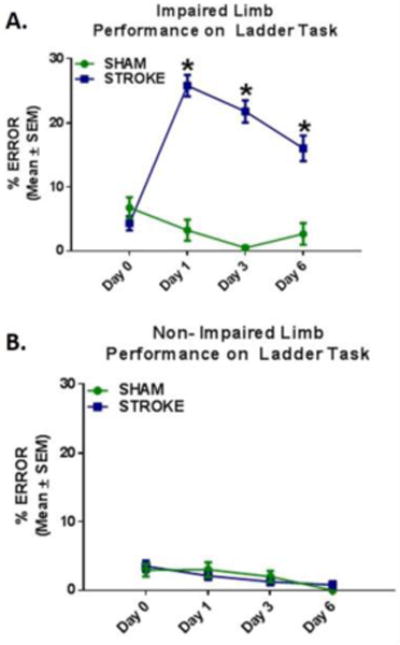
(a) Following injury, animals made significantly more errors with their impaired limb compared to sham controls. (b) There was no difference in errors made with the non-impaired forelimb between stroke and sham animals. Values reported as mean ± SEM. n ≥ 10, *p < 0.005
ET-1 Induced Stroke Disruption in Mitochondria Homeostasis
Decreased respiratory chain gene expression and mtDNA content in ipsilesional motor and sensory cortex
Mitochondrial dysfunction is a major contributor to neuronal death following ischemic stroke [11]. Therefore, we assessed mRNA expression of PGC-1α, and components of the electron transport chain, nuclear-encoded NADH dehydrogenase (ubiquinone) Fe-S protein 1 (NDUFS1), and mitochondrial-encoded cytochrome c oxidase subunit 1 (COX1) and ND1. We observed a non-significant downward trend (p= 0.056) in PGC-1α mRNA expression 24 h post-stroke. (Fig. 2A) NDUFS1 mRNA expression was decreased by 25% at 24 h and did not return to sham levels until 144 h post-stroke. (Fig. 2B) Additionally, there was a robust reduction of COX1 and ND1 transcript levels at 72 h and remained suppressed by 50% at 144 h following injury. (Fig. 2 C–D) Mitochondrial DNA copy number was assessed as a marker of mitochondrial content. There was a persistent suppression of mtDNA copy number to 74%, 71%, and 64% (24, 72, 144 h post-stroke, respectively). (Fig. 2E) Taken together, these findings reveal disruption in transcriptional regulation of mitochondrial proteins involved in oxidative phosphorylation and mitochondrial content following ET-1 induction of cerebral ischemia.
Fig. 2. Decreased respiratory chain gene expression and mtDNA content in ipsilesional motor and sensory cortex.
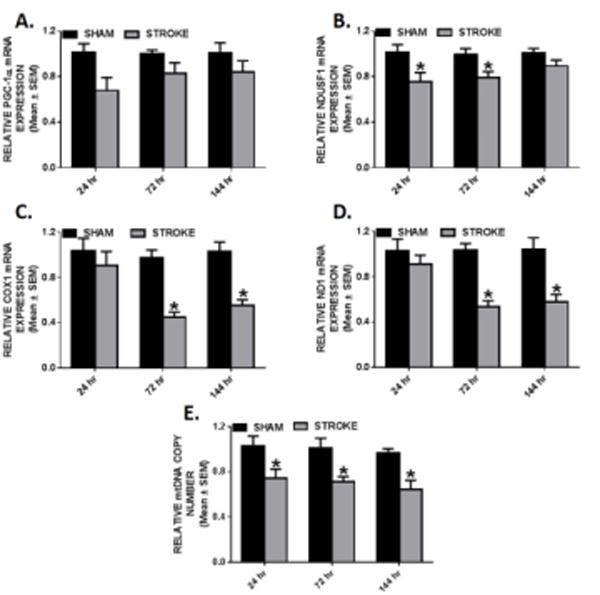
Rats were subjected to either sham or ET-1 treatment. PGC-1α (a), NDUFS1 (b) COX1 (c), and ND1 (d) mRNA expression was measured by qRT-PCR using tubulin as a control gene in stroke and sham animals. mtDNA copy number (e) was measured by qRT-PCR using ND1 for the mtDNA gene and β-actin as the nuclear control gene. These markers were measured in the ipsilesional cortex 24, 72, and 144hr. Values reported as mean ± SEM. n ≥ 10, *p < 0.05
Transitory changes of mitochondrial encoded transcripts in ipsilesional striatum
We screened for mitochondria damage in the ipsilesional striatum following ET-1 ischemic damage due to secondary injury in this area [23]. There were no changes in PGC-1α and NDUFS1 mRNA expression during the 24–144 h injury phase. (Fig. 3A–B) However, we did observe a transitory decrease in COX1 and ND1 mRNA expression at 72 h post-stroke (70% and 53%, respectively). By 144 h, COX1 and ND1 mRNA levels recovered back to sham levels. (Fig. 3C–D) At 24 h, we detected a 25% reduction of mitochondrial DNA copy number in the ipsilesional striatum which returned to sham levels at 72 h. (Fig. 3E) These results reveal that ET-1 induced SMC lesions result in subacute and transitory mitochondrial changes after stroke.
Fig. 3. Transitory changes of mitochondrial encoded transcripts in ipsilesional striatum.
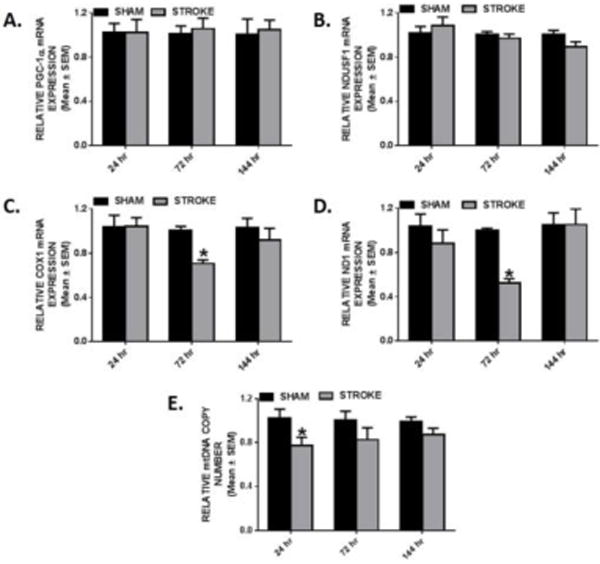
Rats were subjected to either sham or ET-1 treatment. PGC-1α (a), NDUFS1 (b), COX1(c) and ND1 (d) mRNA expression was determined by qRT-PCR using tubulin as a control gene. mtDNA copy number (e) was determined by qRT-PCR, using ND1 for the mtDNA gene and β-actin for the nuclear control gene. These markers were measured in the ipsilesional striatum 24, 72, and 144hr. Values reported as mean ± SEM. n ≥ 10, *p < 0.05
Reduced mitochondrial encoded protein expression in the ipsilesional cortex and striatum
To assess if ET-1 disrupts the translation of mitochondrial genes we also measured protein levels of COX1, ND1, and NDUFS1 at 144 h to determine if translation is chronically reduced. Immunoblot analysis revealed that at 144 h, COX1 and ND1 were depressed to 67% and 76% compared to sham levels in the ipsilesional cortex. (Fig. 4A) COX1 and ND1 protein expression were also decreased in the ipsilesional striatum to 80% and 77%. (Fig. 4B) NDUFS1 remained at sham control levels at this 144 h time point. These data reveal that in our model of cerebral ischemia mitochondrial-encoded genes are more sensitive to transcriptional/translational disruption than nuclear-encoded genes.
Fig. 4. Reduced mitochondrial encoded protein expression in the ipsilesional cortex and striatum.
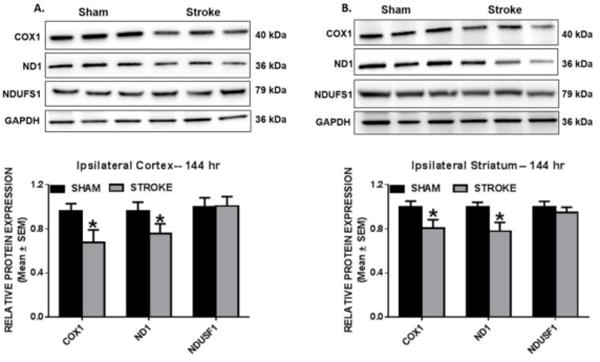
Rats were subjected to either sham or ET-1 treatment. COX1, ND1, and NDUFS1 protein expression was determined by immunoblot analysis. These markers were measured in protein isolated from ipsilesional cortex (a) and striatum (b) 144hr following ET-1 exposure. GAPDH is used as loading control. Values reported as mean ± SEM. n =6–9, *p < 0.05
UCP2 activation in the ipsilesional cortex and striatum
Excessive reactive oxygen species (ROS) generation can induce the up regulation of mitochondrial ROS detoxifying enzymes, manganese superoxide dismutase 2 (SOD2) and the uncoupling protein 2 (UCP2) to neutralize the detrimental effects initiated by oxidative stress. Along with its uncoupling role to mitigate ROS production, UCP2 has been implicated in neuroprotection by suppressing pro- inflammatory cytokines and elevating anti- apoptotic mediator, Bcl2 [27]. Therefore, we measured SOD2 and UCP2 in ipsilesional cortex and striatum. Interestingly, we only observed a modest 17% decrease of SOD2 transcript at 72 h in the cortex and SOD2 mRNA levels remained unchanged in the striatum. (Fig. 5A, C) In contrast, cortical UCP2 transcript levels increased by ~1.5-fold within 24hr and further increased to ~3-fold by 72 h which persisted to 144 h. (Fig. 5B) In the ipsilesional striatum, UCP2 increased 1.5-fold over sham control at 72 h and remained elevated at 144 h. (Fig. 5D) These data reveal that ET-1 mediates sustained increases in UCP2 in cortex and striatum.
Fig. 5. UCP2 activation in the ipsilesional cortex and striatum.
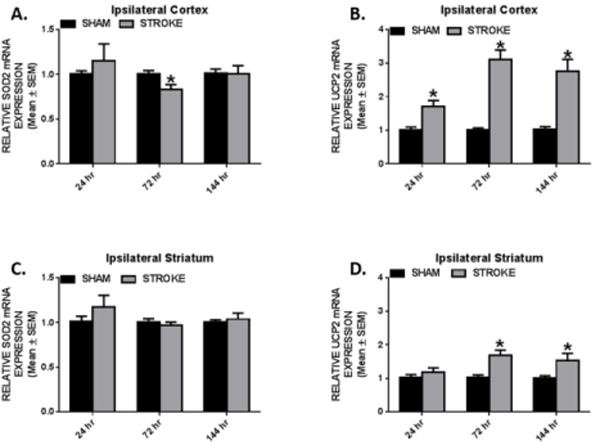
Rats were subjected to either sham or ET-1 treatment. Cortical (a,b) and striatal (c,d) SOD2 and UCP2 mRNA expression was determined by qRT-PCR using tubulin as a control gene. These markers were measured 24, 72, and 144hr following ET-1 exposure. Values reported as mean ± SEM. n ≥ 10, *p < 0.05
ET-1 induced stroke inflammation
Gene expression of inflammatory mediators and macrophages in the ipsilesional cortex and striatum
Since macrophages are known to be involved in innate immune response following ischemic injury [28], we examined the transcriptional expression of F4/80, a surface marker for mature macrophages. We observed a 5-fold increase in cortical F4/80 mRNA expression over sham animals at 72 and 144 h. (Fig. 6A) Additionally, we observed significant 3-, 2- and 1.5-fold increases in cortical interleukin-6 (IL-6) transcripts at 24, 72, and 144 h, respectively. (Fig. 6B) We detected a transitory 1.5-fold increase in F4/80 at 72 h that returned to sham levels by 144 h in the striatum. (Fig. 6C) IL-6 induced 2.5-fold at 24 h and reduced to 1.5-fold increase 72 h in the striatum. (Fig. 6D) Our data demonstrate that mitochondrial changes are occurring in peri-infarct tissue that is undergoing inflammatory responses over the 144 h examined. The pro-inflammatory environment in both the sensorimotor cortex and striatum further demonstrate continued stroke induced pathology in connected brain regions outside the primary stroke region and likely contribute mitochondria disruption.
Fig. 6. Gene expression of inflammatory mediators and macrophages in the ipsilesional cortex and striatum.
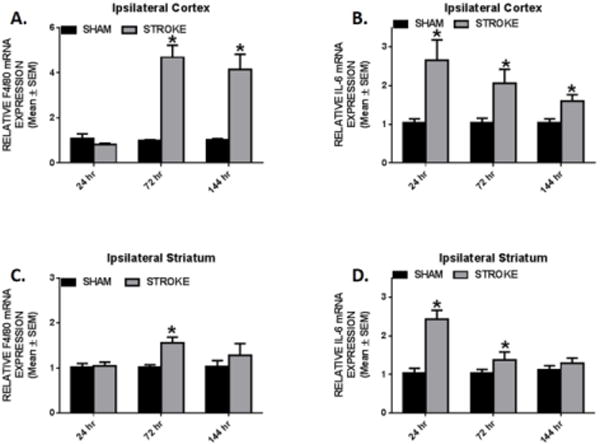
Rats were subjected to either sham or ET-1 treatment. Cortical (a,b) and striatal (c,d) F4/80 and IL6 mRNA expression was determined by qRT-PCR using tubulin as a control gene. These markers were measured 24, 72, and 144hr following ET-1 exposure. Values reported as mean ± SEM. n ≥ 10, *p < 0.05
ET-1 Induced Stroke Cellular Degeneration and Regeneration
Caspase 3 cleavage and GAP43 expression in ipsilateral cortex and striatum
Rodent models of neurodegenerative diseases implicate caspase dependent apoptosis as an important contributor to neuronal and tissue damage [29–32]. Immunoblot analysis was used to assess the level of caspase 3 activation by measuring the cleaved form of caspase 3 following ET-1 induced stroke. Cleaved caspase 3 was detected in the ipsilateral cortex, but not in the ipsilateral striatum 144 h following stroke. (Fig. 7A, 7B) These data demonstrate that there is a range of pathological effects on brain regions neuronally connected to the primary site of stroke and that direct cell death, indicated by increased activation of cleaved caspase 3, may be the consequence of mitochondria disruption.
Fig. 7. Caspase 3 cleavage and GAP43 expression in ipsilateral cortex and striatum.
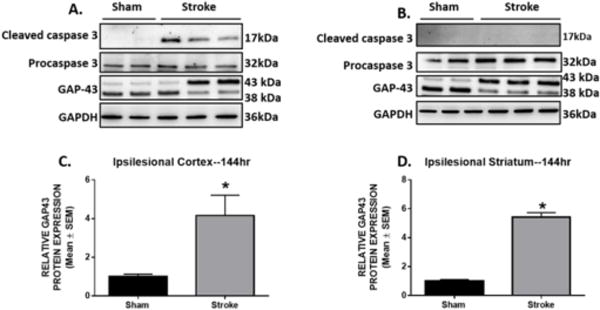
Rats were subjected to either sham or ET-1 surgeries. Cleaved caspase 3, procaspase 3, and GAP-43 protein expression was determined by immunoblot analysis. Protein was isolated from ipsilesional cortex (a) and striatum (b) 144hr post- injury. Cortical (c) and striatal (d) GAP-43 protein expression were quantified. GAPDH is used as loading control. Values reported as mean ± SEM. n= 5–6, *p < 0.05
Sensorimotor cortex and striatum are key areas that undergo neural regrowth and reorganization after following motor cortical damage [23, 33]. We measured axonal regeneration and outgrowth of growth-associated protein 43 (GAP-43). GAP-43 increases during periods of axonal sprouting and within the first week post- stroke in areas surrounding the primary site of injury [34, 35]. In line with previous reports, we observed a 3-fold increase in GAP-43 protein expression in the ipsilesional cortex at 144 h (Fig. 7C). We also detected a 4-fold increase in the striatum at 144 h post–stroke (Fig. 7D). These data reveal that sufficient cell injury occurs in the early stages post-stroke to activate regenerative processes and that this process occurs when mitochondria homeostasis is still disrupted in both striatum and cortex.
DISCUSSION
Several studies have demonstrated mitochondrial dysfunction as a common consequence of cerebral ischemia [12, 36, 37]. Disrupted mitochondrial homeostasis has been linked to detrimental pathways such as ROS production, calcium dysregulation, inflammation, and intrinsic apoptotic cell death [38–41]. These complex pathophysiological pathways have been intimately described following ischemic stroke and it is hypothesized that mitochondria are crucial initiators and targets of these diverse inflammatory and aberrant metabolic pathways. In addition, it is possible but, untested, that mitochondrial dysfunction may be related to reduced neuronal function, altering sensory and behavioral outcomes following stroke. Many of these effects occur immediately post-injury; therefore, elucidation of the pathways linked to mitochondrial dysfunction following insult and over time may provide targets for future pharmacological intervention.
We report mitochondrial disruption, activation of inflammatory pathways, cell death and initiation of regenerative pathways within the first week of ischemic stroke in our focal ET-1 induced rat stroke model. Endothelin-1 injection induces reliable, reproducible, and focal ischemic lesions that mimic the subacute onset of human ischemia, with reduced blood flow in the cortex for 16 h and in the striatum for 7 h demonstrated by Biernaskie et al [42, 43]. Furthermore, ET1- induced strokes produce neuroanatomical focal infarcts in a dose dependent manner. ET-1 is a good model to examine pathway disruptions and ultimately to test potential therapeutics for treatment of stroke. However, as with any stroke model there are limitations. There are ET-1 receptors located on both neurons and glia, and application of ET-1 can alter cell morphology and interfere with the interpretation of neural repair experiments through receptor- mediated signaling effects [44, 45]. We chose to examine the motor cortex and striatum following a sensorimotor lesion to better understand which pathways underwent disruption post stroke, however because we did not examine the effects on individual cell types our study is limited to understanding global effects of ischemia on mitochondria function.
In the cortex, we observed decreased mRNA and protein expression of nuclear and mitochondrial encoded subunits of complex I and IV as well as depletion of mitochondrial DNA number. Concomitantly, mitochondrial DNA copy number was suppressed early and persistently in the ipsilesional cortex. We only observed reduced protein expression in mitochondrial-encoded proteins which may be due to the increased sensitivity of mtDNA damage compared to nuclear DNA. Following ischemic injury, chronic ROS production causes extensive mtDNA damage and the inability to monitor and repair mtDNA damage is due to the less efficient mitochondrial DNA repair mechanisms that become overwhelmed in the presence of excessive oxidative stress [46]. It is expected that mtDNA damage may lead to inactive electron transport chain activity, thereby affecting normal mitochondrial function.
Our study identified mitochondrial deficits in a secondary site of injury, the striatum. Here, we show decreased striatal mitochondrial content, as measured by mtDNA copy number, mtDNA transcripts and protein. We report acute and transient reduction of mitochondrial DNA and mRNA expression of ND1 and COX1 in the striatum. While the biochemical and cellular mechanisms that comprise secondary injury are not entirely understood, we hypothesize that the disturbance in mitochondrial DNA and gene regulation is based on the functional connectively between the cortex and striatum. The striatal changes are transient, with mtDNA and mitochondrial RNA changes recovering to control levels by 72 h and 144 h, respectively, suggesting the damage to the striatum is less severe compared to the primary site of injury and is recoverable. These studies require further investigation into the role of these cellular stress responses and the dynamic interplay of multiple pathophysiological cascades in secondary injury, that go beyond both the initial ischemic insult and the disruption of cerebral circulation [47].
It is important to note that although we do not report a significant decrease in PGC-1α mRNA expression in either the cortex or striatum, we hypothesize that the PGC-1α transcript levels are reduced abruptly following injury and recover rapidly to control levels to regulate compensatory mechanisms such as antioxidant production. Under normal physiological conditions, mitochondrial respiratory complexes are a source of ROS generation [11]. When respiratory enzymes are damaged following injury, these complexes produce an excessive amount of ROS which can lead to protein, lipid, and DNA damage, subsequently contributing to mitochondrial damage and dysfunction. Therefore, we assessed mRNA level of the mitochondrial antioxidant gene, SOD2 as a marker of oxidative stress. Surprisingly, we only observed a decrease in SOD2 transcript at 72 h post-stroke in the cortex, which recovered to sham control levels by 144 h and SOD2 mRNA levels remained unchanged in the striatum. Numerous studies have reported SOD2 is a direct downstream target of PGC-1α [48, 49]; therefore, we predict that SOD2 mRNA levels transiently decreased in response to the modest suppression of PGC-1α in the cortex at 24 h.
Recently, an increasing number of studies have focused on the various physiological and pathological roles of UCP2 [50–53], which has been described to have a role in mediating lipid peroxidation within the mitochondrial matrix due to ROS formation. When UCP2 detects elevated levels of mitochondrial ROS, a feedback loop is activated to induce UCP2 expression in the inner mitochondrial membrane [53]. It is well documented that the primary role of UCP2 is to dissipate the proton gradient across the inner membrane to prevent ATP synthesis and transport superoxide radicals across the inner mitochondrial membrane, thus decreasing the effect of ROS produced by the respiratory chain [51]. Interestingly, UCP2 mRNA expression was persistently increased in both the ispilesional cortex and striatum in our study. UCP2 has also been reported to perform other functions such as regulating neuroinflammation and apoptosis following ischemic stroke. Haines, et al., demonstrated that the overexpression of UCP2 alleviated the ischemia-induced increase in IL-6 mRNA which may reduce the deleterious effects of prolonged inflammation. Overexpression of UCP2 also rescued diminished pro-survival markers such as Bcl-2, cyclin G2, and HSP90 [27]. Furthermore, UCP2 has been documented to be neuroprotective via its involvement in neurogenesis and synaptogenesis, suggesting a role in neuronal growth and development [53, 54]. Future studies are needed to explore the potential roles of UCP2 in our cerebral ischemic model.
Consistent with cortical mitochondrial deficits, the presence of neuroinflammation during the first days of experimental stroke was confirmed by the elevated mRNA expression of macrophage marker, F4/80 and inflammatory mediator cytokine, IL-6. Interestingly, cortical IL-6 transcript levels were induced earlier than F4/80, with a 3-fold increase in expression that remained persistently elevated above sham control levels. Furthermore, the maximal expression of F4/80 and IL-6 in the cortex corresponds to the maximal expression of both transcripts in the striatum, suggesting that neuroinflammation may have a greater role in development of secondary damage post-stroke than previously anticipated. Elevated macrophage and cytokine markers, suggest that cortical and striatal compensatory pathways are insufficient to blunt neuroinflammatory responses following ischemic stroke. This may be due to the potentiation of the activation of cellular injury cascade by prolonged mitochondrial dysregulation in the cortex. Together, these findings support the implication that ischemic injury causes detrimental effects in surrounding tissue days after the initial stroke; thus, these data suggest that supporting or improving mitochondria homeostasis and function in peri-infarct tissue could widen the window of effective pharmacological treatment days after stroke.
Finally, we suggest that cell death in peri-infarct cortex is, in part, a consequence of mitochondrial dysfunction as evidenced by caspase 3 cleavage in the cortex. One of the most common forms of cell death in neurodegeneration is through the intrinsic mitochondrial apoptotic pathway. Following initiation of the intrinsic pathway, cytochrome c is released from the mitochondria and works with other apoptotic factors to process the inactive form of procaspase 3 to the cleaved, active form [55]. Cleaved caspase 3 in turn induces cellular changes including chromatin condensation, DNA fragmentation, and formation of apoptotic bodies, leading finally to cell death [56, 57]. Our study revealed that cerebral ischemia induced caspase 3 cleavage in the peri-infarct region of ipsilateral cortex 144 h post-stroke, in concert with increases in GAP-43 in both cortex and striatum. GAP-43 is associated with neurite outgrowth, axon growth cone formation and axonal sprouting post -injury and may play an important role in experience-dependent plasticity [58, 59]. After onset of stroke, one of the most prominent regenerative events is axonal sprouting in the penumbra, which is accompanied by expression of GAP- 43 [60, 61].
Additionally, GAP-43 is a recently discovered postsynaptic substrate of caspase 3 and caspase 3 has also been demonstrated to be involved in the formation of new synaptic contacts; therefore, it has been hypothesized that caspase 3 and GAP- 43 are part of a common molecular pathway involved in tissue response after stroke [62, 63]. Our data are consistent with this hypothesis as evidenced by caspase 3 cleavage and increased GAP-43 protein expression in our model of cerebral ischemia, which demonstrates that sufficient cell injury occurs in the early stages post-stroke to activate regenerative processes needed to preserve and restore neuronal function.
Conclusion
In conclusion, ET-1-induced focal experimental stroke to the SMC leads to mitochondrial dysregulation, inflammatory cell infiltration, and cell death during the first week of injury which lasts for days following initial injury in peri-infarct cortex and striatum. Further, mitochondrial disruptions occur in accordance with marked motor impairment in the ladder task, which assesses both cortical and striatal injury. We also observed transient alterations in the pathways in the striatum, a secondary site of damage. The presence of mitochondrial suppression in the cortex and striatum depicts how pathogenic mechanisms can affect adjacent cells, intensifying the damaging effects of ischemic stroke. Given the significance of mitochondria in regulation of neuronal function and survival, we propose that more studies are warranted to explore the relationship between mitochondrial function and behavior outcomes following stroke. Future experiments aim to identify therapeutics to treat ischemic stroke by reduction of mitochondrial dysfunction, bioenergetics failure, and inflammation.
Acknowledgments
The authors would like to thank Elyse Clayton for laboratory support and behavioral assistance.
Funding: This study was funded by MUSC Neuroscience Institute Pilot Grant (D.L.A), National Institutes of Health National Institute of General Medical Sciences: GM084147 (R.G.S), the Biomedical Laboratory Research and Development Program of the Department of Veterans Affairs: BX-000851 (R.G.S.), Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health: P20GM12345 (D.L.A), and the National Institutes of Health Ruth L. Kirschstein National Research Service Award: Grant 5T32-DK083262 (W.S.G.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contribution Statement
Whitney Gibbs and Rachel Weber were responsible for running the experiments and analyzing the data. Dr. DeAnna Adkins and Dr. Rick Schnellmann were responsible for the overall direction of the project, supervising progress, and interpreting the experiments. All authors wrote the manuscript.
Conflict of Interest: Whitney Gibbs, Rachel Weber, Rick Schnellman, and DeAnna Adkins declare no conflict of interest.
References
- 1.Towfighi A, Saver JL. Stroke declines from third to fourth leading cause of death in the United States: historical perspective and challenges ahead. Stroke. 2011;42(8):2351–5. doi: 10.1161/STROKEAHA.111.621904. [DOI] [PubMed] [Google Scholar]
- 2.Mozaffarian D, et al. Heart disease and stroke statistics–2015 update: a report from the American Heart Association. Circulation. 2015;131(4):e29–322. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 3.Go AS, et al. Executive summary: heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation. 2013;127(1):143–52. doi: 10.1161/CIR.0b013e318282ab8f. [DOI] [PubMed] [Google Scholar]
- 4.Wang X, et al. White matter microstructure changes induced by motor skill learning utilizing a body machine interface. Neuroimage. 2013;88C:32–40. doi: 10.1016/j.neuroimage.2013.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kothari R, et al. Acute stroke: delays to presentation and emergency department evaluation. Ann Emerg Med. 1999;33(1):3–8. doi: 10.1016/s0196-0644(99)70431-2. [DOI] [PubMed] [Google Scholar]
- 6.Lacy CR, et al. Delay in presentation and evaluation for acute stroke: Stroke Time Registry for Outcomes Knowledge and Epidemiology (S.T.R.O.K.E.) Stroke. 2001;32(1):63–9. doi: 10.1161/01.str.32.1.63. [DOI] [PubMed] [Google Scholar]
- 7.Williams LS, et al. Stroke patients’ knowledge of stroke. Influence on time to presentation. Stroke. 1997;28(5):912–5. doi: 10.1161/01.str.28.5.912. [DOI] [PubMed] [Google Scholar]
- 8.Duchen MR. Mitochondria and calcium: from cell signalling to cell death. J Physiol. 2000;529(Pt 1):57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borgens RB, Liu-Snyder P. Understanding secondary injury. Q Rev Biol. 2012;87(2):89–127. doi: 10.1086/665457. [DOI] [PubMed] [Google Scholar]
- 10.Niizuma K, Endo H, Chan PH. Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J Neurochem. 2009;109(Suppl 1):133–8. doi: 10.1111/j.1471-4159.2009.05897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bayir H, V, Kagan E. Bench-to-bedside review: Mitochondrial injury, oxidative stress and apoptosis–there is nothing more practical than a good theory. Crit Care. 2008;12(1):206. doi: 10.1186/cc6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoppins S. The regulation of mitochondrial dynamics. Curr Opin Cell Biol. 2014;29:46–52. doi: 10.1016/j.ceb.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 13.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–95. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 14.Franklin JL. Redox regulation of the intrinsic pathway in neuronal apoptosis. Antioxid Redox Signal. 2011;14(8):1437–48. doi: 10.1089/ars.2010.3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harmon JL, et al. Striatal Mitochondrial Disruption following Severe Traumatic Brain Injury. J Neurotrauma. 2016 doi: 10.1089/neu.2015.4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stallons LJ, Whitaker RM, Schnellmann RG. Suppressed mitochondrial biogenesis in folic acid-induced acute kidney injury and early fibrosis. Toxicol Lett. 2014;224(3):326–32. doi: 10.1016/j.toxlet.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith JA, et al. Suppression of mitochondrial biogenesis through toll-like receptor 4-dependent mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signaling in endotoxin-induced acute kidney injury. J Pharmacol Exp Ther. 2015;352(2):346–57. doi: 10.1124/jpet.114.221085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adkins DL, Voorhies AC, Jones TA. Behavioral and neuroplastic effects of focal endothelin-1 induced sensorimotor cortex lesions. Neuroscience. 2004;128(3):473–86. doi: 10.1016/j.neuroscience.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 19.Metz GA, I, Whishaw Q. The ladder rung walking task: a scoring system and its practical application. J Vis Exp. 2009;(28) doi: 10.3791/1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nudo RJ. Recovery after brain injury: mechanisms and principles. Front Hum Neurosci. 2013;7:887. doi: 10.3389/fnhum.2013.00887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones TA, et al. Motor skills training enhances lesion-induced structural plasticity in the motor cortex of adult rats. J Neurosci. 1999;19(22):10153–63. doi: 10.1523/JNEUROSCI.19-22-10153.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carmichael ST. Plasticity of cortical projections after stroke. Neuroscientist. 2003;9(1):64–75. doi: 10.1177/1073858402239592. [DOI] [PubMed] [Google Scholar]
- 23.Carmichael ST, Chesselet MF. Synchronous neuronal activity is a signal for axonal sprouting after cortical lesions in the adult. J Neurosci. 2002;22(14):6062–70. doi: 10.1523/JNEUROSCI.22-14-06062.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wills LP, et al. The beta2-adrenoceptor agonist formoterol stimulates mitochondrial biogenesis. J Pharmacol Exp Ther. 2012;342(1):106–18. doi: 10.1124/jpet.112.191528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garrett SM, et al. Agonism of the 5-hydroxytryptamine 1F receptor promotes mitochondrial biogenesis and recovery from acute kidney injury. J Pharmacol Exp Ther. 2014;350(2):257–64. doi: 10.1124/jpet.114.214700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haines B, Li PA. Overexpression of mitochondrial uncoupling protein 2 inhibits inflammatory cytokines and activates cell survival factors after cerebral ischemia. PLoS One. 2012;7(2):e31739. doi: 10.1371/journal.pone.0031739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87(5):779–89. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen M, et al. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med. 2000;6(7):797–801. doi: 10.1038/77528. [DOI] [PubMed] [Google Scholar]
- 30.Ferrer I, et al. Caspase-dependent and caspase-independent signalling of apoptosis in the penumbra following middle cerebral artery occlusion in the adult rat. Neuropathol Appl Neurobiol. 2003;29(5):472–81. doi: 10.1046/j.1365-2990.2003.00485.x. [DOI] [PubMed] [Google Scholar]
- 31.Li M, et al. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science. 2000;288(5464):335–9. doi: 10.1126/science.288.5464.335. [DOI] [PubMed] [Google Scholar]
- 32.Louneva N, et al. Caspase-3 is enriched in postsynaptic densities and increased in Alzheimer’s disease. Am J Pathol. 2008;173(5):1488–95. doi: 10.2353/ajpath.2008.080434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones TAaA DA. Brain Reorganization after Stroke - Stimulating and Training towards Perfection. Journal of Neurophysiology. doi: 10.1152/physiol.00014.2015. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stroemer RP, Kent TA, Hulsebosch CE. Neocortical neural sprouting, synaptogenesis, and behavioral recovery after neocortical infarction in rats. Stroke. 1995;26(11):2135–44. doi: 10.1161/01.str.26.11.2135. [DOI] [PubMed] [Google Scholar]
- 35.Benowitz LI, Carmichael ST. Promoting axonal rewiring to improve outcome after stroke. Neurobiol Dis. 2010;37(2):259–66. doi: 10.1016/j.nbd.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sims NR, Anderson MF. Mitochondrial contributions to tissue damage in stroke. Neurochem Int. 2002;40(6):511–26. doi: 10.1016/s0197-0186(01)00122-x. [DOI] [PubMed] [Google Scholar]
- 37.Anderson MF, Sims NR. Mitochondrial respiratory function and cell death in focal cerebral ischemia. J Neurochem. 1999;73(3):1189–99. doi: 10.1046/j.1471-4159.1999.0731189.x. [DOI] [PubMed] [Google Scholar]
- 38.Chan PH. Mitochondrial dysfunction and oxidative stress as determinants of cell death/survival in stroke. Ann N Y Acad Sci. 2005;1042:203–9. doi: 10.1196/annals.1338.022. [DOI] [PubMed] [Google Scholar]
- 39.Sciamanna MA, et al. Ischemic injury to rat forebrain mitochondria and cellular calcium homeostasis. Biochim Biophys Acta. 1992;1134(3):223–32. doi: 10.1016/0167-4889(92)90180-j. [DOI] [PubMed] [Google Scholar]
- 40.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22(9):391–7. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 41.Sims NR, Muyderman H. Mitochondria, oxidative metabolism and cell death in stroke. Biochim Biophys Acta. 2010;1802(1):80–91. doi: 10.1016/j.bbadis.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 42.Biernaskie J, et al. A serial MR study of cerebral blood flow changes and lesion development following endothelin-1-induced ischemia in rats. Magn Reson Med. 2001;46(4):827–30. doi: 10.1002/mrm.1263. [DOI] [PubMed] [Google Scholar]
- 43.Marchal G, Young AR, Baron JC. Early postischemic hyperperfusion: pathophysiologic insights from positron emission tomography. J Cereb Blood Flow Metab. 1999;19(5):467–82. doi: 10.1097/00004647-199905000-00001. [DOI] [PubMed] [Google Scholar]
- 44.Carmichael ST. Rodent models of focal stroke: size, mechanism, and purpose. NeuroRx. 2005;2(3):396–409. doi: 10.1602/neurorx.2.3.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakagomi S, Kiryu-Seo S, Kiyama H. Endothelin-converting enzymes and endothelin receptor B messenger RNAs are expressed in different neural cell species and these messenger RNAs are coordinately induced in neurons and astrocytes respectively following nerve injury. Neuroscience. 2000;101(2):441–9. doi: 10.1016/s0306-4522(00)00345-6. [DOI] [PubMed] [Google Scholar]
- 46.Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A. 1997;94(2):514–9. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Veenith T, Goon S, Burnstein RM. Molecular mechanisms of traumatic brain injury: the missing link in management. World J Emerg Surg. 2009;4:7. doi: 10.1186/1749-7922-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiong W, et al. NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage. J Clin Invest. 2015;125(4):1433–45. doi: 10.1172/JCI79735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marmolino D, et al. PGC-1alpha down-regulation affects the antioxidant response in Friedreich’s ataxia. PLoS One. 2010;5(4):e10025. doi: 10.1371/journal.pone.0010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wojtczak L, et al. Inhibition by purine nucleotides of the release of reactive oxygen species from muscle mitochondria: indication for a function of uncoupling proteins as superoxide anion transporters. Biochem Biophys Res Commun. 2011;407(4):772–6. doi: 10.1016/j.bbrc.2011.03.098. [DOI] [PubMed] [Google Scholar]
- 51.Brand MD, Esteves TC. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2005;2(2):85–93. doi: 10.1016/j.cmet.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 52.Chen SD, et al. Effects of rosiglitazone on global ischemia-induced hippocampal injury and expression of mitochondrial uncoupling protein 2. Biochem Biophys Res Commun. 2006;351(1):198–203. doi: 10.1016/j.bbrc.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 53.Dietrich MO, Andrews ZB, Horvath TL. Exercise-induced synaptogenesis in the hippocampus is dependent on UCP2-regulated mitochondrial adaptation. J Neurosci. 2008;28(42):10766–71. doi: 10.1523/JNEUROSCI.2744-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Simon-Areces J, et al. UCP2 induced by natural birth regulates neuronal differentiation of the hippocampus and related adult behavior. PLoS One. 2012;7(8):e42911. doi: 10.1371/journal.pone.0042911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vila M, Przedborski S. Targeting programmed cell death in neurodegenerative diseases. Nat Rev Neurosci. 2003;4(5):365–75. doi: 10.1038/nrn1100. [DOI] [PubMed] [Google Scholar]
- 56.Galluzzi L, et al. Targeting post-mitochondrial effectors of apoptosis for neuroprotection. Biochim Biophys Acta. 2009;1787(5):402–13. doi: 10.1016/j.bbabio.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 57.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407(6805):770–6. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 58.Benowitz LI, Routtenberg A. GAP-43: an intrinsic determinant of neuronal development and plasticity. Trends Neurosci. 1997;20(2):84–91. doi: 10.1016/s0166-2236(96)10072-2. [DOI] [PubMed] [Google Scholar]
- 59.Carmichael ST. Emergent properties of neural repair: elemental biology to therapeutic concepts. Ann Neurol. 2016;79(6):895–906. doi: 10.1002/ana.24653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goto S, et al. Increased expression of growth-associated protein GAP-43/B-50 following cerebral hemitransection or striatal ischemic injury in the substantia nigra of adult rats. Brain Res. 1994;647(2):333–9. doi: 10.1016/0006-8993(94)91332-3. [DOI] [PubMed] [Google Scholar]
- 61.Carmichael ST, et al. Growth-associated gene expression after stroke: evidence for a growth-promoting region in peri-infarct cortex. Exp Neurol. 2005;193(2):291–311. doi: 10.1016/j.expneurol.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 62.Han MH, et al. The novel caspase-3 substrate Gap43 is involved in AMPA receptor endocytosis and long-term depression. Mol Cell Proteomics. 2013;12(12):3719–31. doi: 10.1074/mcp.M113.030676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang JY, et al. Caspase-3 cleavage of dishevelled induces elimination of postsynaptic structures. Dev Cell. 2014;28(6):670–84. doi: 10.1016/j.devcel.2014.02.009. [DOI] [PubMed] [Google Scholar]